
Abstract
Pancreatic ductal adenocarcinoma (PDAC) is an aggressive cancer that is most frequently detected at advanced stages, limiting treatment options to systemic chemotherapy with modest clinical responses. Here, we review recent advances in targeted therapy and immunotherapy for treating subtypes of PDAC with diverse molecular alterations. We focus on the current preclinical and clinical evidence supporting the potential of these approaches and the promise of combinatorial regimens to improve the lives of patients with PDAC.
Similar content being viewed by others

Main
PDAC is a devastating disease with a 5-year overall survival (OS) of only 11% (ref. 1). Recent progress in chemotherapy regimens has improved outcomes in resectable PDAC2; however, 80% of patients are diagnosed at an advanced stage, precluding them from curative intent surgery3. The past decade has cemented gemcitabine and fluoropyrimidine-based combination regimens as standard of care chemotherapy for patients with metastatic pancreatic cancer4, although median OS (mOS) rates remain at approximately 1 year (refs. 5,6). In stark contrast to other tumor types, multiple large-scale trials using targeted agents have been unsuccessful for PDAC7,8,9. However, an improved understanding of the biology and genetics of PDAC have spurred the advent of new targeted and immune-based therapies that may be available for patients with PDAC in the near future. There is a critical unmet need to translate our understanding of PDAC biology to the clinic to improve survival and quality of life. Here, we review our current understanding of the molecular features and immune landscape of PDAC to develop targeted and immune-based therapies and improve outcomes for patients with PDAC.
Targeted approaches to PDAC
Identifying key oncogenic drivers and dependencies may yield novel approaches for PDAC. In this section, we review efforts toward the molecular characterization of PDAC and the strategies aimed at targeting potential vulnerabilities.
The mutational landscape of PDAC
Several large-scale genomic efforts have cataloged potential driver mutations in PDAC, enabling genomic-guided clinical decisions and therapeutic development. Early next-generation sequencing (NGS) studies of resected PDAC tumors revealed that KRAS, TP53, CDKN2A and SMAD4 displayed the highest mutation frequencies, with >90% of individuals having oncogenic KRAS mutations10,11. The International Cancer Genome Consortium (ICGC) confirmed these findings and described several structural variations12. In addition, The Cancer Genome Atlas (TCGA) program reported the presence of 20 genes mutated at a frequency of less than 10%, which included chromatin modification genes (such as ARID1A, KMT2D and KMT2C), DNA repair genes (for example, BRCA1, BRCA2 and PALB2) and additional oncogenes (BRAF, MYC, FGFR1 and others)13.
Recently, the ‘Know Your Tumor’ initiative reported that 28% of patients with available clinical outcomes had an actionable mutation, defined as a genetic aberration for which a specific targeted therapy existed14. However, only 7% of these individuals had received a matched, precision-based therapy15. Notably, even though the OS of patients with or without actionable mutations was comparable, the ones who received matched therapies survived longer than those who received unmatched therapy (2.58 years versus 1.32 years, respectively). The survival of patients with an actionable mutation who did not receive targeted therapy and the survival of those with no actionable mutation were comparable. The vast majority of individuals on matched therapies either had BRCA mutations or displayed microsatellite instability-high (MSI-H) status and thus were placed on a poly(ADP-ribose) polymerase (PARP) inhibitor or immune checkpoint blockade (ICB), respectively. In the COMPASS trial, 35 of the 50 evaluable participants with advanced PDAC had progressed on first-line chemotherapy, and, whereas 19 out of 35 went on to receive second-line treatment, these decisions were guided by NGS data in only 5 of those participants16. In the PancSeq prospective program, patients with advanced PDAC underwent a biopsy followed by rapid turnaround whole-exome sequencing to allocate them to genomics-guided therapies17, and 24% (17 out of 71) were treated with off-label use of an approved or experimental agent, highlighting the potential of this integrated multidisciplinary approach. In all of these studies, limitations included the relatively low frequency of actionable alterations beyond BRCA1, BRCA2 or MSI-H and the accessibility of appropriate therapies on or off clinical trials for patients with advanced disease and generally declining performance status. The more recent advent of additional therapies targeting oncogenic driver alterations in both KRAS-wild-type and KRAS-mutated PDAC will likely improve the number of patients who benefit from genotype-directed therapies in the near future.
