
Abstract
Single-photon emitters are crucial building blocks for optical quantum technologies. Hexagonal boron nitride (hBN) is a promising two-dimensional material that hosts bright, room-temperature single-photon emitters. However, photo instability is a persistent challenge preventing practical applications of these properties. Here, we reveal the ubiquitous photobleaching of hBN vacancy emitters. Independent of the source or the number of hBN layers, we find that the photobleaching of a common emission at 1.98 ± 0.05 eV can be described by two consistent time constants, namely a first bleaching lifetime of 5 to 10 s, and a second bleaching lifetime in the range of 150 to 220 s. Only the former is environmentally sensitive and can be significantly mitigated by shielding O2, whereas the latter could be the result of carbon-assisted defect migration. Annular dark-field scanning transmission electron microscopy of photobleached hBN allows for visualizing vacancy defects and carbon substitution at single atom resolution, supporting the migration mechanism along with X-ray photoelectron spectroscopy. Thermal annealing at 850 °C of liquid exfoliated hBN eliminates both bleaching processes, leading to persistent photostability. These results represent a significant advance to potentially engineer hBN vacancy emitters with the photostability requisite for quantum applications.
Similar content being viewed by others

Introduction
Harnessing quantum mechanics for real-world applications is becoming one of the most intriguing and fastest growing research fields. Single-photon emitters (SPEs) are essential in photonic quantum technologies, where quantum states of photons are exploited for information processing and transmission1. Various solid-state single-photon sources have been investigated in three-dimensional crystals, with emitting structures embedded in diamond, silicon carbide, and zinc oxide2. Light emitting from these sources, however, could be easily trapped due to the bulky dielectric environment, leading to reduced efficiency of emission. Color centers in two-dimensional (2D) hosts, such as transition metal dichalcogenides (TMDs)3 and hexagonal boron nitride (hBN)4, could overcome this challenge with significantly reduced dielectric thickness down to a single atomic layer, and benefit additionally from easier integration with on-chip photonics5 and versatile manipulation methods6,7,8. While single-photon emission in TMDs has only been observed at low temperatures, hBN can host bright, room-temperature SPEs4. With such unique photo-physical property, hBN is also advantageous over the one-dimensional SPE host of carbon nanotubes and the zero-dimensional quantum dots, both of which mainly operate at cryogenic temperatures2.
Thin films of hBN hosting SPEs can be prepared via multiple methods. Bulk crystal can be synthesized under high temperature and high pressure9,10, and then exfoliated mechanically11 or in liquid12,13. Atomically thin films can also be directly grown by chemical vapor deposition (CVD)14,15,16,17 or metal organic vapor phase epitaxy (MOVPE)18,19. Among these methods, recent breakthroughs in wafer-scale synthesis of single-crystal hBN monolayers20,21,22 suggest that CVD is one of the most promising approaches for scalable, low-cost and reproducible fabrication of atomically thin hBN, where uniform spectral and spatial distribution of SPEs could be achieved16,23. Emitters in CVD-grown hBN, however, often display spectral diffusion17, intermittency in luminescence (i.e., blinking)16,17, and irreversible loss of emission (i.e., photobleaching)4,16. The fundamental mechanisms underlying these unstable photo-physical behaviors remain elusive, and few ways have been explored to improve the photostability of CVD hBN emitters14,17. On the other hand, liquid exfoliated hBN shows sharp and stable emission after thermal annealing12,13,15. The exfoliation approach, however, produces nanoflakes with hundreds of nanometer in lateral size that are too small for practical applications. In order to develop strategies for scalable fabrication of hBN thin films with photostable emitters, it is important to understand the fundamental physics underlying the aforementioned varying photostability, which is critical for the performance of SPEs2.
In this study, we quantitatively evaluate the photobleaching lifetimes of various hBN samples. By introducing an environmental chamber with a controllable atmosphere and performing laser power dependence measurements, we reveal that the photobleaching in monolayer CVD hBN is dominated by photochemical reactions with O2, corresponding to a bleaching lifetime of approximately 5 s. We then demonstrate that such bleaching can be significantly mitigated by a simple strategy of stacking additional hBN monolayers. We further characterize the photobleached monolayer of CVD hBN with annular dark-field scanning transmission electron microscopy (ADF-STEM) at the emitter position, and achieve high-quality atomic scale imaging of the hBN lattice. We present with high crystallographic detail a rich variety of monovacancy measurements, where carbon and oxygen substitutions in the hBN lattice are observed. Additionally, we observe a second bleaching lifetime in the range of 150 to 220 s in different multilayer samples in the air and in monolayer samples under N2, which is not dependent on external environment. By combining analysis from X-ray photoelectron spectroscopy (XPS), ADF-STEM and previous theoretical predictions, we propose that the second bleaching mechanism is caused by defect migration upon laser excitation, especially when the carbon concentration is high in the hBN lattice.
Results and discussion
Comparing the photostability of emitters in various hBN samples
To study the photostability of emitters in various hBN samples, we compare three of the most common hBN thin films that are commercially available, including monolayer CVD-grown hBN on copper foil (from Grolltex), multilayer CVD-grown hBN on copper foil (from Graphene Supermarket), and multilayer liquid exfoliated hBN nanoflakes suspended in ethanol/water mixture (from Graphene Supermarket). These hBN samples were transferred onto SiO2 substrates (Methods), without any post-treatment, and characterized with time-dependent photoluminescence (PL) measurements using a custom-built confocal spectroscopic microscope15. From our previous study, we find that the emission energy distribution in the visible range is organized into six ubiquitous species across diverse hBN samples15. The six discrete emission species are 2.25 ± 0.03 eV (P1), 2.15 ± 0.06 eV (P2), 1.98 ± 0.05 eV (P3), 1.90 ± 0.04 eV (P4), 1.81 ± 0.04 eV (P5), and 1.69 ± 0.06 eV (P6), respectively. Here in this study, we focus on the emitter species P3 with the emission energy of 1.98 ± 0.05 eV, which is observed in all hBN samples we examined (Supplementary Fig. 1a–c). Emission species P2 is also widely observed, and is the most abundant species in multilayer CVD hBN (Supplementary Fig. 1b).
Representative PL spectra (upper panels) and time-traces (lower panels) of P3 emission in different hBN samples are shown in Fig. 1a–d. In the case of monolayer CVD hBN, the emission signal of a zero-phonon line (ZPL) near 1.98 eV disappears after 5 s (blue arrow), and a new signal briefly appears at 13 s with its emission energy shifted +50 meV from the original (red arrow), which then disappears completely (Fig. 1a). Intriguingly, three different emission behaviors are observed in both monolayer and multilayer CVD samples (Fig. 1a, b): (1) blinking, which is a stochastic switching between on and off emission states, (2) spectral diffusion, which corresponds to the fluctuation in the emission energy of a ZPL, and (3) bleaching, which is the irreversible disappearance of emission signal under continuous excitation. Multilayer liquid exfoliated hBN (as-prepared, and not treated with thermal annealing) exhibits two different emission modes, which we label as type-I and type-II, respectively. Type-I emission in liquid exfoliated hBN exhibits blinking and bleaching (Fig. 1c), similar to the emission in CVD-grown hBN. Type-II emission in liquid exfoliated hBN is relatively stable over a long measurement time (Fig. 1d). It is noteworthy that such photostable emission is only observed in liquid exfoliated samples, and not in CVD samples. From room-temperature second-order correlation measurements, antibunching of P3 emitters is only observed in liquid-exfoliated type-II hBN (Supplementary Fig. 1e), while for P2 emitters antibunching is observed in both multilayer CVD-grown and liquid-exfoliated type-II hBN (Supplementary Fig. 1f, g).

