
Abstract
Isoprene is emitted from the biosphere into the atmosphere, and may strengthen the defense mechanisms of plants against oxidative and thermal stress. Once in the atmosphere, isoprene is rapidly oxidized, either to isoprene-hydroxy-hydroperoxides (ISOPOOH) at low levels of nitrogen oxides, or to methyl vinyl ketone (MVK) and methacrolein at high levels. Here we combine uptake rates and deposition velocities that we obtained in laboratory experiments with observations in natural forests to show that 1,2-ISOPOOH deposits rapidly into poplar leaves. There, it is converted first to cytotoxic MVK and then most probably through alkenal/one oxidoreductase (AOR) to less toxic methyl ethyl ketone (MEK). This detoxification process is potentially significant globally because AOR enzymes are ubiquitous in terrestrial plants. Our simulations with a global chemistry-transport model suggest that around 6.5 Tg yr−1 of MEK are re-emitted to the atmosphere. This is the single largest MEK source presently known, and recycles 1.5% of the original isoprene flux. Eddy covariance flux measurements of isoprene and MEK over different forest ecosystems confirm that MEK emissions can reach 1–2% those of isoprene. We suggest that detoxification processes in plants are one of the most important sources of oxidized volatile organic compounds in the atmosphere.
Similar content being viewed by others

Introduction
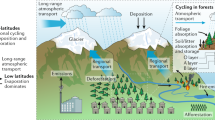
Biogenic volatile organic compounds (BVOC) are thought to account for 90% of the total VOC emission into the Earth’s atmosphere1. Isoprene (C5H8) is the dominant BVOC with an estimated annual flux of 350–800 Tg yr−1 2. Despite this large flux (mainly from broadleaf tree species) the question of why plants emit isoprene is still not fully understood3. Recent hypotheses suggest that isoprene acts as a signaling molecule that, by altering gene expressions, strengthens plant defense mechanisms against oxidative and thermal stress4. Once in the atmosphere isoprene reacts rapidly with OH radicals, which account for 90% of its total sink5. Under typical daytime conditions isoprene is thus converted to oxidized VOC (OVOCs) within 1–2 h6. OH preferentially adds to the terminal carbons of isoprene, forming allyl radicals that in collisions with oxygen react to form isoprene peroxy radicals HO–C5H8O2 (ISOPOO). Subsequent oxidative steps depend critically on the ambient abundance of NOx (NO + NO2). At elevated NOx, ISOPOO predominantly reacts with NO to form methyl vinyl ketone (MVK, yield 30–45%), methacrolein (MACR, yield 20–30%), and isoprene hydroxy nitrate (IHN, yield 13%) plus formaldehyde (HCHO) as the major products7. Under low nitrogen oxide (NOx) conditions the main product of ISOPOO reacting with HO2 is isoprene hydroxy hydroperoxide (ISOPOOH), with the 1,2-ISOPOOH isomer formed in highest yield5. Recently, Nguyen et al.8 applied the eddy covariance (EC) technique to quantify the rapid dry deposition of ISOPOOH and other oxidized BVOC to a temperate forest. They determined a daytime mean deposition velocity (vd) for ISOPOOH + IEPOX of 2.5 cm s−1. Isoprene epoxydiol (IEPOX) is an isomeric photooxidation product of ISOPOOH + OH. ISOPOOH + IEPOX deposition velocities (vd) were then compared with a resistance model described elsewhere9,10 to evaluate whether ISOPOOH + IEPOX is primarily lost to the leaf surface or taken up by the plants through their stomata. The calculated Henry’s law constant for ISOPOOH + IEPOX suggests a very small residual resistance favoring efficient uptake to any liquid surfaces8. However, the vd for ISOPOOH + IEPOX measured by Nguyen et al.8 was too close to both the upper limit for stomata-controlled resistance and the upper limit for deposition without surface resistance to determine which mechanism is dominant. Thus, while direct EC flux measurements quantify the deposition of chemical species to the canopy, the fate of species at the leaf level—and thus any associated ecological impact—cannot be evaluated in this way. In the present study, we combine uptake rates and deposition velocities for 1,2-ISOPOOH and MVK obtained in laboratory experiments on gray poplars (Populus × canescens) with field observations using the EC technique in natural forest settings. Furthermore, we quantify the gene expression and enzyme activity of the detoxifying NADPH-dependent enzyme alkenal/one oxidoreductase (AOR) in poplar leaves and demonstrate the worldwide dissemination of AOR genes across terrestrial plants. Finally, we apply chemistry-transport modeling to assess atmospheric implications of our findings (see “Methods” section for details). This enables the first leaf-to-global scale description of biosphere–atmosphere exchange for major isoprene photooxidation products, as summarized in Fig. 1.

Under low nitrogen oxide (NOx) conditions 1,2-ISOPOOH is preferentially formed, producing epoxides that then react with OH and contribute to aerosol formation74. At high NOx the production of carbonyls such as methyl vinyl ketone (MVK) supports ozone formation5 (panel a). Dry-deposited 1,2-ISOPOOH and MVK (panel b) is instantaneously detoxified within the plant leaf (panels c, d) via the enzyme alkenal/one oxidoreductase (AOR). EC measurements in natural forest settings confirm our modified GEOS-Chem model results (indicated with *) that ~6.5 Tg methyl ethyl ketone (MEK) (corresponding to 1.5% of the isoprene source) is emitted into the atmosphere in this way. This is the single largest known MEK source on a global scale. †Values from ref. 75. ‡Values from ref. 7.
Results
Enclosure experiments
In a laboratory study, we investigated the fate of isoprene oxidation products when exposed to poplar leaves (Fig. 2, Supplementary Figs. 1–4). Poplars occur worldwide, both in natural forests and plantations, and are among the most important boreal deciduous tree species. In plant science, poplars represent the most important model system for tree research and were the first tree species for which the genome was described. Many studies, in particular for VOCs, show that the biological and physiological processes studied in poplars can be transferred to other tree species (e.g. refs. 3,11). Poplar plants several months in age were selected for the present work because they allow gas exchange analyses and deposition experiments to be carried out in a very well-controlled and reproducible manner. Many VOC emission studies have shown that such results can be transferred to older trees and can hence be used for modeling studies on regional and global scales (e.g. refs. 2,12).

Box plots of volume mixing ratios (VMR) are shown in panels a–c and deposition/emission fluxes (Φ) d–f for 1,2-ISOPOOH, MVK, and MEK, respectively. OVOCs were analyzed at the enclosure inlet and subsequently at the enclosure outlet during fumigation of the empty enclosure (BG) and of the darkened/illuminated gray poplar trees. For each box, the red line indicates the median, bars show the minimum/maximum, and the blue box indicates the 25th and 75th percentiles of the sample data (N = 5).
