
Abstract
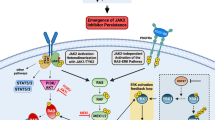
Myeloproliferative neoplasms (MPNs) exhibit a propensity for transformation to secondary acute myeloid leukemia (sAML), for which the underlying mechanisms remain poorly understood, resulting in limited treatment options and dismal clinical outcomes. Here, we performed single-cell RNA sequencing on serial MPN and sAML patient stem and progenitor cells, identifying aberrantly increased expression of DUSP6 underlying disease transformation. Pharmacologic dual-specificity phosphatase (DUSP)6 targeting led to inhibition of S6 and Janus kinase (JAK)–signal transducer and activator of transcription (STAT) signaling while also reducing inflammatory cytokine production. DUSP6 perturbation further inhibited ribosomal S6 kinase (RSK)1, which we identified as a second indispensable candidate associated with poor clinical outcome. Ectopic expression of DUSP6 mediated JAK2-inhibitor resistance and exacerbated disease severity in patient-derived xenograft (PDX) models. Contrastingly, DUSP6 inhibition potently suppressed disease development across Jak2V617F and MPLW515L MPN mouse models and sAML PDXs without inducing toxicity in healthy controls. These findings underscore DUSP6 in driving disease transformation and highlight the DUSP6–RSK1 axis as a vulnerable, druggable pathway in myeloid malignancies.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 digital issues and online access to articles
$119.00 per year
only $9.92 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout








Similar content being viewed by others

Data availability
Sequencing data have been deposited in the Gene Expression Omnibus under accession code GSE214361. Mass cytometry data are available upon request at https://wustl.cytobank.org/. Imaging mass cytometry data are deposited on Figshare at https://doi.org/10.6084/m9.figshare.21514257. ChIP–seq data are available under accession numbers Nfil3 (GSM1437733)26, Atf3 (GSM2663858)29, Fos (GSM1875490 (ref. 25), GSM2663847 (ref. 29)), Fosl2 (GSM1004808 (ref. 24)), H3K27ac (GSM2974670 (ref. 27), GSM851270 (ref. 28)). OHSU BeatAML clinical and expression data were obtained from Tyner et al.21 and http://vizome.org. Expression arrays of CD34+ and CD34− sorted mononuclear cells from patients with AML and normal CD34+ BM cells were obtained from GSE30029 (ref. 22). Expression arrays of CD34+ cells from patients with primary MF and CD34+ NBM were obtained from GSE53482 (ref. 31). Pan-cancer TCGA gene expression and TCGA LAML mutational data were accessed from cBioPortal70. Dropout candidates in AML cell lines (such as RPS6KA1) at a false discovery rate of 10% that were not essential in non-AML lines HT29 and HT-1080 (‘#non-AML = 0’) were identified and plotted from the Tzelepis et al. CRISPR dropout screen37. RPS6KA1 gene dependency data are from DepMap38, and expression data were obtained from the CCLE36. Gene expression of DUSP family genes across AML cell lines and RPS6KA1 expression (public 21Q1; https://depmap.org/portal/download/) were accessed from the CCLE by the Broad Institute. Gene effect scores (CERES) from CRISPR (Avana) public 21Q1 were accessed from the DepMap portal by the Broad Institute. For cell line drug-sensitivity correlations, fedratinib IC50 values of AML cell lines were derived from CancerRxGene and the GDSC1 dataset71 and correlated with CCLE RPS6KA1 mRNA expression. AUC of apitolisib, KW-7-42-1, torin-2, WYE-125132, MK-2206 and PF-4708671 and DUSP6 expression were accessed from the DepMap portal by the Broad Institute. Additional sequencing data were accessed: scRNA-seq of CD34+ cells from a patient with primary MF32 (GSE153319), inducible KLF1 iPSC macrophages33 (GSE125150), murine BM-derived macrophage Klf2-knockout cells34 (GSE149119) and murine Klf2−/− yolk sac erythroid cells35 (GSE27602). All other data supporting the findings of this study are available from the corresponding author on reasonable request. Source data are provided with this paper.
Code availability
R scripts used in this study are available from corresponding author upon request.
References
Levine, R. L. & Gilliland, D. G. Myeloproliferative disorders. Blood 112, 2190–2198 (2008).
Oh, S. T. & Gotlib, J. JAK2 V617F and beyond: role of genetics and aberrant signaling in the pathogenesis of myeloproliferative neoplasms. Expert Rev. Hematol. 3, 323–337 (2010).
Zoi, K. & Cross, N. C. Genomics of myeloproliferative neoplasms. J. Clin. Oncol. 35, 947–954 (2017).
Tefferi, A. Myelofibrosis with myeloid metaplasia. N. Engl. J. Med. 342, 1255–1265 (2000).
Harrison, C. et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N. Engl. J. Med. 366, 787–798 (2012).
Verstovsek, S. et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N. Engl. J. Med. 366, 799–807 (2012).
Vannucchi, A. M. et al. Mutations and prognosis in primary myelofibrosis. Leukemia 27, 1861–1869 (2013).
Dunbar, A. J., Rampal, R. K. & Levine, R. Leukemia secondary to myeloproliferative neoplasms. Blood 136, 61–70 (2020).