Photoluminescence (PL) spectra at time 0 s (upper panels) and time-traces from 0 s to 150 s (lower panels) with zero-phonon lines (ZPLs) near 1.98 eV in as-prepared (a) monolayer CVD, (b) multilayer CVD, and multilayer liquid exfoliated hBN with (c) type-I and (d) type-II emission. In the lower panel of (a), the blue arrow indicates a ZPL disappearing at approximately 5 s, while the red arrow highlights a new emission peak appearing at approximately 13 s, which is shifted +50 meV from the original ZPL, before completely losing emission signal. Representative time-dependent intensity at a fixed emission energy is plotted in blue lines of (e) monolayer CVD, (f) multilayer CVD, and multilayer liquid exfoliated hBN with (g) type-I and (h) type-II emission. Orange lines in (e) to (h) are fitting with a cluster analysis, details of which are presented in Supplementary Note 1. In (e) monolayer CVD hBN, discrete bleaching steps are observed, where each step may correspond to an individual emitter.
When monolayer CVD hBN is under continuous laser excitation in the air, time-traces of intensity at a fixed emission energy show discrete bleaching steps, as shown in Fig. 1e and Supplementary Fig. 2a. Each bleaching step may correspond to an individual emitter, indicating that there could be a few emitters within the laser focus spot (approximately 0.8 μm in diameter). The difference in step heights could arise from varying intensity of each emitter. Meanwhile, multilayer CVD hBN and type-I emission in liquid exfoliated hBN show slower bleaching (Fig. 1f, g). Type-II emission in liquid exfoliated hBN displays a relatively stable time-trace of intensity (Fig. 1h). To analyze the bleaching process quantitatively, we sum up these time-traces of intensity, and obtain an overall decay curve of total intensity (Supplementary Fig. 2b). As shown in Fig. 2a, the total intensity I of monolayer CVD hBN emitters (green line) could be fitted with a single exponential decay function16 (red dashed line):

Since individual time-traces have discrete bleaching steps as shown in Fig. 1e and Supplementary Fig. 2a, the overall decay curves of total intensity are obtained by summing up N time-traces. Here, N = 31, 7, 19 and 6 for (a) monolayer CVD hBN, (b) multilayer CVD hBN, (c) liquid exfoliated hBN with type-I emission and (d) liquid exfoliated hBN with type-II emission, respectively. Red dashed lines and yellow dotted lines are fitting with one exponential decay function, and the sum of two exponential decay functions, respectively. From the one-exponential fitting with 95% confidence interval, the bleaching lifetime in monolayer CVD hBN is 6.6 ± 0.6 s. From the two-exponential fitting with 95% confidence interval, two bleaching lifetimes are extracted for monolayer CVD hBN, multilayer CVD and liquid exfoliated hBN with type-I emission, which are 2.9 ± 0.1 and 55 ± 3.2 s, 11 ± 4.1 and 170 ± 20 s, and 10 ± 1.6 s and 220 ± 30 s, respectively. Liquid exfoliated hBN with type-II emission has a bleaching lifetime longer than 2000 s, which is regarded as photostable.
Here I0 is the maximum intensity at t = 0 s, and τ is the characteristic lifetime of bleaching. From such analysis, τ is estimated to be 6.6 ± 0.6 s in monolayer CVD hBN in the air (with 95% confidence interval). As an alternative method to analyze the bleaching lifetime, a decay time constant can be extracted from the dwell time distribution24, which is estimated to be 4.7 s (Supplementary Fig. 2c). The bleaching lifetimes obtained from these two different methods are in agreement with each other.
In multilayer CVD hBN and type-I emission of liquid exfoliated hBN, the overall decay curves of P3 (1.98 ± 0.05 eV) emission are obtained by summing up individual time-traces in a similar method as described in Supplementary Fig. 2b. The total intensity I (green lines in Fig. 2b, c) can be fitted with the sum of two exponential decay functions (yellow dotted lines in Fig. 2b, c):
Here I0 is the maximum intensity at t = 0 s, \({\tau }_{1}\) and \({\tau }_{2}\) are two distinct bleaching lifetimes, and the ratio r indicates the percentage of decay corresponding to τ1. When the overall decay curves in the multilayer samples are fitted with just one exponential decay function, a large discrepancy appears in the initial 10 s (red dashed lines in Fig. 2b, c). The bleaching lifetimes of \({\tau }_{1}\) and \({\tau }_{2}\) are 11 ± 4.1 s and 170 ± 20 s in multilayer CVD hBN, and 10 ± 1.6 s and 220 ± 30 s in type-I emission of liquid exfoliated hBN, respectively. In both cases, the second bleaching lifetime \({\tau }_{2}\) is more than one order of magnitude larger than the first lifetime \({\tau }_{1}\), which indicates two distinct bleaching mechanisms.
Type-II emission of liquid exfoliated hBN displays a bleaching lifetime longer than 2000 s (Fig. 2d). Since this value is close to the upper limit of the measurement tool (Supplementary Fig. 3), type-II emission in liquid exfoliated hBN is confirmed to be photostable. Interestingly, the ZPLs of type-II emission are also sharper than that of type-I (Supplementary Fig. 1d). The average full width at half maximum (FWHM) of type-I emission in liquid exfoliated hBN is similar to that of monolayer and multilayer CVD hBN (Supplementary Fig. 1d). In another word, ZPLs with FWHM values larger than 100 meV appear to be less photostable, regardless of the sample preparation method (Supplementary Fig. 1d). The bleaching lifetimes of P3 emission in different hBN samples under various conditions are summarized later in Table 1. The observations presented above highlight that the photostability of hBN emitters with the same emission energy is specific to sample preparation method, which may correspond to different structural origins.
Bleaching mechanisms in monolayer CVD hBN
We now investigate the bleaching mechanism of monolayer hBN emitters under different atmospheres, with the assistance of a custom-built environmental chamber. The monolayer sample of CVD-grown hBN was placed in a small sealed chamber with a quartz observation window, and the chamber could be filled with different gases, such as N2, O2 and water vapor (Fig. 3a). Inspired by the fact that many organic fluorophores are degraded from reactions with O2 or water25, we first filled the environmental chamber with N2 to minimize the concentrations of O2 or water vapor. Under such an atmosphere, the bleaching of P3 (1.98 ± 0.05 eV) emission in monolayer CVD hBN is significantly slower (Fig. 3b), compared to the bleaching in the air (Fig. 1a). It should be noted, however, that N2 atmosphere does not mitigate bleaching completely. In fact, by fitting the overall decay curve of eight time-traces, we obtain two bleaching lifetimes of \({\tau }_{1}\) = 5.0 ± 1.1 s and \({\tau }_{2}\,\)= 150 ± 10 s (Fig. 3d). As described in Eq. 2, the ratio r is 0.46 corresponding to the decay process of \({\tau }_{1}\). The value of \({\tau }_{1}\) is almost the same as the bleaching lifetime in the air (i.e., 6.6 ± 0.6 s), while \({\tau }_{2}\) is similar to the second bleaching lifetime of multilayer CVD hBN in the air (i.e., 170 ± 20 s).