Targeting KRAS mutations in PDAC
KRAS, the most frequently mutated oncogene, transmits signals from receptor tyrosine kinases (RTKs) through the MAPK pathway18. Oncogenic KRAS mutations occur in over 90% of human PDAC tumors, with different incidences of specific mutated sites, including G12D (41%), G12V (34%), G12R (16%), Q61H (4%) and G12C (~1%)19,20. Genetically engineered mouse models (GEMMs) confirmed the role of oncogenic Kras in PDAC initiation and tumor maintenance21,22,23 through cancer cell autonomous and non-autonomous mechanisms24. Expression of the oncogenic KrasG12D mutation by a pancreas-specific Cre/loxP system (that is, the Kras;Cre or ‘KC’ model) is sufficient to drive precursor pancreatic intraepithelial neoplasia (PanIN) and PDAC21. Although the penetrance of PDAC is variable in KC mice, expression of dominant-negative Trp53 (KPC model) or loss of Cdkn2a (encoding p16Ink4a and p19Arf) results in complete penetrance of PDAC and liver metastases22,25. Notably, pancreas-specific KrasA146T GEMMs do not exhibit PanIN lesions26, in line with the observation that KRASA146T mutations do not typically occur in human PDAC27, indicating that different Kras alleles confer varying oncogenic properties in PDAC26. Notably, KRAS codon 61 mutations have been associated with decreased downstream phospho-ERK signaling, raising the possibility of an association with improved survival11,28. By contrast, KRASG12D mutations are associated with worse outcomes than all other KRAS mutations and wild-type KRAS status in resected PDAC29.
Despite the high frequency of KRAS mutations in PDAC, developing RAS inhibitors has been challenging owing to a lack of drug-accessible pockets amenable to high-affinity binding30. However, recent advances in drug development have ushered in an era of mutation-specific KRAS-targeting tools. The KRASG12C mutation represents just 1% of PDAC cases31 but is the first KRAS-mutant protein that could be effectively targeted with a specific small-molecule inhibitor, ARS-1620. This inhibitor was developed through structure-based drug design, and it covalently binds the inactive GDP-bound state of KRASG12C, hindering the exchange of GDP for GTP and thereby preventing KRAS activation32. In vitro studies demonstrated that ARS-1620 elicits potent, dose-dependent inhibition of phospho-ERK, phospho-S6 and phospho-AKT in KRASG12C-mutated, but not KRASG12D- or KRASG12V-mutated, cell lines. ARS-1620 treatment had antitumor efficacy in in vitro and in vivo models of PDAC. Thus, ARS-1620 provided the first preclinical proof-of-concept evidence for specific KRASG12C inhibition.
Later, X-ray crystallography studies led to the identification of a cryptic pocket in KRASG12C (H95/Y96/Q99) that was exploited for the development of the first potent clinical-grade KRASG12C inhibitor, AMG-510 (sotorasib)33,34. Sotorasib proceeded to clinical trials and demonstrated significant radiographic responses in patients with KRASG12C-mutated lung adenocarcinoma33. A recent phase 2 clinical trial demonstrated durable clinical benefit, leading to sotorasib becoming the first US Food and Drug Administration (FDA) fasttrack-approved RAS inhibitor35. The multicenter, open-label, phase 1/2 CodeBreak 100 clinical trial continues to assess the safety and response rates of sotorasib in a variety of KRASG12C-mutated advanced solid cancers (NCT03600883)36. Preliminary data demonstrate promising activity in PDAC, with six of eight evaluable participants with PDAC achieving stable disease and three experiencing approximately a 30% reduction in disease burden at 4.3 months median follow-up37.
A separate KRASG12C inhibitor, MRTX849 (adagrasib), was simultaneously reported38, and demonstrated efficacy across preclinical models with radiographic responses in both KRASG12C-mutated lung and colon adenocarcinomas in a proof-of-concept clinical study. In early results from the KRYSTAL-1 trial, ten patients with previously treated KRASG12C-mutated metastatic PDAC treated with adagrasib were reported39. All evaluable individuals demonstrated clinical benefit, including five of ten partial responses (PRs). Progression-free survival (PFS) for this small cohort was 6.6 months, a remarkable observation given that the response rate of second-line chemotherapy for advanced PDAC is only around 10%, with a median PFS of less than 3 months40. These preliminary results need to be validated in larger cohorts, and ongoing clinical trials are testing adagrasib as a monotherapy or in combination with other agents, including inhibitors of EGFR, SHP2 and other targets (NCT03785249 and NCT04330664)41,42.
A small-molecule inhibitor specific for KRASG12D, MRTX1133, is currently undergoing preclinical development, with an investigational new drug application pending43. In early preclinical work using a panel of KRASG12D-mutated PDAC cell line xenograft and patient-derived xenograft (PDX) models, 73% (8 out of 11) of these models demonstrated in vivo tumor regression following treatment with MRTX1133 (refs. 44,45). Several other groups also have KRASG12D drug development programs underway. Given that the KRASG12D mutation is the most common KRAS alteration in PDAC with a prevalence of 41% (refs. 19,20), KRASG12D inhibitors have the potential to make a substantial impact on PDAC treatment.