Patel, A. A. & Odenike, O. Genomics of MPN progression. Hematology Am. Soc. Hematol. Educ. Program 2020, 440–449 (2020).
Rampal, R. et al. Genomic and functional analysis of leukemic transformation of myeloproliferative neoplasms. Proc. Natl Acad. Sci. USA 111, E5401–E5410 (2014).
Jayavelu, A. K. et al. Splicing factor YBX1 mediates persistence of JAK2-mutated neoplasms. Nature 588, 157–163 (2020).
Stivala, S. et al. Targeting compensatory MEK/ERK activation increases JAK inhibitor efficacy in myeloproliferative neoplasms. J. Clin. Invest. 129, 1596–1611 (2019).
Keyse, S. M. Dual-specificity MAP kinase phosphatases (MKPs) and cancer. Cancer Metastasis Rev. 27, 253–261 (2008).
Bermudez, O., Pages, G. & Gimond, C. The dual-specificity MAP kinase phosphatases: critical roles in development and cancer. Am. J. Physiol. Cell Physiol. 299, C189–C202 (2010).
Ahmad, M. K., Abdollah, N. A., Shafie, N. H., Yusof, N. M. & Razak, S. R. A. Dual-specificity phosphatase 6 (DUSP6): a review of its molecular characteristics and clinical relevance in cancer. Cancer Biol. Med. 15, 14–28 (2018).
Fisher, D. A. C. et al. Mass cytometry analysis reveals hyperactive NFκB signaling in myelofibrosis and secondary acute myeloid leukemia. Leukemia 31, 1962–1974 (2017).
Fisher, D. A. C. et al. Cytokine production in myelofibrosis exhibits differential responsiveness to JAK–STAT, MAP kinase, and NFκB signaling. Leukemia 33, 1978–1995 (2019).
Daver, N., Schlenk, R. F., Russell, N. H. & Levis, M. J. Targeting FLT3 mutations in AML: review of current knowledge and evidence. Leukemia 33, 299–312 (2019).
Edwards, D. K. T. et al. CSF1R inhibitors exhibit antitumor activity in acute myeloid leukemia by blocking paracrine signals from support cells. Blood 133, 588–599 (2019).
Testa, U. et al. Elevated expression of IL-3Rα in acute myelogenous leukemia is associated with enhanced blast proliferation, increased cellularity, and poor prognosis. Blood 100, 2980–2988 (2002).
Tyner, J. W. et al. Functional genomic landscape of acute myeloid leukaemia. Nature 562, 526–531 (2018).
de Jonge, H. J. et al. Gene expression profiling in the leukemic stem cell-enriched CD34+ fraction identifies target genes that predict prognosis in normal karyotype AML. Leukemia 25, 1825–1833 (2011).
Bialkowska, A. B., Yang, V. W. & Mallipattu, S. K. Kruppel-like factors in mammalian stem cells and development. Development 144, 737–754 (2017).
Ciofani, M. et al. A validated regulatory network for TH17 cell specification. Cell 151, 289–303 (2012).
Eichenfield, D. Z. et al. Tissue damage drives co-localization of NF-κB, Smad3, and Nrf2 to direct Rev-erb sensitive wound repair in mouse macrophages. eLife 5, e13024 (2016).
Fang, B. et al. Circadian enhancers coordinate multiple phases of rhythmic gene transcription in vivo. Cell 159, 1140–1152 (2014).
Link, V. M. et al. Analysis of genetically diverse macrophages reveals local and domain-wide mechanisms that control transcription factor binding and function. Cell 173, 1796–1809 (2018).
Shen, Y. et al. A map of the cis-regulatory sequences in the mouse genome. Nature 488, 116–120 (2012).
Tong, A. J. et al. A stringent systems approach uncovers gene-specific mechanisms regulating inflammation. Cell 165, 165–179 (2016).
The Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 368, 2059–2074 (2013).
Norfo, R. et al. miRNA–mRNA integrative analysis in primary myelofibrosis CD34+ cells: role of miR-155/JARID2 axis in abnormal megakaryopoiesis. Blood 124, e21–e32 (2014).
Parenti, S. et al. Mutated clones driving leukemic transformation are already detectable at the single-cell level in CD34-positive cells in the chronic phase of primary myelofibrosis. NPJ Precis. Oncol. 5, 4 (2021).
Lopez-Yrigoyen, M. et al. Genetic programming of macrophages generates an in vitro model for the human erythroid island niche. Nat. Commun. 10, 881 (2019).
Sweet, D. R. et al. Myeloid Kruppel-like factor 2 is a critical regulator of metabolic inflammation. Nat. Commun. 11, 5872 (2020).
Redmond, L. C. et al. Kruppel-like factor 2 regulated gene expression in mouse embryonic yolk sac erythroid cells. Blood Cells Mol. Dis. 47, 1–11 (2011).
Barretina, J. et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607 (2012).
Tzelepis, K. et al. A CRISPR dropout screen identifies genetic vulnerabilities and therapeutic targets in acute myeloid leukemia. Cell Rep. 17, 1193–1205 (2016).
Meyers, R. M. et al. Computational correction of copy number effect improves specificity of CRISPR–Cas9 essentiality screens in cancer cells. Nat. Genet. 49, 1779–1784 (2017).