a Schematic of a custom-built environmental chamber with a quartz observation window, which could be filled with different gases. b Time-trace of P3 emission in monolayer CVD hBN under a N2 atmosphere. c Initial 10 s of overall decay curves of P3 emission in monolayer CVD hBN under N2 (blue line), N2 with saturated water vapor (green line), air (yellow line) and O2 (red line). The number of time-traces (N) included in each decay curve is 8 (blue, N2), 20 (green, N2 with saturated water vapor), 31 (yellow, air) and 36 (red, O2), respectively. d Intensity decay curve (blue line) of monolayer CVD hBN under N2 (N = 8) is fitted with a single exponential decay function (red dashed line) and two exponential decay functions (yellow dashed line), where the single-decay-fit shows larger discrepancy. From the two-exponential fit with 95% confidence interval, we obtain two bleaching lifetimes of \({\tau }_{1}\) = 5.0 ± 1.1 s and \({\tau }_{2}\) = 150 ± 10 s. e Schematic of laser power dependence measurement, with monolayer CVD hBN on SiO2 substrate in the air. f Illustration of a three-energy-system with ground singlet state (G), excited singlet state (S) and excited triplet state (T). Photobleaching reactions could occur from both excited states, with microscopic rate constants of kbS and kbT. Rate constants of ke, kf and kIC correspond to excitation, fluorescent emission and internal conversion, respectively. Additionally, kISC and kT correspond to intersystem crossing and depopulation from T to G, respectively. g Beaching rate 1/\(\tau\), i.e., the inverse of bleaching lifetime, is linearly dependent on the power density of the incident laser, which could be well explained by the three-energy-system in (f). For the three data points in the lower power density range, each is extracted from the sum of ~100 time-traces; and for the other three, each is extracted from the sum of 20 to 30 time-traces. The error bars correspond to 95% confidence intervals. h Four-layer stack (right) is constructed by repeatedly transferring monolayer CVD hBN onto the same substrate. i Total intensity of four-layer stack (green line) in the air (N = 27) is fitted with a single exponential decay function (red dashed line) and two exponential decay functions (yellow dashed line), where the latter is a better fit. The two bleaching lifetimes are 11 ± 3.9 s and 150 ± 20 s, respectively.
When saturated water vapor at room temperature is introduced into the N2 atmosphere, the decay of total intensity is almost the same in the first 10 s (Fig. 3c), and changes slightly later (Supplementary Fig. 5 and Supplementary Note 2 for longer time scale). These findings suggest that water vapor is not playing a significant role. We then conducted the bleaching experiment in an O2 rich atmosphere (i.e., 84% O2 and 16% N2), where the bleaching decay curve is almost the same as that in the air (Fig. 3c). While oxygen sources from the SiO2 substrate are known to quench fluorescent carbon nanotubes26, we find that the substrate material does not affect the bleaching lifetime of monolayer hBN significantly. When atomic layer deposited Al2O3 is underneath the hBN sample, the bleaching lifetime is almost the same as that on bare SiO2 substrate (Supplementary Fig. 6). Such Al2O3 coating has been utilized to suppresses the blinking in multilayer CVD hBN at cryogenic temperatures17. We would like to point out that O2 could be adsorbed on the top of hBN, and even get trapped between the hBN thin film and the substrate. The observations presented above indicate that O2 plays a key role in the photobleaching, and the bleaching rate is saturated with the concentration of O2 in the air.
To further elucidate the bleaching mechanism of monolayer CVD hBN, we performed power dependence measurements of the bleaching rate (i.e., the inverse of the bleaching lifetime τ) in the air (Fig. 3e). Confocal PL spectroscopy was used for measurements when the laser power density (i.e., excitation irradiance) is larger than 40 μW μm−2. In the range where the irradiance is lower than the minimum (i.e., 40 μW μm−2) of the confocal PL tool, wide-field imaging was conducted, as described in Supplementary Fig. 4. From these measurements, the bleaching rate 1/τ in the air is found to be linearly dependent on the power density of the excitation laser (Fig. 3g). Such a linear relationship is often observed in the photochemical bleaching of organic dyes, and could be well explained by a simple three-level energy system27, including the ground singlet state (G), excited singlet state (S) and excited triplet state (T), as illustrated in Fig. 3f. Assuming that photobleaching reactions could occur from all excited states (both singlet and triplet states), the overall bleaching rate could be described by taking into account different microscopic transition rates between the three energy levels (Supplementary Fig. 7 and Supplementary Note 3 for a description of the reaction model). Note that laser-induced thermal heating alone is excluded from possible bleaching mechanisms here, since the maximum temperature increase is estimated to be smaller than 100 °C (Supplementary Fig. 8). The analysis above reinforces our hypothesis that bleaching in monolayer CVD hBN is dominated by an irreversible photochemical reaction between emitting defects and O2 in the air.
We next demonstrate that emitters in monolayer CVD hBN can be stabilized by a simple strategy of stacking. By transferring monolayer hBN thin films repeatedly onto the same substrate, we constructed a four-layer stack of hBN with random lattice alignment (Fig. 3h). Here, we choose atomically thin hBN itself as the protecting layer because of the airtightness from hBN stacking28. This strategy also avoids possible filtering effects or external PL signals from other materials29 that could modify the emission of hBN. The overall decay curve of the four-layer hBN stack in the air is presented in Fig. 3i, which could be fitted with the sum of two exponential decay functions. The bleaching in the four-layer stack is significantly slower compared to that of single layer in the air (Table 1). Interestingly, the extracted bleaching lifetimes of 11 ± 3.9 s and 150 ± 20 s are similar to those of multilayer CVD hBN (Table 1). When a single exponential function is used for the fitting, there is a large discrepancy in the initial 10 s. The better fit with two bleaching lifetimes can be interpreted in the following way: While the bottom three layers are protected from reaction with O2 in the air, the top layer is in direct contact with the atmosphere. In fact, the ratio r = 0.36 of the four-layer stack, which corresponds to the faster decay process, is close to the expected 0.365 (i.e., 0.25 + 0.25*0.46). Here, 0.25 corresponds to the top layer, and 0.25*0.46 corresponds to the bottom layer, using the ratio r = 0.46 from the case of N2, where O2 could be trapped between hBN and the substrate (Table 1). It is noteworthy that for the layer directly in contact with the air, there is still a significant difference between monolayer (approximately 5 s) and multilayer systems (approximately 10 s). This could be explained by the stabilizing effect of bond distortions—once there is a neighboring layer, nitrogen edge atoms near vacancies could form interlayer bonds that minimize the energy30.