Beyond allele-selective inhibitors of KRAS, additional small molecules are being developed that target RAS isoforms more broadly. For instance, RMC-6236 was recently reported as a novel clinical-grade pan-RAS inhibitor. This molecule bridges the active, GTP-bound form of RAS to an adjacent chaperone molecule (cyclophilin A), thus forming an inactive ‘tricomplex’ molecule and preventing RAS interactions with downstream signaling partners46. In a recent preclinical report, this molecule demonstrated a reduction in tumor volume across KRAS genotypes, including G12D, G12V and G12R mutations47. Furthermore, in immunocompetent PDAC mouse models, RMC-6236 treatment increased intratumor CD45+ cell infiltration and decreased numbers of monocytic myeloid-derived suppressor cells (moMDSCs). Additionally, RMC-6236 synergized with anti-PD-1 ICB, leading to durable complete responses in vivo.
SOS1 is a key guanine nucleotide exchange factor for KRAS, which positively regulates its activity at catalytic and allosteric sites48. A selective oral small-molecule SOS1 inhibitor, BI-3406, was recently reported to disrupt SOS1–KRAS interaction independent of the KRAS mutation. This compound markedly reduced tumor growth and GTP–RAS levels across KRAS-driven cancer models49. Importantly, combined BI-3406 and MEK inhibitor trametinib treatment led to the regression of established KRAS-mutated xenografts through prevention of MAPK feedback reactivation. The clinical SOS1 inhibitor candidate BI-1701963 is currently being investigated in a phase 1 clinical trial of KRAS-mutated advanced solid tumors alone or in combination with trametinib (NCT04111458)50.
Another approach to targeting mutant KRAS in PDAC uses the same lipid nanoparticle-encapsulated mRNA-based vaccine strategy that was used to develop the vaccines for severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)51. This mRNA-based vaccine (named mRNA-5671/V941) encodes the four major KRAS mutations seen in cancers: G12D, G12V, G13D and G12C. KRAS epitopes spanning the mutant amino acid have been shown to be presented on multiple alleles of the human version of the major histocompatibility complex class I (human leukocyte antigen; HLA) suggesting that direct T cell recognition of mutant KRAS is a feasible strategy52,53. A phase 1 clinical trial is underway and aims to enroll 100 patients with KRAS-mutated advanced non-small cell lung cancer (NSCLC), colorectal cancer or PDAC to be treated with mRNA-5671/V941 alone or in combination with the PD-1 antibody pembrolizumab (NCT03948763 (ref. 54); Fig. 1 and Table 1). Finally, a phase 1 trial is underway at MD Anderson Cancer Center to target the KRASG12D allele using synthetic short interfering RNA packaged within mesenchymal stem cell-derived exosomes (iExosomes; NCT03608631)55,56.

Relative frequencies of different KRAS missense mutations in PDAC. Investigative and FDA-approved precision-based therapies against denoted KRAS mutation sites are shown. Therapeutic strategies are also illustrated for KRAS-wild-type PDAC based on the presence of alternative driver aberrations. Asterisks indicate FDA-approved molecules with tissue-agnostic indications against the noted molecular alteration.
Tackling resistance to KRAS inhibition
Despite these encouraging clinical trial results, it is critical to understand the mechanisms that subvert the efficacy of KRAS inhibition. Studies evaluating multiple KRASG12C lung cancer cell lines, xenografts and human-derived xenografts treated with ARS-1620 found newly synthesized KRASG12C through increased EGFR and AURKA signaling57. Indeed, ARS-1620 treatment in combination with either EGFR or AURKA small-molecule inhibitors resulted in markedly enhanced suppression of tumors in vivo compared to ARS-1620 monotherapy. In the clinical setting, the combination of sotorasib with the pan-HER inhibitor afatinib in heavily pretreated patients with KRASG12C-mutated NSCLC (including those on prior KRASG12C inhibitor therapy) resulted in a 30% PR rate and 73% disease control rate58. A large phase 1/2 clinical trial is underway aiming to assess combinations of sotorasib with targeted therapy or immunotherapy in various KRASG12C-mutated tumor types (NCT04185883)59. Similarly, adagrasib showed increased efficacy in vivo when combined with inhibitors of SHP2, EGFR, mTOR and CDK4/CDK6 (ref. 38). Multiple mechanisms of resistance to adagrasib have been identified in patients with lung or colorectal tumors, including KRAS mutations or amplifications, RTK–RAS–MAPK pathway alterations and histological transformation60. Notably, analysis of circulating tumor DNA indicated that multiple mechanisms of resistance had emerged simultaneously in several individuals. Tricomplex inhibitors have demonstrated the ability to overcome second-site KRAS-activating mutations following adagrasib treatment61. Future clinical trials assessing the response to KRAS small-molecule inhibitors in PDAC will need to strongly consider the early adoption of concomitant targeting of escape pathways, an approach that has proven effective in targeting other MAPK components in different cancer types62.