The experimental setup is illustrated in Supplementary Fig. 1. Gray poplar plants were placed in an enclosure system and fumigated with synthetic air containing ~450 ppm CO2 and ~8 ppbv 1,2-ISOPOOH. Before starting the plant fumigation experiments, we fumigated the empty cuvette with 1,2-ISOPOOH to condition the inner surfaces consisting of Teflon-coated glass. Subsequently, the 1,2-ISOPOOH loss to the surface of the empty enclosure was measured for each individual experiment (see also Supplementary Table 1). The OVOC composition of the inlet and outlet air of the plant enclosure was analyzed with a selective reagent ion time-of-flight mass spectrometer (SRI-TOF-MS). Details of the measurement method (see also Supplementary Table 2), SRI-TOF-MS instrument calibration (see also Supplementary Table 3), and plant material (Supplementary Table 4) can be found in “Methods” section.
The deposition velocity vd,i (cm s−1) is commonly used to describe trace gas deposition of a substance i to vegetation from the atmosphere13 and is defined as the ratio between the flux Φi (representing the amount of compound i deposited to a unit surface area per unit time in µg cm−2 s−1) and the local concentration ci (µg cm−3).
Deposition fluxes Φi to the plant surfaces inside the enclosure system were calculated from the difference in trace gas mixing ratios between the outlet of the enclosure (cout,i) and outlet of the empty enclosure (cout,i,BG) taking into account the single-sided leaf area (LA) of the enclosed plant and the gas flow (see Supplementary Methods - Enclosure experiment). In this way, we calculated an average 1,2-ISOPOOH deposition velocity of vd = 0.12 ± 0.04 cm s−1 under dark conditions (Fig. 2). The observed daytime value with open stomata (Supplementary Fig. 5) is 0.79 ± 0.25 cm s−1, close to the 24-h average simulated vd of 0.76 cm s−1 from Nguyen et al.8. Almost all deposited 1,2-ISOPOOH thus enters through open stomata into the plant’s inner space, where it will dissolve on wetted surfaces due to its large effective solubility (H* = 1.7 × 106 M atm−1). Supplementary Fig. 6 shows an experiment with a poplar plant exposed to elevated 1,2-ISOPOOH in the presence of light. Stomatal closure is observed, likely due to stress. Upon stomatal closure, 1,2-ISOPOOH levels in the enclosure increase, indicating that stomatal uptake is most probably the dominant deposition process, and light-induced plant surface uptake is less likely. We further determine from these experiments that 50 ± 15% of deposited 1,2-ISOPOOH is released as the volatile carbonyl MVK (H* = 44 M atm−1) while the other 50% is converted to MEK (H* = 21 M atm−1), which is likewise released back into the atmosphere.
Organic hydroperoxides are inherently unstable species. Homolytic cleavage of the weak peroxy bond (O–OH) is a common decomposition mechanism, and can be catalyzed by metals14. Recently, we have shown that 1,2-ISOPOOH is efficiently converted to MVK and HCHO on clean stainless steel surfaces15,16. We find here from separate laboratory experiments (Supplementary Fig. 7) that a range of different metals catalyze the conversion of 1,2-ISOPOOH to MVK even at room temperature. Chevallier et al.17 studied “Fenton-like” reactions of methylhydroperoxides with Fe2+ in aqueous solutions. They identified Fe2+ + ROOH → Fe3+ + RO + OH as the dominant first reaction step. The alkoxy (RO) radicals further rearrange in water solution, and react with dissolved oxygen to form peroxy radicals which decompose, leading to the formation of aldehydes and other products. Assuming that the same reaction mechanism occurs when ISOPOOH dissolves in the liquid phase of the apoplast, low-valence transition metals present in plant cell walls and also dissolved in the apoplast18 could catalyze via Fenton-type reactions the conversion of 1,2-ISOPOOH to MVK + HCHO + HO2 in plant leaves. Since MVK (in contrast to formaldehyde) has relatively low solubility, it would then be released into the atmosphere through open stomata as its concentration builds up in the liquid phase of the apoplast. In biological systems peroxide functional groups (ROOH) serve as reactive oxygen intermediates that cause oxidative damage and cell death19. For example, Polle and Junkermann20 found hydroxymethyl hydroperoxide (HMHP) to inhibit peroxidase activity in plant apoplast. Plants exposed to harmful compounds in this way typically release stress-induced volatiles such as methyl salicylate (MeSA). MeSA is an airborne cue inducing pathogen resistance within and between plants21, while also enhancing indirect plant defenses against herbivores by attracting their natural enemies22. Furthermore, unstressed poplar varieties in general are weak sesquiterpene (SQT) emitters23, but SQT emissions are known to increase under various type of abiotic and biotic stresses24,25. Here we observe a significant increase in emissions of both MeSA and SQT during 1,2-ISOPOOH fumigation (Supplementary Fig. 8), but only under daylight conditions when stomata are open. Our laboratory experiments therefore indicate that stomatal uptake of 1,2-ISOPOOH causes severe stress to poplar plants and triggers self-defense mechanisms to mitigate oxidative damage. The results also indicate that upon deposition to plants, 1,2-ISOPOOH is converted as a first step to MVK. The exact reaction mechanism of this first step is not known. However, we speculate that the Fenton-type reactions discussed above involving transition metals present in plant cell walls and dissolved in the apoplast18 catalyze the conversion of 1,2-ISOPOOH to MVK. As an α,β-unsaturated carbonyl and reactive electrophilic species, MVK itself is toxic to plant tissue due to its high reactivity and ability to form Michael adducts with the thiol and amino groups of biomolecules26,27. Previous MVK and MACR deposition experiments using poplars, three different Quercus species and houseplants have found both compounds to be immediately lost upon entry through open stomata28,29,30. Tani et al.29 observed that the uptake rate correlates strongly with the stomatal conductance. Further, Cappellin et al.31,32 found that red oak (a strong isoprene emitter) converts MVK to MEK. Here we find based on additional MVK fumigation experiments with gray poplar plants (Supplementary Fig. 4) that upon stomatal opening MVK fumigation leads to a significant increase in stress-related MeSA and SQT emissions (Supplementary Fig. 8). Furthermore, the MVK detoxification mechanism is highly efficient: all (100 ± 5%) deposited MVK is released as MEK into the atmosphere (Supplementary Fig. 4). As shown by Tani et al.29,33 and Cappellin et al.32. MEK undergoes leaf uptake with significant lower rates than MVK. Average deposition velocities for MVK of vd = 0.044 ± 0.045 and 0.22 ± 0.17 cm s−1 during dark and light conditions, respectively, are derived here, demonstrating the importance of stomatal fluxes for this process. Poplar fumigation experiments with the 4,3-ISOPOOH isomer did not result in any MEK production. This finding is expected and supports the above proposed mechanism because 4,3-ISOPOOH is converted on metal surfaces to MACR rather than MVK15,16.