The second bleaching lifetime of the four-layer stack, 150 ± 20 s, is the same as the longer bleaching lifetime of single layer under N2 (Table 1). The consistency of the second lifetime, which is more than 10 times larger than the first bleaching lifetime under each condition, indicates that there is a second bleaching mechanism distinct from the first. The second bleaching process may also exist in monolayer hBN in the air, but with a small percentage (i.e., a large r), and thus becomes negligible. Due to good stacking qualities between layers in hBN31, intercalation and diffusion of the external molecules, such as O2 and water vapor in the air, are highly unlikely28. If emitters surrounded by other hBN layers could bleach by reacting with the external molecules, multilayer CVD and liquid exfoliated hBN should have similar bleaching lifetimes, which contradicts our observations (Table 1). Accordingly, we deduce that the second bleaching mechanism of hBN, which is observed in multilayer CVD samples in the air, type-I emission of liquid exfoliated hBN in the air, and monolayer CVD hBN under N2, is independent of external environment.
The experimental results from stacking also support our conclusion that photobleaching can be significantly mitigated by shielding O2, consistent with the fact that emitters in multilayer hBN are more stable than those in monolayer hBN (Table 1). Note that the average emitter lifetime of P3 is increased by more than 20 times from both stacking and introducing N2, as shown in the one-exponential model comparison in Table 1. This ~ 200 s lifetime is still insufficient for practical applications and we envision that our stacking strategy could be further improved by constructing a tri-layer stack, where a monolayer with photostable emitters (that have the photostability for the intrinsic bleaching mechanism as we discuss later) is sandwiched by two neighboring hBN monolayers that are defect-free. Such tri-layer stack will have the advantages of efficient light extraction due to the extremely thin dielectric thickness, and high photostability as a result of the protection layers. With the recent breakthroughs in hBN synthesis, where wafer-scale production of single-crystal hBN monolayers has been achieved20,21,22, this proposed tri-layer stack could be realized in the near future.
ADF-STEM of monolayer CVD hBN
After a systematic investigation of the bleaching mechanisms by controlling the external environment, we employ ADF-STEM imaging to characterize the intrinsic properties of hBN thin films. ADF-STEM of the same monolayer CVD hBN (from Grolltex) enables a detailed analysis of atomic-scale defect structures with high signal-to-noise ratio. The monolayer hBN was first suspended on a holey carbon support film with 2 µm-diameter holes (Fig. 4a), and emitters were located in a low-power, wide-field photoluminescence measurement (Fig. 4b). We then performed hyperspectral mapping of the whole hBN-covered grid, from which a representative ZPL near 1.98 eV is shown in Fig. 4c (red lines). Note that monolayer hBN suspended on a TEM grid has much weaker photoluminescence (PL) signal from emitters than that on a SiO2/Si substrate. Consistent with the statistics shown in Supplementary Fig. 1a, emitters in the monolayer hBN (from Grolltex) covering the grid are exclusively P3, and they all bleached out after 5 s (which is the exposure time) when a spectrum is taken.

False-color optical micrograph (a) and wide-field photoluminescence imaging (b) of monolayer CVD hBN, suspended on a holey carbon film of TEM grid. Scale bars in (a) and (b) correspond to 20 μm. c Confocal photoluminescence spectrum, showing a zero-phonon line (+) near 1.98 eV (P3) and one-phonon sideband (*) from the suspended hBN monolayer, and a Raman response (o) from the carbon film of the grid. Light-colored and dark-colored lines correspond to the raw and smoothed data, respectively. P3 emission is in red, and the background spectrum in black. In (a) and (b), the circle denotes the position of the background measurement, and the dotted square represents the position of the P3 spectrum. Note that only P3 emission is observed in the hBN covering this TEM grid. d Zoomed-out ADF-STEM image of a hole in the grid, which is the same position as the dotted square in (a) and (b). Scale bar corresponds to 1 µm. e ADF-STEM image of a typical clean region of hBN. The green circle indicates a point defect. Scale bar corresponds to 2 nm. f Representative ADF-STEM analysis of monovacancies missing boron with atomic-scale resolution, showing experimental images (left column), simulated images (middle column) and their corresponding atomic structures (right column). The top row presents a monovacancy with NNN innermost edge atoms, while the bottom row shows OCC replacing NNN. g Averaged intensity of two consecutive frames under high magnification, showing a monovacancy missing boron, which has two O replacing inner most edge N, with higher uniformity in the image by noise reduction. Scale bar corresponds to 2 Å in (f) and 5 Å in (g), respectively. h A representative image of vacancy-free region under high magnification, the white arrow indicates the direction of line 1, 2 and 3. i Line profile analysis corresponding to line 1, 2, 3 in (h). The y-axis values indicate the intensity. j, ADF-STEM image from (h) with annotations, from the analysis in (i). Scale bars correspond to 5 Å in (h) and (j).
After photobleaching, the hBN-covered grid was characterized by ADF-STEM, with focus on the same spot where P3 emission was observed (Fig. 4d). To maximize the percentage of STEM coverage, we focused on one emitter position (shown in Fig. 4a, b). Note that high-coverage and high-resolution under transmission electron microscopy are two conflicting requirements that cannot be achieved at the same time. To address this challenge, we have implemented a combined low-resolution and high-resolution technique. We first searched for vacancy defects under low-magnification, and only switched to high-resolution mode when a possible defect shown up. An example of clean areas of hBN is presented in Fig. 4e, where we can locate one defect under relatively low-magnification. While most of the examined hBN areas are vacancy-free, we have found 22 vacancies in total, where 15 are monovacancies (Supplementary Table 1). The majority of monovacancies are missing boron (VB), and only one is missing nitrogen (VN). Although vacancy defects could be generated in-situ during the imaging process30, our experimental beam conditions were optimized to minimize the number of such events (Supplementary Fig. 9).
Among the VB monovacancies, 12 of them exhibit sufficiently high quality with atomic scale resolution, allowing the accurate assignment of atom species (Fig. 5). Due to the elastic scattering of electrons by Coulomb interaction, the intensity of an atom in ADF-STEM is directly related to its atomic number32. We thus achieve high contrast imaging of boron (B), carbon (C), nitrogen (N), and oxygen (O) atoms by using long camera lengths, and collecting as many low-angle scattered electrons as possible, outside of the central bright field disk. This leads to increased signal compared to the normal high-angle annular dark-field (HAADF) conditions. Line profile analysis of the relative intensities of different atoms is presented in Supplementary Note 4. A comparison between experimental and simulation results (Supplementary Figs. 10, 11, 12, and 13) shows a consistent contrast trend following Z1.85, where Z is the atomic number. O atom shows higher intensity than B and N atoms, while the intensity of C atom lies in the middle between that of B and N (Fig. 4f).