Kamakura, S. et al. Hes binding to STAT3 mediates crosstalk between Notch and JAK–STAT signalling. Nat. Cell Biol. 6, 547–554 (2004).
Wong, G. W., Knowles, G. C., Mak, T. W., Ferrando, A. A. & Zuniga-Pflucker, J. C. HES1 opposes a PTEN-dependent check on survival, differentiation, and proliferation of TCRβ-selected mouse thymocytes. Blood 120, 1439–1448 (2012).
Furukawa, T. et al. AURKA is one of the downstream targets of MAPK1/ERK2 in pancreatic cancer. Oncogene 25, 4831–4839 (2006).
Mullally, A. et al. Physiological Jak2V617F expression causes a lethal myeloproliferative neoplasm with differential effects on hematopoietic stem and progenitor cells. Cancer Cell 17, 584–596 (2010).
Pikman, Y. et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 3, e270 (2006).
Celik, H. et al. JARID2 functions as a tumor suppressor in myeloid neoplasms by repressing self-renewal in hematopoietic progenitor cells. Cancer Cell 34, 741–756 (2018).
Kong, T. et al. Pevonedistat targets malignant cells in myeloproliferative neoplasms in vitro and in vivo via NFκB pathway inhibition. Blood Adv. 6, 611–623 (2022).
Celik, H. et al. A humanized animal model predicts clonal evolution and therapeutic vulnerabilities in myeloproliferative neoplasms. Cancer Discov. 11, 3126–3141 (2021).
Fisher, D. A. C., Fowles, J. S., Zhou, A. & Oh, S. T. Inflammatory pathophysiology as a contributor to myeloproliferative neoplasms. Front. Immunol. 12, 683401 (2021).
Koschmieder, S. et al. Myeloproliferative neoplasms and inflammation: whether to target the malignant clone or the inflammatory process or both. Leukemia 30, 1018–1024 (2016).
Hasselbalch, H. C. & Bjorn, M. E. MPNs as inflammatory diseases: the evidence, consequences, and perspectives. Mediators Inflamm. 2015, 102476 (2015).
Arora, D. et al. Expression of protein-tyrosine phosphatases in acute myeloid leukemia cells: FLT3 ITD sustains high levels of DUSP6 expression. Cell Commun. Signal. 10, 19 (2012).
Molina, G. et al. Zebrafish chemical screening reveals an inhibitor of Dusp6 that expands cardiac cell lineages. Nat. Chem. Biol. 5, 680–687 (2009).
Lai, A. Z. et al. Dynamic reprogramming of signaling upon Met inhibition reveals a mechanism of drug resistance in gastric cancer. Sci. Signal. 7, ra38 (2014).
Kesarwani, M. et al. Targeting c-FOS and DUSP1 abrogates intrinsic resistance to tyrosine-kinase inhibitor therapy in BCR–ABL-induced leukemia. Nat. Med. 23, 472–482 (2017).
Chan, L. N. et al. Signalling input from divergent pathways subverts B cell transformation. Nature 583, 845–851 (2020).
Chae, H. D. et al. RSK inhibitor BI-D1870 inhibits acute myeloid leukemia cell proliferation by targeting mitotic exit. Oncotarget 11, 2387–2403 (2020).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Luo, W., Friedman, M. S., Shedden, K., Hankenson, K. D. & Woolf, P. J. GAGE: generally applicable gene set enrichment for pathway analysis. BMC Bioinformatics 10, 161 (2009).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005).
Satija, R., Farrell, J. A., Gennert, D., Schier, A. F. & Regev, A. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 33, 495–502 (2015).
Tirosh, I. et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196 (2016).
Korotkevich, G. et al. Fast gene set enrichment analysis. Preprint at bioRxiv https://doi.org/10.1101/060012 (2021).
Qiu, X. et al. Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods 14, 979–982 (2017).
Bendall, S. C. et al. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science 332, 687–696 (2011).
Bandyopadhyay, S., Fowles, J. S., Yu, L., Fisher, D. A. C. & Oh, S. T. Identification of functionally primitive and immunophenotypically distinct subpopulations in secondary acute myeloid leukemia by mass cytometry. Cytometry B Clin. Cytom. 96, 46–56 (2019).
Bandyopadhyay, S., Fisher, D. A. C., Malkova, O. & Oh, S. T. Analysis of signaling networks at the single-cell level using mass cytometry. Methods Mol. Biol. 1636, 371–392 (2017).
Catena, R., Montuenga, L. M. & Bodenmiller, B. Ruthenium counterstaining for imaging mass cytometry. J. Pathol. 244, 479–484 (2018).
Kelly, L. M. et al. FLT3 internal tandem duplication mutations associated with human acute myeloid leukemias induce myeloproliferative disease in a murine bone marrow transplant model. Blood 99, 310–318 (2002).
Cerami, E. et al. The cBio Cancer Genomics Portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404 (2012).
Yang, W. et al. Genomics of Drug Sensitivity in Cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 41, D955–D961 (2013).