Enzymatic reactions within plant tissues
Several studies have shown that enzymatic reduction of α,β-unsaturated ketones takes place in plant suspension cells and cytosolic fractions of yeast34,35. In particular, Yamauchi et al.36 identified an NADPH-dependent acrolein-reducing enzyme in cucumber leaves that catalyzes an AOR reaction of the α,β-unsaturated bond. We characterized the in vitro kinetic properties of the poplar AOR (EC 1.3.1.74) enzyme to which we attribute the in planta conversion of MVK to MEK. We obtain Michaelis–Menten constants (substrate concentration at half-maximal enzyme velocity) of 0.049 and 14.15 mM for NADPH and MVK, respectively, and a temperature optimum at 35 °C (Supplementary Fig. 9). This biochemical capability to reduce MVK to MEK is globally present in terrestrial plants, as a phylogenetic survey of genes coding for NADPH-dependent alkenal/on-oxidoreductases (AOR) demonstrates (Fig. 3, Supplementary Fig. 10, Supplementary Table 5). One gene encoding AOR is localized in the chloroplast (AORchl), and two more are located in the cytosol (AORcyt-I and AORcyt-II; Fig. 3b and Supplementary Fig. 10). The distribution of AORchl and AORcyt in plants is independent of isoprene and monoterpene emissions (Fig. 3a): AOR genes are ubiquitous in all land plants including mosses, clubmosses, gymnosperm, and flowering plants (Supplementary Fig. 10). Our gene expression analyses for gray poplar show that AORchl is predominantly expressed in leaf tissue, while AORcyt-II is expressed in the stem (phloem and xylem) (Fig. 3b). Under normal environmental conditions37 the total gene expression rates of AORchl are more than six times those of AORcyt-II. We analyzed in situ AOR activity in leaf extracts from poplar plants fumigated with either 1,2-ISOPOOH or MVK (Supplementary Fig. 11). The fact that we do not see any change to the in situ AOR activity as a result of exposure to 1,2-ISOPOOH and MVK (Supplementary Fig. 11) may be due to the fact that our observation period was too short and missed a stress-induced response. However, AOR also belongs to a group of proteins that are post-translationally modified (PTM) by S-nitrosylation at certain cysteine residues under stress (Supplementary Fig. 12)38, which may change in vitro enzyme activity. In planta, i.e. the intact leaves as we used for the gas exchange analyses, the AOR activity is variable depending on actual leaf temperature, light energy providing electrons in the form of NADPH for the reduction of MVK to MEK, and the presence of MVK, which is provided by deposition or degradation of 1,2-ISOPOOH. Overall, the phylogenetic analysis and the measurements of AOR activity in our model tree system imply that green biomass across the globe is able to convert MVK to MEK.

The maximum likelihood (ML) phylogenetic tree a shows the occurrence of AORchl, AORcyt-I, and AORcyt-II genes in dominant plant species. Asterisks and coloring of the branch labels indicate species emitting predominantly isoprene, monoterpenes, or both. Numbers at tree branches indicate node bootstrap support. Green branch coloring indicates the presence of plastid-targeting peptides (TP) in the corresponding AOR sequences. Basal nodes of the AORchl, AORcyt-I, and AORcyt-II ortholog clusters are labeled with solid circles. The scale bar below the tree shows branch length. Box plots b show tissue-specific expression of AORchl and AORcyt-II genes in gray poplar (Populus × canescens). Error bars show the minimum/maximum.
Impact on regional and global budgets of OVOCs
We performed global simulations using the GEOS-Chem chemical transport model (CTM) (v12.1.1, https://doi.org/10.5281/zenodo.2249246) to assess how dry deposition of 1,2-ISOPOOH and MVK (as the main isoprene oxidation products) impact the atmospheric MEK budget (see “Methods” section for model details). A global emission of 416 Tg isoprene is simulated by the model for 2017, leading to the photochemical production of 91.3 Tg MVK and 167 Tg 1,2-ISOPOOH (Fig. 4). By default, GEOS-Chem uses a modified resistance-based approach from Wesely9 to calculate dry deposition velocities. The performance of this approach depends on knowledge about atmospheric stability, surface conditions, and the solubility and reactivity factors ƒ0 for compounds of interest. The stomatal fraction of 1,2-ISOPOOH dry deposition thus simulated by the model generally ranges from <5% to 30% over land (Supplementary Fig. 13), which does not agree with our findings here (vd = 0.79 cm s−1 during daytime versus only 0.12 cm s−1 in the dark). We therefore question how accurately the default model scheme is able to separate stomatal versus non-stomatal deposition for OVOCs. Instead, we prescribe the modeled 1,2-ISOPOOH and MVK deposition velocities over land based on the results from our laboratory measurements employing constant daytime (nighttime) deposition velocities of 0.22 (0.044) cm s−1 for MVK and 0.79 (0.12) cm s−1 for 1,2-ISOPOOH. Over oceans the default model scheme is used. Since the laboratory chambers were well-mixed, this corresponds to an assumption that canopy uptake for these compounds is controlled by surface and molecular diffusion resistance, an assumption strongly supported by prior work8. Figure 4 shows the resulting simulated global deposition of MVK (3.7 Tg yr−1) and 1,2-ISOPOOH (8.8 Tg yr−1). Assuming based on our measurements that 100% of dry deposited MVK and 50% of dry deposited 1,2-ISOPOOH undergoes conversion to MEK, the model yields a global MEK flux of 6.5 Tg yr−1 (1.5% of isoprene emissions, Supplementary Fig. 14), making this mechanism the largest known MEK source to the atmosphere39. In turn, a fraction of that MEK will return to the ecosystem via further dry deposition (e.g. refs. 29,32,33).

Simulations were performed by the GEOS-Chem CTM for 2017 (see “Methods” section for model details). Reconciling the model deposition velocities using our experimentally-obtained vd, and assuming that 100% of dry deposited MVK and 50% of dry deposited 1,2-ISOPOOH undergoes conversion in plants, reveals a global MEK source of 6.5 Tg yr−1 (and recycling 1.5% of the isoprene flux).