A nomenclature is introduced to label the heteroatom substitutions, which enumerates an edge of the innermost three N atoms clockwise as 1 to 3 highlighted in bold type, and the remaining B and N atoms as 4 to 12.
Monovacancies have been widely discussed as potential structural origins of luminescence in hBN4,19,33,34. We are now able, for the first time, to present a rich variety of them with detailed atomic-level-analysis. We introduce a nomenclature to label the heteroatom compositions of VB, which enumerates the inner three N atoms clockwise as 1 to 3 and the remaining B and N atoms as 4 to 12 along the vacancy edge (Fig. 5). Intriguingly, a majority of VB monovacancies, 10 out of 12, show heteroatom substitution of the innermost N atoms, including C and O atoms (Fig. 5). C substitution also occurs in the remaining N and B edge atoms. The observed heteroatom insertions imply that the innermost N atoms of a boron monovacancy may be chemically reactive, which could explain why the innermost N can be easily replaced with O and C. Surprisingly, a VB monovacancy with oxygen atoms substituting two inner-most nitrogen atoms (NOO), remains unchanged after two consecutive high-resolution scans (Fig. 4g), indicating that this oxygen-rich vacancy structure is relatively stable. The frequent O substitution of the innermost N atoms raises the possibility that such structures are the product of photobleaching reactions, since ADF-STEM was performed after the photobleaching experiment in the air.
Another interesting question worth further investigation is whether the innermost N atoms of a monovacancy could be protected by hydrogen34,35. When a boron is removed from the hBN lattice, dangling bonds could form with unpaired electrons36, which should be highly reactive and sensitive to the environment. On the other hand, if some or all of these N atoms are protected by hydrogen, a meta-stable or stable structure could form. The amount of hydrogen formation could be associated with specific sample preparation method. With the current ADF-STEM technique, however, it is extremely hard to identify the existence of hydrogen atoms. When there is an additional atomic layer of hBN, e.g., in multilayer samples, interlayer bonds could help stabilize vacancy structures30.
Interestingly, C and O insertion are also observed in vacancy-free regions. C could substitute both B and N atoms (Fig. 4h–j), whereas O has only been observed substituting N atoms (Supplementary Fig. 14). These observations are consistent with theoretical predictions of heteroatom insertion34. The frequent C substitution in both vacancy defects and vacancy-free regions is coherent with the fact that C replacing B or N would induce negligible lattice mismatch. The C-C structures embedded in the BN lattice (Fig. 4h–j) agree with earlier theoretical predictions of hybrid B-C-N structures37. Such hybrid B-C-N structures have been attracting considerable research attention due to their visible-light photocatalysis effects38. Note that among the 22 vacancy defects (Supplementary Table 1) we observe, C substitution along vacancy edges is observed in 9 of 12 VB monovacancies (Fig. 5), and a divacancy with hybrid structures (Supplementary Fig. 15a–d). C insertion in the lattice is also observed in the proximity to a VB monovacancy (Supplementary Fig. 15e–g). This raises the question whether C is related to vacancy formation during CVD growth.
To briefly summarize, atomic-scale ADF-STEM results enable the identification of specific forms of carbon insertion in the hBN lattice, and additionally reveal a rich variety of atomic compositions of VB. The above findings from ADF-STEM highlight the versatility of possible defect structures hBN can host, which has important implications not only in engineering emitters for photonic applications2, but also in tuning electronic properties of 2D devices39. We believe that our attempt to link photo-physical properties with atomic-scale characterization of hBN thin films will inspire extensive studies in the near future. Accuracy of colocalizing the ADF-STEM and PL could be improved further, e.g., by combining a customized TEM grid with smaller hole sizes and super-resolution PL imaging techniques35. Only through such persistent efforts will we be able to unambiguously determine the structural origins of quantum emission in hBN.
XPS analysis and thermal annealing effect
To advance our understanding of heteroatom insertion in the hBN lattice, we conduct XPS analysis of various hBN samples. XPS is a surface-sensitive approach to identify different elements and their chemical states in contrast to elemental analysis by inductively coupled plasma technique (Supplementary Note 5). Before XPS measurements, sample surfaces were first sputtered with argon (Ar) ion beam to remove adsorbed hydrocarbons from the environment. When the Ar sputtering time increases, the atomic concentration of C in multilayer CVD sample decreases and then reaches a stable value (Supplementary Fig. 16). After Ar sputtering for 1 min (Methods), CVD hBN still contains a detectable amount of C (Fig. 6c), i.e., 8.3 at%, whereas liquid exfoliated hBN does not exhibit C 1 s signal (Fig. 6f). We, therefore, exclude the role of adventitious C in our XPS results after Ar cleaning. Since the Ar sputtering inevitably removes several layers of hBN, we focus on multilayer samples for XPS characterization. From peak deconvolution of the XPS spectra (Supplementary Note 6), a small amount of B-O is observed in both CVD (Fig. 6a) and liquid exfoliated hBN (Fig. 6d). By contrast, C 1 s signal is only detected in CVD samples, where sp2 C-C bonds, i.e., graphene-like domains, are the mostly likely forms of carbon substitution (Fig. 6c and Supplementary Fig. 18). These findings are consistent with ADF-STEM imaging of monolayer CVD hBN, where O is only observed to substitute N, while C is observed to replace both N and B atoms. In fact, when the C concentration is high, atomic layers of hybridized boron nitride and graphene domains can be grown by CVD, with methane and ammonia borane as precursors40.

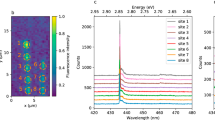
XPS spectra of hBN samples that are pre-cleaned with Ar ion sputtering, showing B 1 s (a, d), N 1 s (b, e), and C 1 s (c, f). While (a) to (c) represent multilayer CVD hBN, (d) to (f) are results of multilayer liquid exfoliated hBN. The C concentration is higher in the CVD sample, mainly in the form of sp2 C-C, which corresponds to graphene domains. Note that C-N is different from C-(N)3, with details in Supplementary Fig. 18 and Supplementary Table 3. g Photoluminescence (PL) spectra of type-I emission in liquid exfoliated hBN, before (pink) and after (yellow) annealing at 850 ˚C in Ar for 30 min. h PL spectra of type-II emission in liquid exfoliated hBN, with sharper zero-phonon lines (ZPLs), before and after annealing at 850 ˚C in Ar for 30 min. i Number of each emitter species Pi (i = 1, 2, 3, 4) in liquid exfoliated hBN, before (left bars) and after (right bars) 850 ˚C annealing in Ar. Pink and yellow colors indicate broad ZPLs, corresponding to type-I emission. Blue and green indicate sharp ZPLs, corresponding to type-II emission. Before annealing, P3 has a large percentage of type-I emitters, which disappear almost completely after annealing. All type-II emitters remain the same after annealing, regardless of the emission species Pi (i = 1, 2, 3, 4). More details of thermal annealing experiments are presented in Supplementary Note 9.