Acknowledgements
This work was supported by NIH grants R01HL134952 (S.T.O.), R01HL147978 (G.A.C.) and T32HL007088 (J.S.F.) and by Canadian Institutes of Health Research grants PJT-156233 (S.H.) and PJT-438303 (S.H.). Additional support was provided by the Leukemia and Lymphoma Society Translational Research Program (S.T.O.), the MPN Research Foundation (S.T.O.), the When Everyone Survives Foundation (S.T.O.), the Edward P. Evans Foundation (G.A.C.), the Gabrielle’s Angel Foundation (G.A.C.), a Canderel Rising Star Summer Studentship (K.Y.) and a Canadian Research Chair in Functional Genomics (S.H.). G.A.C. is a scholar of the Leukemia and Lymphoma Society. Technical support was provided by the Alvin J. Siteman Cancer Center Tissue Procurement Core Facility, the Biostatistics Shared Resource, the Flow Cytometry Core, the Barnes-Jewish Hospital, the Institute of Clinical and Translational Sciences and the Immunomonitoring Laboratory, which are supported by an NCATS Clinical and Translational Sciences Award (UL1 TR002345) and NCI Cancer Center Support Grant P30CA91842. Additional support was provided by the Barnard Cancer Institute. The Immunomonitoring Laboratory is also supported by the Andrew M. and Jane M. Bursky Center for Human Immunology and Immunotherapy Programs. We thank D. Bender, R. Lin and K. Link for assistance with mass cytometry experiments. We are grateful to A. Mullally (DFCI) for providing Jak2V617F knockin mice and to R. Levine (MSKCC) for providing the MPLW515L retroviral construct. We thank M. Fulbright for assistance with mouse colony management. We thank T. Ley for sharing TCGA LAML data. We thank F. Gao for assistance with biostatistical analysis. We thank A. Vogt for helpful discussions related to BCI. We thank the Genetic Perturbation Service of the Goodman Cancer Research Centre at McGill University for access to and preparation of functional genetic tools.
Author information
Authors and Affiliations
Contributions
T.K., A.B.A.L., K.Y., L.Y., D.A.C.F., L.P.D.L.F., M.C.F. and J.S.F. performed experiments. A.Z.W., M.J.C., H.C., G.A.C. and S.T.O. provided technical and clinical support. M.B.R. evaluated histopathology. T.K., S.H. and S.T.O. designed and supervised experiments. T.K. and S.T.O. wrote the manuscript. All authors read and approved of the manuscript.
Corresponding author
Ethics declarations
Competing interests
S.T.O. has served as a consultant for Kartos Therapeutics, CTI BioPharma, Celgene–Bristol Myers Squibb, Disc Medicine, Blueprint Medicines, PharmaEssentia, Constellation, Geron, AbbVie, Sierra Oncology and Incyte. G.A.C. has received research funding from Incyte. All other authors disclose no competing interests.
Peer review information
Nature Cancer thanks Richard Moriggl, Rebekka Schneider and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Elevated DUSP6 expression in patients with sAML compared to MPN.
a) Heatmap of top differentially expressed genes from microarray analysis in lin- CD34 + cells from sAML patients (n = 14) compared to MF patients (n = 6), and reference expression in healthy donor bone marrow (NBM; n = 5). b) DUSP6 expression from CD34 + cells from NBM (n = 31 patients), AML bone marrow CD34 + subfraction (n = 46 patients), and AML bone marrow CD34- subfraction (n = 44 patients) from GSE30029. DUSP6 values represent quartile normalized, log-transformed values. Statistics were assessed by two-tailed Student’s t test. Data are presented as mean values + /- s.d. c) Immunofluorescence of bone marrow from additional MF and sAML patients, and healthy donors. White arrows denote DUSP6-positive cell staining. IF image acquired from one section. Scale bar: 50 µM. d) Imaging mass cytometry analysis of PBMC cell pellets from normal donor peripheral blood (LRS2), MF (MF20), or sAML (sAML1) patients. Individual images show overlap of indicated channels as denoted, acquired from one section. Scale bar = 16 µM.
Extended Data Fig. 2 Serial patient CD34 + scRNA sequencing shows elevated DUSP6 along MPN to sAML progression.
a) Gene set enrichment analysis of top altered Hallmark pathways in serial CD34 + samples at the sAML stage compared to MPN stage of three patients. b) Violin plots showing relative expression of top shared differentiate expressed genes in sAML vs MPN disease states from three patients, and healthy donors (N34, N39). c) Venn diagram showing shared candidates identified in the top 1000 differentially expressed genes from two sAML stages relative to their chronic MF stage(s) with violin plots highlighting gene expression of shared transcription factors at sAML vs MF stage(s). d) ChIP tracks of key transcription factors identified from scRNA-seq showing occupancy at the Mus musculus Dusp6 locus across various tissue samples. e) Heatmap of pearson correlations between DUSP6 and top, shared candidates across five databases. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. f) KLF2 identified as the top correlating gene, and KLF1 identified among the bottom 10 correlating genes, with DUSP6 in patient 381812 (MF and sAML) scRNA-seq.