For comparison, we performed a GEOS-Chem base-case run (Supplementary Fig. 15) in which the default Wesely scheme was used to simulate 1,2-ISOPOOH and MVK dry deposition. Assuming that only the stomatal component of deposition leads to MEK then yields a global source of 3 Tg yr−1. We view this as a lower limit given the apparent underestimate of OVOC stomatal uptake in the default model. On the other hand, assuming that all dry deposited MVK and 50% of all dry deposited 1,2-ISOPOOH undergoes conversion results in an MEK source of 14 Tg yr−1 (upper limit).
Eddy covariant (EC) VOC flux measurements
To evaluate these findings, we performed EC flux measurements of isoprene and MEK at two sites. The first is the SMEAR II station in Hyytiälä (Finland) (Supplementary Fig. 16), which is surrounded by low isoprene-emitting plants (mainly Scots pine Pinus sylvestris) with an average daytime isoprene flux of 1 nmol m−2 s−1 in spring. Details of the eddy covariant VOC flux measurements can be found in “Methods” section and Supplementary Methods - Eddy covariance VOC flux measurements. The second EC flux measurements were performed at PROPHET (US) (Supplementary Fig. 17), which is a high isoprene emission site in a mixed deciduous/coniferous forest. Typical daytime isoprene flux values at PROPHET reach ~10 nmol m−2 s−1 (Supplementary Fig. 17). Flux data for isoprene and MEK were obtained using the EC method, correlating fast mixing ratio changes (~10 Hz) with vertical wind velocities. The high sensitivity achieved by the PTR3-TOF for MEK (>12,000 cps/ppbv) enabled the first accurate measurement of these fluctuations at the low isoprene-emission site. In general, the recent development of very sensitive, fast, and quantitative mass spectrometry-based detectors such as PTR-QiTOF and PTR3-TOF40 was essential for EC measurements of MEK. We find that MEK emissions are 1.8% and 0.9% those of isoprene, at these sites, respectively. This is in excellent agreement with the model best-estimate (1.5%; see also Supplementary Fig. 14) and supports the global importance of this mechanism for plant oxidative protection and for the biogenic MEK atmospheric source.
Discussion
Ketones are an important class of OVOC with sufficiently long atmospheric lifetimes to be transported to the upper troposphere. MEK, like acetone, photolyzes in the near UV region; as a result, these ketones provide an important source of HOx (OH + HO2) radicals in the dry upper troposphere41,42. Degradation of MEK in the lower atmosphere generates toxic and photo-active compounds such as acetaldehyde, a peroxyacetylnitrate (PAN) precursor, and formaldehyde43. Global sources of MEK, the second most abundant atmospheric ketone, are still not fully understood. Singh et al.39 presented a first assessment of MEK sources and sinks based on aircraft measurements over the Pacific Ocean. Based on their derived MEK atmospheric burden and an assumed 7-day lifetime due to reaction with OH and photolysis, they inferred a total global source of 11 Tg yr−1. They further estimated MEK sources of 1–3 Tg yr−1 each from hydrocarbon oxidation and biomass burning, and of 0–1 Tg yr−1 from oceanic emissions. The latter estimate is supported by more recent analysis by Brewer et al.44. A residual unexplained source of 7 Tg yr−1 was then attributed by Singh et al.39 to direct biogenic emissions. Our work combining dedicated laboratory, field, and model analyses derives a global biogenic MEK source of 6.5 Tg yr−1, and provides a new biochemical and genetic basis for understanding the processes driving plant–atmosphere exchange of 1,2-ISOPOOH, MVK and MEK, and for modeling this exchange at regional and global scales.
Plants possess the ability to absorb OVOCs from the atmosphere and actively convert toxic chemicals to less toxic species, which can then be re-emitted into the atmosphere. Here we show that following uptake the toxic isoprene oxidation products 1,2-ISOPOOH and MVK are converted and re-emitted as MEK. The genes coding for the responsible enzyme alkenal/an oxidoreductase (AOR) are ubiquitously distributed in the plant kingdom. For MEK this process leads to an atmospheric input of about 6.5 Tg, the single largest known term in the global MEK budget and re-emitting 1.5% of the original isoprene emissions. Our findings thus support and provide a mechanistic underpinning to understand the biogenic emissions inferred by Singh et al.39, and implicate detoxification as the most important source process in the global MEK budget. The global MEK estimate has uncertainties due to the underlying up-scaling errors. These uncertainties include those linked with the enclosure measurements as well as AOR activity changes with temperature. We found in enclosure measurements that stomatal uptake is the dominant route, but leaf surface conversion on wet surfaces may also occur under real world conditions. However, for many years scientists have used plant enclosure experiments in combination with field-based flux measurements to better asses global BVOC emissions. For example, Guenther et al.45 compared BVOC flux estimates from enclosure and ambient measurements and found agreement to within a factor of two. These findings suggest a significant modification of OVOCs at the biosphere–atmosphere interface. Traditional dry deposition schemes that are widely used in atmospheric chemistry models do not capture the uptake process, and new mechanistic approaches should be developed to capture the complex behavior of bi-directional exchange at the biosphere–atmosphere interface.
Methods
SRI-TOF-MS
The SRI-TOF-MS is a custom-built instrument that represents an advanced version of the PTR-TOF-MS system described by Graus et al.46, and features the capability to switch between different reagent ions within seconds. Additionally, we replaced the standard metal drift rings of the reaction chamber with chemically inert conductive PEEK rings. This feature and the high flow rate through the reaction chamber (~800 ml min−1 instead of 10–30 ml min−1 in standard PTR-MS instruments) were essential for minimizing metal-catalyzed decomposition of 1,2-ISOPOOH as observed in standard PTR-MS instruments15. The OVOC composition of the inlet and outlet air of the plant enclosure system was sequentially analyzed by SRI-TOF-MS operated with ammonia (NH4+) and occasionally with nitronium (NO+) or hydronium (H3O+) reagent cations. The SRI-TOF-MS was operated at a constant temperature (35 °C) and pressure (2.3 mbar) in the drift tube. Different drift voltages of 250, 300, and 500 V resulting in E/N values of 45 Td (NH4+-mode), 54 Td (NO+-mode), and 90 Td (H3O+-mode), respectively, were used (E is the electric field strength, while N is the number gas density; 1 Td = 10−17 V cm2). Raw data analysis was performed with the PTR-TOF-Data Analyzer47 and the data processing routine described in Breitenlechner et al.40. For details about chemical ionization mechanism and calibration of the SRI-TOF-MS see the Supplementary Methods—Enclosure experiments and Supplementary Table 4.