We further confirm that carbon could be easily introduced in a common CVD growth method of hBN thin films (without methane, only with ammonia borane precursor). We synthesized hBN from a lab-built low-pressure CVD reactor, and typical hBN crystals with triangular morphology are observed in scanning electron microscopy (SEM) and optical microscopy (Supplementary Fig. 20a, b). Besides the triangular hBN crystals, however, we also find polygonal sheets that are approximately 40 µm in lateral size (indicated by the arrows in Supplementary Fig. 20a, b). Raman spectroscopy of these regions exhibits typical graphene response (D, G and 2D peaks), confirming that the polygonal sheets are indeed graphene (Supplementary Fig. 20c). Such formation is very likely attributed to the presence of organic impurity in the borane-ammonia precursor, which can act as the precursor for graphene growth. Since most of CVD-grown hBN are synthesized with borane-ammonia precursors, the existence of graphene islands or similar forms of carbon insertion could be a ubiquitous phenomenon in CVD hBN samples, which also agrees with theoretical predictions that phase-separated hBN and graphene islands are preferred to form when B, C and N are all present37.
In the XPS characterization of liquid exfoliated hBN, no C 1 s peak is detected after Ar cleaning (Fig. 6f). It should be noted, however, that the detection limit of XPS is 0.1–1 atomic %. Hence, liquid exfoliated samples could still contain low concentrations of carbon. Additionally, N-C and N-H have similar peak positions in the XPS response (Fig. 6b, Supplementary Table 3), a small amount of C could therefore exist in the form of N-C in liquid exfoliated hBN. Since liquid exfoliation of hBN is carried out in a water/ethanol mixture, the edge of exfoliated nanoflakes as well as vacancies in the flakes could be functionalized by N-H, as indicated in Fig. 6e. Interestingly, photostable and narrowband ZPLs have been predominantly observed in liquid exfoliated hBN after thermal annealing, as shown in Supplementary Figs. 21a–c and 22a–d, as well as in our previous study15. The annealing process involves heating hBN samples in Ar gas under 1 Torr at 850 °C for 30 min. Similar heat treatment has been widely applied to liquid exfoliated hBN since the pioneering work by ref. 4.
To advance the understanding of high-temperature Ar annealing effect, we performed bleaching lifetime analysis from overall intensity decay of emitters in multilayer CVD-grown and liquid-exfoliated hBN (Supplementary Note 8), as summarized in Table 1. We find that the most stable emitters are only observed in liquid-exfoliated samples, not even in annealed CVD hBN. The lifetimes of emitters that survive the annealing process are longer, regardless of the emitter species. It should be noted, however, that P2 and P3 emitters behave differently both before and after annealing (Table 1), which indicates that these two emission species could have different structural origins. Additionally, we measured PL spectra at exactly the same locations of liquid-exfoliated hBN nanoflakes, which were drop-cast onto a SiO2 substrate, before and after the annealing process (Supplementary Note 9). In as-prepared liquid exfoliated hBN, we observe two different emission modes at the same emission energies, as presented in Fig. 1. Type-I emitters exhibit blinking and bleaching behaviors, similar to those in CVD-grown hBN. Type-II emitters, on the contrary, are photostable and have sharper ZPLs than type-I (Supplementary Fig. 1). Notably, the photostability of type-II emission appears to be independent of the emission energy, both before (Supplementary Fig. 23) and after Ar annealing (Supplementary Fig. 24). Most of type-I emitters disappeared after Ar annealing, while all type-II emitters remained the same after the annealing process (Fig. 6g–i and Supplementary Figs. 27–32). This indicates that type-I and type-II emitters could have different structural origins, despite exhibiting the same emission energies. We emphasize again that photostable emitters (i.e., type-II) are only observed in liquid exfoliated hBN, which are unaffected by high temperature Ar annealing.
In a pure hBN lattice, migration barriers of VB monovacancies were predicted to be approximately 2.3–3.1 eV, corresponding to annealing temperatures of 840–1110 K34,41. We therefore propose that the disappearance of type-I emitters in liquid exfoliated hBN after Ar annealing at 850 °C (i.e., 1123 K) could arise from vacancy migrations. Very recently, defect migration induced self-healing has been observed in plasma-treated graphene42. Ouyang, et al. simulated migrations of vacancies at the interface between hBN and graphene43, and predicted that the existence of a graphene domain will reduce migration barriers and accelerate defect migration, resulting from the trend of C-C < C-B < C-N < B-N in the bonding strength. Monovacancies tend to aggregate at the interface, forming divacancies or even vacancy clusters43. It was also predicted that C atoms in the hBN lattice tend to migrate to the edge of vacancies43.
We would like to point out that carbon-substituted monovacancies and a hybrid carbon-divacancy are observed in ADF-STEM characterization of photobleached CVD hBN (Fig. 5 and Supplementary Fig. 15). The photon energy of 2.33 eV from laser excitation in our photoluminescence experiment could activate vacancy migration in time-dependent measurements. From our experimental observations and previous theoretical predictions, we propose that C impurities near the emitting defects may cause bleaching in hBN by changing the structures of emitting defects upon laser excitation. The final product of such structural change could be a carbon-modified defect, or an enlarged vacancy defect from coalescence that is no longer emitting. The second bleaching mechanism that is ubiquitously observed in CVD-grown hBN is very likely associated with high C concentration and their specific substitution in the hBN lattice, and thus not dependent on the external environment.
We note that the accurate role of C in defect formation might be more complicated. Recent studies of C-rich hBN19,44,45 show that introducing carbon into hBN could increase emitter densities. The photostability of these carbon-induced emitters under continuous laser excitation, however, were not discussed. Koperski, M., et al. studied ZPLs from carbon-doped bulk hBN crystals, which correspond to two types of ZPLs with emission energy of 1.54 eV and 2 eV in the cryogenic temperature range, respectively44. It should be noted that the ZPLs of these doping-induced emitters are very broad (i.e., almost unrecognizable) at room temperature44. In the study from Mendelson, N., et al., the emission energies of ZPLs they focused on are near 2.15 eV, which we label as P2 (in our study). When the carbon implantation concentration is increased, the ZPLs become broader19, looking similar to the broad ZPLs from CVD hBN samples (in our study) that exhibit sp2 carbon signal from XPS. At low carbon implantation concentration, the ZPLs are narrow and sharp19, which look similar to the ZPLs from liquid-exfoliated hBN (in our study) that has non-detectable carbon from XPS characterization. It is, therefore, highly possible that carbon concentration plays a key role in determining the performance of hBN single-photon emitters. Our findings reveal that optimizing C concentration, and preventing the formation of graphene domains during hBN synthesis is critical in the scalable and reproducible fabrication of hBN with photostable and narrow-band emitters.