Extended Data Fig. 3 Identification of distinct subpopulations from CD34+ scRNA-seq.
a) TotalSeq surface protein detection and mRNA features to guide distinct subpopulation identification. MEP (n = 235 cells); GMP/MKP (n = 651 cells); MLP (n = 867 cells); HSC (n = 1803 cells); CMP/ERP (n = 287 cells); ERP (n = 969 cells); ERYTHROID (n = 611 cells); GMP (n = 2352 cells); GMPgran (n = 532 cells); GMPmono (n = 507 cells); CLP (n = 420 cells). b) Schematic and relative quantification of distinct subpopulations identified from 381812. c) UMAP analysis and violin plot of DUSP6, KLF2, and KLF1 of subpopulations from N34, N39, 374024, and 145790. Additional trajectory analysis of patients 374024 and 145790 along disease progression from MPN to sAML.
Extended Data Fig. 4 Exploration of DUSP6 regulation by KLF1 and KLF2 across additional models.
a) Schematic of a separate serial CD34 + scRNA-seq dataset of a primary MF patient at multiple timepoints along transformation to sAML. As per source publication by Parental et al., sample T1 (PMF) was collected at chronic MPN phase, after which the patient was treated with ruxolitinib for 8 months at which sample T2 (treatment PMF) was collected, and then after 11 months of ruxolitinib treatment at sAML diagnosis (T3; sAML). Dataset investigated: GSE153319. b) Violin plots of DUSP6, KLF2, and KLF1 expression at different disease timepoints from Parenti et al. CD34 + scRNA-seq in (a). c) Relative fold change of DUSP6, KLF2, and KLF1 expression at sAML timepoint (T3) compared to PMF (T1) in across identified cell populations from Parenti et al. CD34 + scRNA-seq in (a). d) KLF2 (left) and KLF1 (right) expression from CD34 + cells from NBM (n = 5), MF (n = 14), and sAML (n = 6) patient samples. DUSP6 values represent RMA from microarray. Statistics were assessed by two-tailed Student’s t test. Data are presented as mean values + /- s.d. e) qRT-PCR of DUSP6 after KLF2 knockdown in HEL cells. DUSP6 mRNA expression normalized to ACTB for each group and then normalized to pLKO vector control. n = 3 independent experiments in each group. Statistics were assessed by two-tailed Student’s t test. Data are presented as mean values + /- SD. f) Immunoblot of DUSP6 expression after KLF2 knockdown in HEL cells utilizing shRNA or pLKO control vector. Immunoblot representative of 3 independent experiments. g) Cell viability assay of HEL cells after KLF2 knockdown relative to control vector. Cells were grown for 96 hours and viability was normalized to the pLKO control vector. n = 6 independently treated cell cultures pooled from two independent experiments per condition. Mean and standard deviation presented. Statistics were assessed by two-tailed Student’s t test. h) DUSP6 expression in KLF1 inducible iPSC-derived macrophages relative to control. Dataset investigated: GSE125150. i) Klf2 expression in myeloid-specific Klf2 knockout murine bone marrow derived macrophages with Mann-Whitney U comparison. Dataset investigated: GSE149119. j) Multi-Chip Significance score (S-score) of Dusp6 in Klf2 knockout murine embryonic yolk sac erythroid cells. Multi-Chip Significance score (S-score) and expression comparison p-value of Dusp6 (probe 1415834_at) between WT and Klf2-/- samples were identified. Per source publication, probe with absolute S-score values greater or equal to 2.00 were considered to be significant. Dataset investigated: GSE27602.
Extended Data Fig. 5 Functional characterization of DUSP6 in MPN/AML.
a) Enrichment plots of top upregulated Hallmark pathways in DUSP6high vs DUSP6low patients by GSEA. b) Relative mRNA expression of DUSP family genes across 35 AML cell lines from the Cancer Cell Line Encyclopedia. Boxplots represent min to max ranges with median, 25th, and 75th percentiles. c) Relative mRNA expression of DUSP family genes in HEL cells from the Cancer Cell Line Encyclopedia. d) Cell viability curves of HEL cells treated with BCI or trametinib across multiple drug doses. Cells were treated for 72 hours and viability was normalized to control. N = 6 independently treated cell cultures pooled from two independent experiments at each drug dose. Mean and standard deviation presented. e) Immunoblot of HEL cells treated with increasing doses of BCI or the MEK inhibitor trametinib. Cells were treated at their indicated drug dose for 24 hours. Immunoblot representative of two experiments. f) Phospho-STAT3 and phospho-STAT5 flow cytometry of HEL cells treated with 1 µM of BCI or control for 24 hours. n = 1 independently treated cell culture. g) Immunoblot profiling of different signaling pathways altered by BCI and trametinib. HEL cells were treated with 1 µM BCI or 1 µM trametinib for 24 hours. Immunoblot representative of 2 independent experiments. h) Hallmark gene set enrichment analysis showing top altered pathways by normalized enrichment score (NES) and enrichment plots of E2F targets and G2M checkpoint from RNA-seq of HEL cells treated with 1 µM of BCI, or DMSO control for 24 hours. i) Immunblot of HEL and UKE-1 cells treated with 1 µM BCI for 24 hours. Immunoblot representative of 3 independent experiments. j) Immunoblot of HEL cells ectopically expressing DUSP6 or GFP control vector. Immunoblot representative of 3 independent experiments. k) Cell viability assay of HEL cells ectopically expressing DUSP6 or GFP control vector. n = 6 independently treated cell cultures pooled from two independent experiments for each condition and grown for 96 hours with viability normalized to the control vector. Mean and standard deviation presented. Statistics were assessed by two-tailed Student’s t test. l) Cell viability assay of HEL cells ectopically expressing DUSP6 or GFP control vector treated with 300 nM BCI. Cells were plated at n = 6 independently treated cell cultures pooled from two independent experiments grown for 96 hours with viability from normalized to control treatment from each group. Mean and standard deviation presented. Statistics were assessed by two-tailed Student’s t test. m) Representative images of lin- CD34 + colonies grown in MethoCult H4034 Optimum for 12 days in 0.5 µM BCI or RPMI control. Samples plated in duplicate. Images acquired from one field of view representative of two plates/condition. Scale bar: 1000 µM.