PTR3-TOF
In the recently developed PTR3-TOF instrument40 a discharge ion source is coupled to a contact-free inlet system through the novel reaction chamber at 80 mbar. The inlet system is operated at high sample gas flow rates. The PTR3 front portion is coupled to a TOF from TOFWERK mass analyzer. The instrument has sensitivities of up to 20,000 cps/ppbv and a mass resolution of ~8000 m/Δm. VOCs were ionized via reactions with H3O+(H2O)n primary ions. Flux data for isoprene and MEK were obtained using the EC method, correlating fast mixing ratio changes (~10 Hz) with vertical wind velocities. The high sensitivity achieved by the PTR3-TOF for MEK (>12,000 cps/ppbv) enables more accurate measurement of these fluctuations than was previously possible. Calibration details are described in the Supplementary Methods—Eddy covariance VOC flux measurements.
Plant material
In the present study we used 4-month-old gray poplars (Populus × canescens INRA clone 7171-B4; syn. P. tremula × P. alba (Aiton.) Smith). Gray poplar shoots had been amplified by micropropagation on half-concentrated MS medium and further cultivated at the Helmholtz Zentrum München to an age of 3 months as described elsewhere48. Before being used in the experiments, the poplar plants were grown in a greenhouse at the Institute of Microbiology of the University of Innsbruck for 1–4 weeks under natural light conditions. One day prior to the experiment, the plants were transferred to the laboratory to adapt to the light conditions (12-h photoperiod with an approximate PAR of 400 µmol photons m−2 s−1). All plants were well watered and showed no visible illness symptoms.
Determination AOR activity
To determine apparent in vitro AOR activities in poplar leaf protein extracts, we deep-froze three mature leaves (leaf # 9–11, below the apex) from each poplar plant with liquid nitrogen at the following experimental phases: (1) before the experiments, (2) immediately after the experiment, i.e., after 24 h of ISOPOOH fumigation (12 h illuminated and 12 h in the dark), and (3) 24 h after the end of the enclosure experiment. The apparent in vitro alkenal/one oxidoreductase (AOR) activity was assessed as described in Yamauchi et al.49. Frozen leaf material was ground in liquid nitrogen using a dismembrator ball mill (B. Braun Biotech International, Melsungen, Germany). 200 mg of leaf powder were extracted in 4 ml of plant extraction buffer (100 mM Tris/HCl, pH 8.0, 20 mM MgCl2, 100 mM CaCl2, 1.5% PEG1500 (w/v), 5% (v/v) glycerol, 0.1% (v/v) Tween-20, and 20 mM DTT) with 200 mg polyvinylpolypyrrolidone (PVPP), stirred for 15 min on ice (4 °C) and centrifuged for 15 min at 20,000 × g. The supernatant was purified on Sephadex G-25 PD-10 columns (GE Healthcare, Solingen, Germany) equilibrated with enzyme buffer (50 mM Tris/HCl, 20 mM MgCl2, 5% (v/v) glycerol, 2 mM DTT)50. Protein concentrations were determined by the Bradford assay using a Roti-Quant Kit (Carl Roth, Karlsruhe, Germany). Kinetic properties of AOR were initially characterized with changing concentrations of NADPH and MVK and at changing assay temperatures (Supplementary Fig. 9). AOR activities were finally measured in crude protein extracts in the presence of 0.15 mM NADPH (under a saturating NADPH concentration; 3-times Michaelis–Menten constant (0.049 mM)), monitoring changes at A340 nm with a substrate concentration of 30 mM MVK (dissolved in enzyme buffer at a saturating MVK concentration of 2-times Michaelis–Menten constant (14.15 mM)) against a control without MVK according to Yamauchi et al.49 in a final assay volume of 1 mL. From extracts of 7 (1,2-ISOPOOH fumigation) and 5 (MVK fumigation) biological replicates three technical replicates were measured. A description of the construction of the AOR phylogenetic tree and AOR gene expression analysis can be found in the Supplementary Methods—Construction of AOR phylogenetic trees and AOR gene expression analysis.
EC flux measurements
The first EC flux measurement site is situated at the SMEAR II station in Hyytiälä, Finland. Vegetation around the site is dominated by Scots pine (P. sylvestris) with a canopy height of ~15 m. The ground level vegetation consists of lingonberry (Vaccinium vitis-idaea), blueberry (V. myrtillus), and mosses (Pleurozium scheberi, Dicramum polysetum)51. Flux data for isoprene and MEK were obtained with the PTR3 using the eddy covariance (EC) method during spring 2016. The PTR3 sampled air through a specially designed 5 m long tube with a high flow rate on top of a 35 m tower. Wind speed measurements were taken 0.5 m above the inlet opening by a METEK USA-1 sonic anemometer at 20 Hz. Wind direction and speed were cross checked against the SMEAR II station instrumentation.
The second EC flux measurement site is situated at the University of Michigan Biological Station (UMBS). Isoprene, MEK, and other VOCs were measured by PTR-QiTOF as part of the PROPHET-AMOS field study in July 201652,53. The 34 m PROPHET tower (45.559°N, 84.715°W, 232 m elevation) is located in a mixed deciduous/coniferous forest (canopy height ~23 m) with an upper canopy dominated by aspen, birch, and red oak, and a lower canopy consisting mainly of white pine, red maple, beech, and red oak54,55. Net above-canopy VOC fluxes were measured each hour by eddy covariance throughout the campaign. Sampling, instrument operation, calibration procedures, data processing, and QA/QC are described in detail elsewhere52.
GEOS-Chem model
GEOS-Chem (v12.1.1, https://doi.org/10.5281/zenodo.2249246; www.geos-chem.org) is a global 3D CTM driven by assimilated meteorological fields (Goddard Earth Observation System Forward Processing product; GEOS-FP) from the NASA Global Modeling and Assimilation Office (GMAO). The GEOS-FP data have native horizontal resolution of 0.25° latitude × 0.3125° longitude with 72 vertical layers. For the year 2017 global simulations presented here, we degrade the horizontal resolution to 2° × 2.5° and use a 15-min transport time step. We use the TPCORE advection algorithm56, convective mass fluxes from the GEOS-FP archive57, and the non-local boundary layer mixing scheme described by Lin and McElroy58.