Conclusions
In conclusion, we have uncovered a key to improve photostability of emitters in hBN, which is shielding O2 from the environment and optimizing carbon substitution in the hBN lattice. We find that photobleaching of hBN emitters in direct contact with the air is dominated by photochemical reactions of emitting defects with O2. To mitigate such bleaching, we demonstrate two simple yet effective strategies of shielding O2 by using a N2 atmosphere or stacking additional hBN layers. The average emitter lifetime is increased by more than 20 times by such strategies. By comparing photostability of emitters in diverse hBN samples exhibiting similar emission energies, we observe a second, slower bleaching process independent of the external environment. Our experimental results of ADF-STEM, XPS, and thermal annealing, combined with previous theoretical predictions, strongly suggest that the second bleaching process is due to carbon-assisted vacancy migration upon laser excitation. With atomic-scale imaging of ADF-STEM, we identify configurations of carbon-substitution in the hBN lattice, and provide detailed information of monovacancy compositions, paving the way for more in-depth studies of structural origins of hBN quantum emission. The knowledge we gained about defect engineering in hBN will also be insightful to broader research fields, such as photocatalysis effect of B-C-N materials, tuning electronic properties for integrated 2D devices, and future sensing applications based on 2D quantum materials.
Methods
Sample preparation
All multilayer liquid exfoliated hBN was purchased from Graphene Supermarket, with hBN nanoflakes suspended in ethanol/water solution. The solution was drop-cast onto SiO2 (300 nm) / Si substrate and dried at 100 °C. All multilayer and monolayer CVD-grown hBN (on copper foil) were obtained from Graphene Supermarket and Grolltex, respectively. CVD hBN film was spin-coated with poly(methyl methacrylate) (950 PMMA A4, MicroChem) at 2000 rpm for 2 min, dried at 70 °C for 30 min, and etched in ammonium persulfate solution (APS-100, Transene) for 1 h to dissolve the copper foil. The PMMA protected hBN thin film was then rinsed thoroughly by deionized water, and transferred onto the SiO2 (300 nm) / Si or Al2O3 (10 nm) / SiO2 (300 nm) / Si substrate. Afterwards, the sample was dried at 80 °C for 1 h and baked at 180 °C for 15 min to stretch wrinkles. Finally, the PMMA layer was removed by acetone, and the hBN sample was rinsed gently with isopropyl alcohol and water, and dried at 80 °C for 1 h. Four-layer stack of hBN was constructed by repeating the transfer process of monolayer hBN four times. To measure the bleaching lifetime under N2, O2 rich, and water vapor atmospheres, monolayer CVD hBN transferred on a substrate was fixed inside a small lab-built environmental chamber with quartz window for optical measurements. Ar thermal annealing experiment was carried out in a tube furnace at 850 °C for 30 min, with 50 sccm Ar flow under 1 Torr vacuum.
Optical measurements
All measurements were conducted at room temperature46. For excitation, a continuous-wave 532 nm laser was used. A custom-built confocal spectroscopic microscope is equipped with high-precision piezoelectric scanning stage. The power density of the excitation laser is fixed at approximately 900 μW/μm2 unless specified otherwise. The photoluminescence signal was collected by a spectrometer (Pylon 100BR eXcelon). Wide-field imaging is achieved with a EMCCD camera (Andor iXon).
Material characterization
For the annular dark-field scanning transmission electron microscopy (ADF-STEM), monolayer CVD hBN (from Grolltex) was transferred onto a TEM grid with holey carbon film (Quantifoil). Imaging was conducted using an aberration-corrected JEOL ARM200CF STEM equipped with a CEOS probe corrector operated at an accelerating voltage of 80 kV. For X-ray photoelectron spectroscopy (XPS), liquid exfoliated hBN solution (from Graphene Supermarket) was drop-cast on SiO2 substrates, and multilayer CVD hBN (from Graphene Supermarket) on copper foil was used as purchased (without the transfer process onto SiO2 substrates). XPS spectra were obtained by using ULVAC-PHI, PHI VersaProbe II with mono-chromated Al Kα source. hBN were cleaned with Ar ion sputtering (500 eV, 500 nA for 1 min) before XPS.
Data availability
All data generated or analyzed during this study are included in this published article (and its supplementary information files).
References
Eisaman, M. D., Fan, J., Migdall, A. & Polyakov, S. V. Single-photon sources and detectors. Rev. Sci. Instrum. 82, 071101 (2011).
Aharonovich, I., Englund, D. & Toth, M. Solid-state single-photon emitters. Nat. Photonics 10, 631–641 (2016).
He, M. et al. Single quantum emitters in monolayer semiconductors. Nat. Nanotechnol. 10, 497–502 (2015).
Tran, T. T., Bray, K., Ford, M. J., Toth, M. & Aharonovich, I. Quantum emission from hexagonal boron nitride monolayers. Nat. Nanotechnol. 11, 37–41 (2016).
Peyskens, F., Chakraborty, C., Muneeb, M., Thourhout, D. V. & Englund, D. Integration of single photon emitters in 2D layered materials with a silicon nitride photonic chip. Nat. Commun. 10, 4435 (2019).
Mak, K. F. & Shan, J. Photonics and optoelectronics of 2D semiconductor transition metal dichalcogenides. Nat. Photonics 10, 216–226 (2016).
Grosso, G. et al. Tunable and high-purity room temperature single-photon emission from atomic defects in hexagonal boron nitride. Nat. Commun. 8, 705 (2017).
Noh, G. et al. Stark tuning of single-photon emitters in hexagonal boron nitride. Nano Lett. 18, 4710–4715 (2018).
Tran, T. T. et al. Quantum emission from defects in single-crystalline hexagonal boron nitride. Phys. Rev. Appl. 5, 034005 (2016).
Martínez, L. J. et al. Efficient single photon emission from a high-purity hexagonal boron nitride crystal. Phys. Rev. B 94, 121405 (2016).
Vogl, T., Campbell, G., Buchler, B. C., Lu, Y. & Lam, P. K. Fabrication and deterministic transfer of high-quality quantum emitters in hexagonal boron nitride. ACS Photonics 5, 2305–2312 (2018).
Jungwirth, N. R. et al. Temperature dependence of wavelength selectable zero-phonon emission from single defects in hexagonal boron nitride. Nano Lett. 16, 6052–6057 (2016).
Tran, T. T. et al. Robust multicolor single photon emission from point defects in hexagonal boron nitride. ACS Nano 10, 7331–7338 (2016).
Chejanovsky, N. et al. Structural attributes and photodynamics of visible spectrum quantum emitters in hexagonal boron nitride. Nano Lett. 16, 7037–7045 (2016).
Kozawa, D. et al. Discretized hexagonal boron nitride quantum emitters and their chemical interconversion. Nanotechnology 34, 115702 (2023).
Stern, H. L. et al. Spectrally resolved photodynamics of individual emitters in large-area monolayers of hexagonal boron nitride. ACS Nano 13, 4538–4547 (2019).
Li, X. et al. Nonmagnetic quantum emitters in boron nitride with ultranarrow and sideband-free emission spectra. ACS Nano 11, 6652–6660 (2017).
Chugh, D. et al. Flow modulation epitaxy of hexagonal boron nitride. 2D Mater. 5, 045018 (2018).
Mendelson, N. et al. Identifying carbon as the source of visible single photon emission from hexagonal boron nitride. Nat. Mater. 20, 321–328 (2020).
Lee, J. S. et al. Wafer-scale single-crystal hexagonal boron nitride film via self-collimated grain formation. Science 362, 817–821 (2018).