Extended Data Fig. 6 Suppression of signaling and cytokine production in primary samples by BCI assessed by mass cytometry.
a) tSNE dimensional reduction clustering of distinct subpopulations from sAML4 and altered signaling upon BCI, TPO induction, or combination treatment post mass cytometry analysis. Samples were treated with 1 µM BCI for 4 hours, 20 ng/mL TPO for 1 hour, or combination. tSNE plots of sAML4 representative of plots from 2 MF, 3 sAML, and 2 normal patient samples. b) TPO-induced signaling across different subpopulations from sAML4. Patient samples were treated with 20 ng/mL TPO for 1 hour. Signals were normalized to the control treatment and reported as median Arcsinh ratio. tSNE plots of sAML4 representative of plots from 3 sAML patients. c) TPO-induced cytokine production across different subpopulations from sAML5. Patient samples were treated with 20 ng/mL TPO for 4 hour. Signals were normalized to the control treatment and reported as 90 percentile Arcsinh ratio. tSNE plots of sAML5 representative of plots from 3 sAML patients. d) Heatmap and dot plots of altered cytokine production of CD14 + monocytes from bone marrow (NBM40) and peripheral blood (NPB LRS2) of healthy donors, and PBMCs from MF and sAML patients by mass cytometry. Unique patient samples were treated with 1 µM BCI for 4 hours, 20 ng/mL TPO for 4 hour, or combination. Signals were normalized to the control treatment of each individual patient sample and reported as 90th percentile Arcsinh ratio. Basal cytokine expression in CD14 + cells from MF and sAML are also presented (left panel) and are normalized to the NBM/NPB within each individual CyTOF run to control for batch effect: run 1 - sAML4 and sAML6 normalized to NBM40; run 2 - MF20 and MF102 normalized to NPB LRS2; run 3- MF40 and sAML5 normalized to NBM40. e) Dot plot of MIP-1β/CCL4 in CD123 + and CD16 + monocyte populations from sAML5. Samples were treated with 1 µM BCI for 4 hours, 20 ng/mL TPO for 4 hours, or combination. Signals were normalized to the control treatment and reported as 90 percentile Arcsinh ratio. n = 1 independent experiment with sample sAML5.
Extended Data Fig. 7 Functional characterization of RSK1 in MPN/AML.
a) Heatmap of inhibitors of upstream regulators of S6 activity and the pearson correlation of their area under curve (AUC) with DUSP6 expression in AML cell lines. b) CRISPR dropout screen showing RPS6KA1 as an essential gene in AML. Candidates were identified if meeting criteria of FDR < 10% and whose inhibition affected # of AML lines but neither of non-AML lines. Data retrieved from Tzelepis et al. c) RPS6KA1 expression across 10,071 patient samples representing 31 distinct cancer subtypes from the TCGA Pan-Cancer cohort. Expression values provided as log2 (value +1). See additional information in methods. Boxplots represent min to max ranges with median, 25th, and 75th percentiles. d) Immunoblot analysis of RPS6KA1 knockdown by shRNA or control vector in HEL cells. Immunoblot representative of 5 independent experiments. e) Cell viability assay of HEL cells after RPS6KA1 knockdown relative to control vector. Cells were grown for 96 hours and viability was normalized to the pLKO control vector. n = 6 independently treated cell cultures pooled from two independent experiments per construct. Mean and standard deviation presented. Statistics were assessed by two-tailed Student’s t test. f) Heatmap of altered signaling pathways of lin- CD34 + cells from unique normal bone marrow donors and peripheral blood of MF patients by mass cytometry. Patient samples were treated with 5 µM BI-D1870 for 4 hours, 20 ng/mL TPO for 1 hour, or combination. Signals were normalized to the control treatment of each individual patient sample and reported as 90th percentile Arcsinh ratio. g) Ridge plot of RPS6KA1 expression from CD34 + scRNA-seq of N34, N39, and 381812 at MF and sAML stages. h) RPS6KA1 expression from CD34 + cells from NBM (n = 5), MF (n = 14), and sAML (n = 6) patient samples. RPS6KA1 values represent RMA from microarray. Statistics were assessed by two-tailed Student’s t test. i) Downregulation of HES1 expression from RNA sequencing of HEL cells treated for 4 hours with 1 µM BCI vs DMSO control. n = 2 independently treated cell cultures. j) qRT-PCR of HES1 in HEL cells treated with 1 µM BCI for 24 hours. HES1 mRNA expression normalized to ACTB. n = 3 independent experiments. Statistics were assessed by two-tailed Student’s t