Wet deposition in GEOS-Chem is as described by Amos et al.59, and includes convective scavenging as well as rainout and washout by large-scale precipitation. The default dry deposition in GEOS-Chem employs the resistance-in-series scheme formulated by Wesely9 and subsequently updated and implemented by Wang et al.60. Deposition velocities are computed in the model as a function of aerodynamic, boundary layer and surface resistances, with the first two computed from relevant molecular and meteorological parameters as detailed previously60. Surface resistances incorporate stomatal, cuticular, lower canopy, and ground surface pathways, and are likewise derived according to local environmental conditions60,61. Relevant parameters for 1,2-ISOPOOH and MVK include H* values of 1.7 × 106 and 44 M/atm, respectively, and reactivity (f0) values of 1.0 for both species.
The GEOS-Chem chemical mechanism (publicly available online via https://doi.org/10.5281/zenodo.2249246) includes comprehensive HOx–NOx–VOC–ozone–halogen chemistry coupled to aerosols and incorporates current JPL/IUPAC recommendations with recent updates for isoprene chemistry62,63, peroxyacetyl nitrate64, and Criegee chemistry65. Photolysis frequencies are calculated for 18 wavelength bins spanning 177–850 nm using the Fast-JX algorithms developed by Bian and Prather66,67. Biogenic VOC emissions are computed online based on the Model of Emissions of Gases and Aerosols from Nature (MEGANv2.1)2 as described by Hu et al.68. Specifically, emissions for each model grid cell are computed from vegetation-specific emission factors and multiplicative non-dimensional activity factors accounting for emission dependencies on light, temperature, LA, and leaf age2. Biogenic MEK fluxes are computed separately by applying our laboratory measured-yields (100% and 50%, respectively) to the modeled MVK and ISOPOOH deposition fluxes as detailed in the main text. Global anthropogenic emissions of VOCs, CO, NH3, NOx, SO2, and aerosols are from the Community Emissions Data System (CEDS) inventory69 overwritten by regionally specific inventories for North America70, Asia71, and Africa72. Biogenic soil NOx emissions are from Hudman et al.73.
Data availability
The data that support the findings of this study are openly available in Zenodo (https://doi.org/10.5281/zenodo.4010848).
Code availability
We performed global simulations using the GEOS-Chem chemical transport model (CTM) (v12.1.1, https://doi.org/10.5281/zenodo.2249246), which is freely available at www.geos-chem.org.
References
Goldstein, A. H. & Galbally, I. E. Known and unexplored organic constituents in the Earth’s atmosphere. Environ. Sci. Technol. 41, 1514–1521 (2007).
Guenther, A. B. et al. The model of emissions of gases and aerosols from nature version 2.1 (MEGAN2.1): an extended and updated framework for modeling biogenic emissions. Geosci. Model Dev. 5, 1471–1492 (2012).
Sharkey, T. D., Wiberley, A. E. & Donohue, A. R. Isoprene emission from plants: why and how. Ann. Bot. 101, 5–18 (2008).
Lantz, A. T., Allman, J., Weraduwage, S. M. & Sharkey, T. D. Isoprene: new insights into the control of emission and mediation of stress tolerance by gene expression. Plant Cell Environ. 42, 2808–2826 (2019).
Wennberg, P. O. et al. Gas-phase reactions of isoprene and its major oxidation products. Chem. Rev. 118, 3337–3390 (2018).
Atkinson, R. & Arey, J. Gas-phase tropospheric chemistry of biogenic volatile organic compounds: a review. Atmos. Environ. 37, 197–219 (2003).
Teng, A. P., Crounse, J. D. & Wennberg, P. O. Isoprene peroxy radical dynamics. J. Am. Chem. Soc. 139, 5367–5377 (2017).
Nguyen, T. B. et al. Rapid deposition of oxidized biogenic compounds to a temperate forest. Proc. Natl. Acad. Sci. USA 112, E392–E401 (2015).
Wesely, M. L. Parameterization of surface resistances to gaseous dry deposition in regional-scale numerical models. Atmos. Environ. 23, 1293–1304 (1989).
Wesely, M. A review of the current status of knowledge on dry deposition. Atmos. Environ. 34, 2261–2282 (2000).
Baldwin, I. T. & Schultz, J. C. Rapid changes in tree leaf chemistry induced by damage: evidence for communication between plants. Science (80-.) 221, 277–279 (1983).
Monson, R. K. et al. High productivity in hybrid-poplar plantations without isoprene emission to the atmosphere. Proc. Natl. Acad. Sci. USA 117, 1596–1605 (2020).
Seinfeld, J. H. & Pandis, S. N. Atmospheric Chemistry and Physics: from Air Pollution to Climate Change (John Wiley & Sons, 2012).
Fenton, H. J. H. LXXIII—oxidation of tartaric acid in presence of iron. J. Chem. Soc. Trans. 65, 899–910 (1894).
Rivera-Rios, J. C. et al. Conversion of hydroperoxides to carbonyls in field and laboratory instrumentation: observational bias in diagnosing pristine versus anthropogenically controlled atmospheric chemistry. Geophys. Res. Lett. 41, 8645–8651 (2014).
Bernhammer, A. K., Breitenlechner, M., Keutsch, F. N. & Hansel, A. Technical note: conversion of isoprene hydroxy hydroperoxides (ISOPOOHs) on metal environmental simulation chamber walls. Atmos. Chem. Phys. 17, 4053–4062 (2017).
Chevallier, E., Jolibois, R. D., Meunier, N., Carlier, P. & Monod, A. “Fenton-like” reactions of methylhydroperoxide and ethylhydroperoxide with Fe2+ in liquid aerosols under tropospheric conditions. Atmos. Environ. 38, 921–933 (2004).
Sattelmacher, B. The apoplast and its significance for plant mineral nutrition. New Phytol. 149, 167–192 (2001).
Mittler, R. Oxidative stress, antioxidants and stress tolerance. Trends Plant Sci. 7, 405–410 (2002).
Polle, A. & Junkermann, W. Inhibition of apoplastic and symplastic peroxidase activity from Norway Spruce by the photooxidant hydroxymethyl hydroperoxide. Plant Physiol. 104, 617–621 (1994).
Shulaev, V., Silverman, P. & Raskin, I. Airborne signalling by methyl salicylate in plant pathogen resistance. Nature 385, 718–721 (1997).
Rowen, E., Gutensohn, M., Dudareva, N. & Kaplan, I. Carnivore attractant or plant elicitor? Multifunctional roles of methyl salicylate lures in tomato defense. J. Chem. Ecol. 43, 573–585 (2017).