Wang, L. et al. Epitaxial growth of a 100-square-centimetre single-crystal hexagonal boron nitride monolayer on copper. Nature 570, 91–95 (2019).
Chen, T. et al. Wafer-scale single-crystal hexagonal boron nitride monolayers on Cu (111). Nature 579, 219–223 (2020).
Mendelson, N. et al. Engineering and tuning of quantum emitters in few-layer hexagonal boron nitride. ACS Nano 13, 3132–3140 (2019).
Zhang, W. et al. Single-molecule imaging reveals transforming growth factor-b-induced type II receptor dimerization. Proc. Natl Acad. Sci. 106, 15679–15683 (2009).
Demchenko, A. P. Photobleaching of organic fluorophores: quantitative characterization, mechanisms, protection. Methods Appl. Fluoresc. 8, 022001 (2020).
Tayo, B. O. & Rotkin, S. V. Charge impurity as a localization center for singlet excitons in single-wall nanotubes. Phys. Rev. B 86, 125431 (2012).
Eggeling, C., Widengren, J., Rigler, R. & Seidel, C. A. M. Photobleaching of fluorescent dyes under conditions used for single-molecule detection: evidence of two-step photolysis. Anal. Chem. 70, 2651–2659 (1998).
Holler, J. et al. Air tightness of hBN encapsulation and its impact on Raman spectroscopy of van Der Waals materials. 2D Mater. 7, 015012 (2020).
Lorchat, E. et al. Filtering the photoluminescence spectra of atomically thin semiconductors with graphene. Nat. Nanotechnol. 15, 283–288 (2020).
Alem, N. et al. Probing the out-of-plane distortion of single point defects in atomically thin hexagonal boron nitride at the picometer scale. Phys. Rev. Lett. 106, 126102 (2011).
Constantinescu, G., Kuc, A. & Heine, T. Stacking in bulk and bilayer hexagonal boron nitride. Phys. Rev. Lett. 111, 036104 (2013).
Krivanek, O. L. et al. Atom-by-atom structural and chemical analysis by annular dark-field electron microscopy. Nature 464, 571–574 (2010).
Huang, B., Xiang, H., Yu, J. & Wei, S. Effective control of the charge and magnetic states of transition-metal atoms on single-layer boron nitride. Phys. Rev. Lett. 108, 206802 (2012).
Weston, L., Wickramaratne, D., Mackoit, M., Alkauskas, A. & Van de Walle, C. G. Native point defects and impurities in hexagonal boron nitride. Phys. Rev. B 97, 214104 (2018).
Comtet, J. et al. Direct observation of water-mediated single-proton transport between hBN surface defects. Nat. Nanotechnol. 15, 598–604 (2020).
Turiansky, M. E., Alkauskas, A., Bassett, L. C. & Van de Walle, C. G. Dangling bonds in hexagonal boron nitride as single-photon emitters. Phys. Rev. Lett. 123, 127401 (2019).
Mazzoni, M. S. C., Nunes, R. W., Azevedo, S. & Chacham, H. Electronic structure and energetics of BxCyNz layered structures. Phys. Rev. B 73, 073108 (2006).
Huang, C. et al. Carbon-doped BN nanosheets for metal-free photoredox catalysis. Nat. Commun. 6, 7698 (2015).
Hong, J., Jin, C., Yuan, J. & Zhang, Z. Atomic defects in two‐dimensional materials: from single‐atom spectroscopy to functionalities in opto-/electronics, nanomagnetism, and catalysis. Adv. Mater. 29, 1606434 (2016).
Ci, L. et al. Atomic layers of hybridized boron nitride and graphene domains. Nat. Mater. 9, 430–435 (2010).
Zobelli, A., Ewels, C. P., Gloter, A. & Seifert, G. Vacancy migration in hexagonal boron nitride. Phys. Rev. B 75, 094104 (2007).
Vinchon, P., Glad, X., Robert Bigras, G., Martel, R. & Stafford, L. Preferential self-healing at grain boundaries in plasma-treated graphene. Nat. Mater. 20, 49–54 (2021).
Ouyang, B., Meng, F. & Song, J. Energetics and kinetics of vacancies in monolayer graphene boron nitride heterostructures. 2D Mater. 1, 035007 (2014).
Koperski, M. et al. Midgap radiative centers in carbon-enriched hexagonal boron nitride. Proc. Natl Acad. Sci. 117, 13214–13219 (2020).
Chejanovsky, N. et al. Single-spin resonance in a van der Waals embedded paramagnetic defect. Nat. Mater. 20, 1079–1084 (2021).
Bommer, A. & Becher, C. New insights into nonclassical light emission from defects in multi-layer hexagonal boron nitride. Nanophotonics 8, 11 (2019).
Acknowledgements
This work was funded by the Army Research Office via award no. 64655-CH-ISN to the Institute for Soldier Nanotechnologies. The authors acknowledge support from 2015 US Office of Naval Research Multi University Research Initiative (MURI) grant on Foldable and Adaptive Two-Dimensional Electronics (FATE) at MIT, Harvard University and University of Southern California. We appreciate characterization support from The MIT Center for Materials Science and Engineering. D.K. is supported by JSPS KAKENHI Grant Number JP22H01893 from Japan Society for the Promotion of Science. Y.Z. acknowledges the National Science Foundation (CBET-1511526). V.B.K. is supported by The Swiss National Science Foundation (projects no. P2ELP3_162149 and P300P2_174469). M.K. acknowledges support by the German Research Foundation (DFG) Research Fellowship KU 3952/1-1. Microfabrication for this work was performed at the Harvard University Center for Nanoscale Systems (CNS), a member of the National Nanotechnology Coordinated Infrastructure Network (NNCI), which is supported by the National Science Foundation under NSF award no. 1541959.
Author information
Authors and Affiliations
Contributions
S.X.L., T.I., and M.S.S. conceptualized the idea, and formulated the overarching research goals and aims. T.I. and S.X.L. acquired, curated, and analyzed optical experimental results. T.I. prepared all samples purchased from vendors. G.H. grew hBN in a lab-built CVD chamber. D.K. assisted in building the optical measurement setup. Y.Z. assisted in building the environmental chamber. Y.W. and J.H.W. performed ADF-STEM imaging. H.P. conducted ADF-STEM simulation. S.X.L., H.P., and J.H.W. analyzed and interpreted the ADF-STEM results. V.B.K. conducted XPS measurements. T.I. and S.X.L. analyzed and interpreted the XPS results. G.H., Y.Z., M.K., Z.Y., and S.F. assisted in the interpretation of experimental results. S.X.L., T.I., and M.S.S. wrote the original draft, with input from all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Materials thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editor: Aldo Isidori.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, S.X., Ichihara, T., Park, H. et al. Prolonged photostability in hexagonal boron nitride quantum emitters. Commun Mater 4, 19 (2023). https://doi.org/10.1038/s43246-023-00345-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s43246-023-00345-8
This article is cited by
-
Electronic and optical properties of quantum emitters in h-BN
Applied Physics A (2023)