test. Data are presented as mean values + /- s.d. k) Immunoblot of HES1 in HEL cells after DUSP6 knockdown by shRNA or control vector. Immunoblot representative of 3 independent experiments. l) qRT-PCR of HES1 in HEL cells after DUSP6 knockdown by shRNA or control vector. HES1 mRNA expression normalized to ACTB for each group and then normalized to pLKO vector. n = 3 independent experiments. Statistics were assessed by two-tailed Student’s t test. Data are presented as mean values + /- s.d. m) Immunoblot of HEL cells after HES1 knockdown by shRNA or control vector. Immunoblot representative of 2 independent experiments. n) Cell viability curve of HEL cells after ectopic expression of RPS6KA1 or GFP control treated with increasing concentrations of BI-D1870. Cells were treated for 96 hours and viability was normalized to the control treatment from each group. n = 6 independently treated cell cultures pooled from two independent experiments per construct. Mean and standard deviation presented. o) Cell viability assay of HEL cells treated with 1 µM BI-D1870, 300 µM BCI or combination, and UKE-1 cells treated with 2 µM BI-D1870, 200 µM BCI, or combination. Cells were treated for 72 hours and viability was normalized to the control treatment. n = 6 independently treated cell cultures pooled from two independent experiments at each drug dose. Mean and standard deviation presented. p) Hallmark gene set enrichment analysis showing top altered pathways from RNA-seq of HEL cells treated for 24 hours with 10 µM BI-D1870 + 1 µM BCI compared to DMSO control (left) and 10 µM BI-D1870 + 1 µM BCI compared to 10 µM BI-D1870 alone (right). q) Dot plot of mass cytometry analysis of lin- CD34 + cells from MF103 treated with 1 µM BCI for 4 hours, 5 µM BI-D1870 for 4 hours, 20 ng/mL TPO for 1 hour, or combination. Signals of key phosphorylated proteins were normalized to the control treatment and reported as 90th percentile Arcsinh ratio.
Extended Data Fig. 8 DUSP6 mediates response to JAK2 inhibitors.
a) Correlation of DUSP6 expression and fedratinib IC50 in 18 AML cell lines. DUSP6 expression obtained from the CCLE database and fedratinib IC50 obtained from the GDSC1 collection. b) Cell viability assay of HEL parental or HEL Fed-P cells treated with BCI at the indicated doses. Cells were treated for 72 hours and viability was normalized to the control treatment from each group. n = 6 independently treated cell cultures pooled from two independent experiments at each drug dose. Mean and standard deviation presented. c) Cell viability assay of HEL cells treated with BCI, fedratinib, or combination. Cells were treated for 72 hours at the indicated drug doses and viability was normalized to the control treatment. n = 6 independently treated cell cultures pooled from two independent experiments at each drug dose. Mean and standard deviation presented.
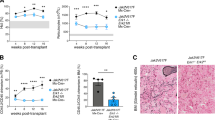
Extended Data Fig. 9 BCI alleviates disease burden across MPN and sAML mouse models.
a) WBC subpopulation counts, platelet counts, and body weight from Jak2 transplanted mice treated with vehicle (n = 9) or BCI (n = 10) across multiple timepoints. Statistics were assessed by two-way ANOVA comparing vehicle to BCI. Data are presented as mean values + /- s.d.. b) Representative gross spleen of Jak2 mice treated with vehicle or BCI at endpoint. c) Hematocrit, white blood cell (WBC) counts and differentials, and platelets counts of wildtype primary mice treated with vehicle (n = 4) or 25 mg/kg BCI (n = 5) following weekly schedule of 5 days on, 2 days off treatment across multiple timepoints. Liver, spleen, and body weights collected at endpoint. Two-way ANOVA and two-tailed Student’s t test statistical analysis resulted in non-significant values across all comparisons between vehicle and BCI treatment. Data are presented as mean values + /- s.d. d) Representative flow cytometry analysis of peripheral blood from CD45.1 mice showing engraftment of MPL W515 GFP + CD45.2 cells. e) WBC subpopulation counts, platelet counts, and normalized spleen weight from MPL W515 MF model of transplanted mice treated with vehicle (n = 8) or BCI (n = 8) across multiple timepoints. Statistics were assessed by two-way ANOVA for white count differential comparisons between vehicle and BCI and two-tailed Student’s t test for normalized spleen weight. Data are presented as mean values + /- s.d.
Extended Data Fig. 10 Humanized PDX models of MF and sAML.