Kesselmeier, J. & Staudt, M. Biogenic volatile organic compounds (VOC): an overview on emission, physiology and ecology. J. Atmos. Chem. 33, 23–88 (1999).
Schnitzler, J.-P., Louis, S., Behnke, K. & Loivamäki, M. Poplar volatiles—biosynthesis, regulation and (eco)physiology of isoprene and stress-induced isoprenoids. Plant Biol. 12, 302–316 (2010).
Ghirardo, A. et al. Urban stress-induced biogenic VOC emissions and SOA-forming potentials in Beijing. Atmos. Chem. Phys. 16, 2901–2920 (2016).
Alméras, E. et al. Reactive electrophile species activate defense gene expression in Arabidopsis. Plant J. 34, 205–216 (2003).
Esterbauer, H., Schaur, R. J. & Zollner, H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic. Biol. Med. 11, 81–128 (1991).
Karl, T. et al. Efficient atmospheric cleansing of oxidized organic trace gases by vegetation. Science (80-.) 330, 816–819 (2010).
Tani, A., Tobe, S. & Shimizu, S. Uptake of methacrolein and methyl vinyl ketone by tree saplings and implications for forest atmosphere. Environ. Sci. Technol. 44, 7096–7101 (2010).
Tani, A. & Hewitt, C. N. Uptake of aldehydes and ketones at typical indoor concentrations by houseplants. Environ. Sci. Technol. 43, 8338–8343 (2009).
Cappellin, L. et al. Field observations of volatile organic compound (VOC) exchange in red oaks. Atmos. Chem. Phys. 17, 4189–4207 (2017).
Cappellin, L., Loreto, F., Biasioli, F., Pastore, P. & McKinney, K. A mechanism for biogenic production and emission of MEK from MVK decoupled from isoprene biosynthesis. Atmos. Chem. Phys. 19, 3125–3135 (2019).
Tani, A., Tobe, S. & Shimizu, S. Leaf uptake of methyl ethyl ketone and croton aldehyde by Castanopsis sieboldii and Viburnum odoratissimum saplings. Atmos. Environ. 70, 300–306 (2013).
Wanner, P. & Tressl, R. Purification and characterization of two enone reductases from Saccharomyces cerevisiae. Eur. J. Biochem. 255, 271–278 (1998).
Kergomard, A., Renard, M. F., Veschambre, H., Courtois, D. & Petiard, V. Reduction of α,β-unsaturated ketones by plant suspension cultures. Phytochemistry. 27, 407–409 (1988).
Yamauchi, Y., Hasegawa, A., Mizutani, M. & Sugimoto, Y. Chloroplastic NADPH-dependent alkenal/one oxidoreductase contributes to the detoxification of reactive carbonyls produced under oxidative stress. FEBS Lett. 586, 1208–1213 (2012).
Georgii, E. et al. The systems architecture of molecular memory in poplar after abiotic stress. Plant Cell 31, 346–367 (2019).
Vanzo, E. et al. Facing the future: effects of short-term climate extremes on isoprene-emitting and nonemitting poplar. Plant Physiol. 169, 560–575 (2015).
Singh, H.B. et al. Analysis of the atmospheric distribution, sources, and sinks of oxygenated volatile organic chemicals based on measurements over the Pacific during TRACE-P. J. Geophys. Res. D 109, (2004) https://doi.org/10.1029/2003JD003883.
Breitenlechner, M. et al. PTR3: an instrument for studying the lifecycle of reactive organic carbon in the atmosphere. Anal. Chem. 89, 5824–5831 (2017).
Singh, H. B., Kanakidou, M., Crutzen, P. J. & Jacob, D. J. High concentrations and photochemical fate of oxygenated hydrocarbons in the global troposphere. Nature 378, 50–54 (1995).
Brewer, J. F. et al. Atmospheric photolysis of methyl ethyl, diethyl, and propyl ethyl ketones: temperature-dependent UV absorption cross sections. J. Geophys. Res. Atmos. 124, 5906–5918 (2019).
Jenkin, M. E., Saunders, S. M. & Pilling, M. J. The tropospheric degradation of volatile organic compounds: a protocol for mechanism development. Atmos. Environ. 31, 81–104 (1997).
Brewer, J. F. et al. Evidence for an oceanic source of methyl ethyl ketone to the atmosphere. Geophys. Res. Lett. 591, 5022–5024 (2020).
Guenther, A. et al. Estimates of regional natural volatile organic compound fluxes from enclosure and ambient measurements. J. Geophys. Res. Atmos. 101, 1345–1359 (1996).
Graus, M., Müller, M. & Hansel, A. High resolution PTR-TOF: quantification and formula confirmation of VOC in real time. J. Am. Soc. Mass Spectrom. 21, 1037–44 (2010).
Müller, M., Mikoviny, T., Jud, W., D’Anna, B. & Wisthaler, A. A new software tool for the analysis of high resolution PTR-TOF mass spectra. Chemom. Intell. Lab. Syst. 127, 158–165 (2013).
Vanzo, E. et al. Modulation of protein S -nitrosylation by isoprene emission in poplar. Plant Physiol. 170, 1945–1961 (2016).
Yamauchi, Y., Hasegawa, A., Taninaka, A., Mizutani, M. & Sugimoto, Y. NADPH-dependent reductases involved in the detoxification of reactive carbonyls in plants. J. Biol. Chem. 286, 6999–7009 (2011).
Schnitzler, J. P. et al. Biochemical properties of isoprene synthase in poplar (Populus × canescens). Planta 222, 777–786 (2005).
Ilvesniemi, H., Giesler, R., Van Hees, P., Magnussson, T. & Melkerud, P. A. General description of the sampling techniques and the sites investigated in the Fennoscandinavian podzolization project. Geoderma 94, 109–123 (2000).
Millet, D. B. et al. Bidirectional ecosystem-atmosphere fluxes of volatile organic compounds across the mass spectrum: how many matter?. ACS Earth Space Chem. 2, 764–777 (2018).
Alwe, H. D. et al. Oxidation of volatile organic compounds as the major source of formic acid in a mixed forest canopy. Geophys. Res. Lett. 46, 2940–2948 (2019).
Bryan, A. M. et al. Forest-atmosphere BVOC exchange in diverse and structurally complex canopies: 1-D modeling of a mid-successional forest in northern Michigan. Atmos. Environ. 120, 217–226 (2015).
Palik, B. J. & Pregitzer, K. S. A comparison of presettlement and present-day forests on two bigtooth aspen-dominated landscapes in Northern Lower Michigan. Am. Midl. Nat. 127, 327 (1992).
Lin, S.-J. & Rood, R. B. Multidimensional flux-form semi-Lagrangian transport schemes. Mon. Weather Rev. 124, 2046–2070 (2002).
Wu, X., Deng, L., Song, X. & Zhang, G. J. Coupling of convective momentum transport with convective heating in global climate simulations. J. Atmos. Sci. 64, 1334–1349 (2007).
Lin, J.-T. & McElroy, M. B. Impacts of boundary layer mixing on pollutant vertical profiles in the lower troposphere: Implications to satellite remote sensing. Atmos. Environ. 44, 1726–1739 (2010).
Amos, H. M. et al. Gas-particle partitioning of atmospheric Hg(II) and its effect on global mercury deposition. Atmos. Chem. Phys. 12, 591–603 (2012).
Wang, Y., Logan, J. A. & Jacob, D. J. Global simulation of tropospheric O3–NOx–hydrocarbon chemistry: 2. Model evaluation and global ozone budget. J. Geophys. Res. Atmos. 103, 10727–10755 (1998).
Silva, S. J. & Heald, C. L. Investigating dry deposition of ozone to vegetation. J. Geophys. Res. Atmos. 123, 559–573 (2018).
Travis, K. R. et al. Why do models overestimate surface ozone in the Southeast United States? Atmos. Chem. Phys. 16, 13561–13577 (2016).
Fisher, J. A. et al. Organic nitrate chemistry and its implications for nitrogen budgets in an isoprene- and monoterpene-rich atmosphere: constraints from aircraft (SEAC 4 RS) and ground-based (SOAS) observations in the Southeast US. Atmos. Chem. Phys. 16, 5969–5991 (2016).
Fischer, E. V. et al. Deolal, atmospheric peroxyacetyl nitrate (PAN): a global budget and source attribution. Atmos. Chem. Phys. 14, 2679–2698 (2014).
Millet, D. B. et al. A large and ubiquitous source of atmospheric formic acid. Atmos. Chem. Phys. 15, 6283–6304 (2015).
Bian, H. & Prather, M. J. Fast-J2: accurate simulation of stratospheric photolysis in global chemical models. J. Atmos. Chem. 41, 281–296 (2002).
Eastham, S. D., Weisenstein, D. K. & Barrett, S. R. H. Development and evaluation of the unified tropospheric-stratospheric chemistry extension (UCX) for the global chemistry-transport model GEOS-Chem. Atmos. Environ. 89, 52–63 (2014).
Hu, L. et al. Isoprene emissions and impacts over an ecological transition region in the U.S. Upper Midwest inferred from tall tower measurements. J. Geophys. Res. 120, 3553–3571 (2015).
Hoesly, R. M. et al. Historical (1750–2014) anthropogenic emissions of reactive gases and aerosols from the Community Emissions Data System (CEDS). Geosci. Model Dev. 11, 369–408 (2018).
EPA. 2011 National Emissions Inventory Data & Documentation https://www.epa.gov/air-emissions-inventories/2011-national-emission-inventory-nei-report (2015).
Li, M. et al. MIX: a mosaic Asian anthropogenic emission inventory under the international collaboration framework of the MICS-Asia and HTAP. Atmos. Chem. Phys. 17, 935–963 (2017).
Marais, E. A. & Wiedinmyer, C. Air quality impact of Diffuse and Inefficient Combustion Emissions in Africa (DICE-Africa). Environ. Sci. Technol. 50, 10739–10745 (2016).
Hudman, R. C. et al. Steps towards a mechanistic model of global soil nitric oxide emissions: implementation and space based-constraints. Atmos. Chem. Phys. 12, 7779–7795 (2012).
Paulot, F. et al. Unexpected epoxide formation in the gas-phase photooxidation of isoprene. Science (80-.) 325, 730–733 (2009).
Berndt, T., Hyttinen, N., Herrmann, H. & Hansel, A. First oxidation products from the reaction of hydroxyl radicals with isoprene for pristine environmental conditions. Commun. Chem. 2, 21 (2019).
Acknowledgements
The authors would like to thank Bernhard Mentler (UIBK) for developing the ISOPOOH and IEPOX measurement protocol using different reagent ions, Ulrich Junghans (Helmholtz) for growing the sample plants, Frank Keutsch & Jean Rivera-Rios (Harvard University) for providing the 1,2-ISOPOOH standard and Holger Kopacka (UIBK) for the NMR analysis of the samples. We thank Xin Chen (UMN) for assistance with the model simulations, along with Steven Bertman (WMU) and the entire PROPHET-AMOS team for their support and contributions to the field campaign. We thank Martin Breitenlechner (UIBK) for contributions to the Hyytiälä field campaign. We thank the Institute of Microbiology from the University of Innsbruck for sharing the greenhouse facility. This study was supported by the Austrian Federal Ministry of Education, Science and Research within Sparkling Science (grant SPA 05/080—AiR and grant SPA 06/222—CHAMPIONS) and the University of Innsbruck. VOC flux measurements at Hyytiälä were supported by the European Commission under Horizon 2020 (H2020-INFRAIA-2014-2015, grant 654109, ACTRIS). VOC measurements during PROPHET were supported by the US National Science Foundation (NSF grants AGS‐1428257 and AGS‐1148951). GEOS‐Chem model development and simulations were supported by the US National Aeronautics and Space Administration (NASA grant NNX14AP89G). Computing resources at UMN were provided by MSI (www.msi.umn.edu).
Author information
Authors and Affiliations
Contributions
A.H., E.C., and J.-P.S. designed the experiment. E.C. led and performed the laboratory measurements and data analysis, E.M.P. assisted with the lab measurements and data analysis, L.F. assisted with data analysis. I.Z. and J.-P.S. measured AOR activities. T.N., E.G., and J.-P.S. generated the AOR phylogenesis. D.B.M., H.D.A., and L.F. contributed with eddy flux data. D.B.M. ran the GEOS-Chem model and analyzed the results. T.K. and M.K. took part in data discussion and manuscript improvement. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Primary handling editor: Heike Langenberg
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Canaval, E., Millet, D.B., Zimmer, I. et al. Rapid conversion of isoprene photooxidation products in terrestrial plants. Commun Earth Environ 1, 44 (2020). https://doi.org/10.1038/s43247-020-00041-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s43247-020-00041-2
This article is cited by
-
Cryptogamic organisms are a substantial source and sink for volatile organic compounds in the Amazon region
Communications Earth & Environment (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