a) Percentage of myeloid cells from hCD45+ PB (left) and CD71 + CD235a + from hCD45- BM (right) from NSGS mice transplanted with CD34 + cells ectopically expressing control (n = 9 mice) and DUSP6 (n = 10 mice) at endpoint. Both statistics assessed by two-tailed non-parametric Mann-Whitney U test after testing for normality by Shapiro-Wilk test. b) DUSP6 overexpression PDX with CD34 + cells from a second MF patient (MF106). Plots show percentage of human CD45 (hCD45) in peripheral blood and bone marrow of transplanted mice ectopically expressing control (n = 5) or DUSP6 (n = 5) across multiple timepoints, and spleen and liver weights of mice at endpoint normalized by mouse weight. %hCD45 in PB statistics assessed by two-way ANOVA incorporating weeks 4-8 post transplant. %hCD45 in BM, and normalized spleen and liver weights statistics were assessed by two-tailed Student’s t test. Data are presented as mean values + /- s.d. c) Kaplan-Meier survival analysis of mice from control or DUSP6 cohorts assessed by log-rank test. d) Colony assay of CD34 + cells from sAML15 after transduction with shRNAs targeting DUSP6 or control vector. Sorted cells were grown in MethoCult H4034 Optimum for 12 days. Samples were plated in triplicate (n = 3 replicates). Statistics were assessed by Two-tailed Student’s t test. Data are presented as mean values + /- s.d. e) CD34 + healthy donor normal bone marrow (NBM) PDX model. Cells were transduced with 2 independent shRNAs targeting DUSP6 or control and transplanted into NSGS mice. Plots show percentage of human CD45 (hCD45) in peripheral blood and bone marrow of transplanted mice treated ectopically expressing control (n = 5), shDUSP6 #1 (n = 5), or shDUSP6 #2 (n = 5) across multiple timepoints, and spleen and liver weights of mice at endpoint normalized by mouse weight. %hCD45 in PB statistics assessed by two-way ANOVA with Dunnett’s multiple comparisons test with control. %hCD45 in BM, and normalized spleen and liver weights statistics were assessed by two-tailed Student’s t test with Dunnett’s multiple comparisons test with control. f) Normalized spleen and liver weights from mice at end-point from sAML14 CD34 + PDX. Mice were treated with vehicle (n = 7), 25 mg/kg BCI (n = 8), 90 mg/kg ruxolitinib (n = 7), or combination (n = 7). Data are presented as mean values + /- s.d. g) tSNE dimensional reduction clustering of mouse and human CD45 + cells from bone marrow of sAML14 PDX mice. h) sAML PDX14 mass cytometry analysis showing percentage of CD123 + CD33 + leukemic cells gated from hCD45+ cells from 3 mice in each treatment group. Statistics were assessed by one-way ANOVA with Dunnett’s multiple comparison test. i) Erythroblast progenitors gated from hCD45+ cells. Statistics were assessed by one-way ANOVA with Dunnett’s multiple comparison test. j) SPADE tree cluster algorithm showing CD123 + and CD71 + populations from vehicle and BCI treated groups. k) UMAP clustering showing CD123 + and CD71 + populations. l) Schematic of the CD34 + healthy donor PDX model. CD34 + cells were isolated from BMMCs from normal donors, pooled, and transplanted into NSGS mice. Mice were treated with vehicle (n = 6), 25 mg/kg BCI (n = 6), 90 mg/kg ruxolitinib (n = 6), or combination (n = 6) following weekly schedule of 5 days on, 2 days off treatment starting on day 38. m) Plots show % hCD45 in peripheral blood and bone marrow of transplanted mice treated with vehicle or BCI across multiple timepoints and spleen weights of mice at endpoint normalized by mouse weight %hCD45 in PB statistics were assessed by two-way ANOVA comparing vehicle vs each individual treatment group with Dunnett’s multiple comparison test. %hCD45 in BM, and spleen and liver weights statistics were assessed by one-way ANOVA with Dunnett’s multiple comparison test. Data are presented as mean values + /- s.d.
Supplementary information
Supplementary Information
Supplementary Fig. 1
Supplementary Tables
Supplementary Tables 1–10.
Source data
Source Data Fig. 1
Statistical source data.
Source Data Fig. 3
Unprocessed western blots.
Source Data Fig. 3
Statistical source data.
Source Data Fig. 4
Unprocessed western blots.
Source Data Fig. 4
Statistical source data.
Source Data Fig. 5
Unprocessed western blots.
Source Data Fig. 5
Statistical source data.
Source Data Fig. 6
Statistical source data.
Source Data Fig. 7
Statistical source data.
Source Data Fig. 8
Statistical source data.
Source Data Extended Data Fig. 1
Statistical source data.
Source Data Extended Data Fig. 2
Statistical source data.
Source Data Extended Data Fig. 3
Statistical source data.
Source Data Extended Data Fig. 4
Unprocessed western blots.
Source Data Extended Data Fig. 4
Statistical source data.
Source Data Extended Data Fig. 5
Unprocessed western blots.
Source Data Extended Data Fig. 5
Statistical source data.
Source Data Extended Data Fig. 6
Statistical source data.
Source Data Extended Data Fig. 7
Unprocessed western blots.
Source Data Extended Data Fig. 7
Statistical source data.
Source Data Extended Data Fig. 8
Statistical source data.
Source Data Extended Data Fig. 9
Statistical source data.
Source Data Extended Data Fig. 10
Statistical source data.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Kong, T., Laranjeira, A.B.A., Yang, K. et al. DUSP6 mediates resistance to JAK2 inhibition and drives leukemic progression. Nat Cancer 4, 108–127 (2023). https://doi.org/10.1038/s43018-022-00486-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s43018-022-00486-8
This article is cited by
-
Decoding leukemia at the single-cell level: clonal architecture, classification, microenvironment, and drug resistance
Experimental Hematology & Oncology (2024)
-
MAPK-negative feedback regulation confers dependence to JAK2V617F signaling
Leukemia (2023)