Genetic inhibition of KRAS or pharmacologic inhibition of downstream MAPK signaling results in decreased glycolysis and mitochondrial function in human PDAC cells, with an increased dependency on autophagy63,64. Indeed, combining ERK or MEK inhibitors with the autophagy inhibitor hydroxychloroquine leads to marked reduction of PDAC growth in vitro and in vivo. Multiple early-phase clinical trials are underway aimed at seeking a signal of efficacy by combining ERK or MEK inhibitors with hydroxychloroquine in patients with metastatic PDAC (NCT03825289, NCT04132505 and NCT04386057)65,66,67.
A useful preclinical system to model potential mechanisms of bypassing KRAS inhibition is the doxycycline-regulated KPC GEMM, where oncogenic Kras expression is dependent on doxycycline exposure (iKPC model)23,68. In this model, tumors initially regress following doxycycline withdrawal only to relapse 4–5 months later despite the absence of oncogenic Kras expression69. The surviving tumor cells had decreased reliance on glycolysis and a high dependency on oxidative phosphorylation, supporting the use of oxidative phosphorylation inhibitors to tackle Kras-independent ‘escaper cells’. Further analysis of escaper-cell subpopulations revealed the acquisition of a mesenchymal phenotype that achieved independence from MAPK signaling through the Smarcb1–Myc signaling network70. These escaper cells displayed an anabolic phenotype featuring increased protein metabolism, thus rendering them vulnerable to alterations in the cellular proteostatic machinery, the endoplasmic reticulum stress response and heat shock protein 90 (ref. 71). Kras loss in this model did not induce reactivation of canonical MAPK signaling, but a proportion of tumors underwent an amplification and overexpression of the YAP transcriptional regulator, driving DNA replication and cell cycle progression72. These relapsed KrasG12D-independent tumors also assumed mesenchymal features. Together, these data provide evidence for multiple signaling hubs accounting for putative mechanisms by which PDAC cancer cells may subvert the effect of KRAS inhibition and therefore point to specific strategies to overcome potential escape mechanisms in clinical studies.
Precision approaches for KRAS-wild-type PDAC
Approximately 8–10% of PDAC tumors do not contain KRAS mutations11,13. Patients with KRAS-wild-type tumors have a relatively better prognosis than those with KRAS-mutated tumors29,73. Patients with wild-type KRAS often have alternative oncogenic mutations, most frequently activating BRAF alterations that lead to constitutive MAPK pathway activation and occur in approximately 2% of all PDAC cases74,75. BRAF activation may occur as a result of activating in-frame deletions or activating point mutations, such as the V600E mutation (BRAFV600E) with preclinical models and limited human experience, suggesting that the former may be sensitive to MEK inhibition17,76. In a PDAC GEMM, expression of BRAFV600E in pancreatic progenitor cells produced PanINs with low incidence of progression to PDAC77. Similar to the KPC GEMM, the addition of the dominant-negative Trp53 mutation to BRAFV600E resulted in full penetrance. In addition, a MEK inhibitor combined with gemcitabine chemotherapy showed regression of orthotopically implanted human-derived xenografts78. Despite these findings, a phase 2 clinical trial of 160 patients with metastatic PDAC treated with gemcitabine, or gemcitabine plus the MEK inhibitor trametinib, failed to show any significant differences in OS, PFS or ORR across KRAS-mutated or KRAS-wild-type individuals, although BRAF mutation status was not reported79. To apply a precision medicine approach to KRAS-wild-type patients with PDAC, BRAF mutation status must be confirmed and patients enrolled into BRAF- and MEK-targeting trials. A phase 2 multicenter, single-arm clinical trial is underway in which patients with advanced PDAC and containing a BRAFV600E mutation will be treated with the BRAF inhibitor encorafenib and the MEK inhibitor binimetinib with ORR as the primary endpoint (NCT04390243)80. Dual BRAF–MEK inhibition is approved for BRAFV600E metastatic melanoma and improves both PFS and OS compared to BRAF inhibition alone81. This strategy uses MEK inhibition to suppress MAPK escape mechanisms during BRAF inhibition82. Furthermore, a combined analysis of the ICGC and TCGA datasets revealed that 4.2% of KRAS-wild-type PDAC tumors had in-frame deletions in BRAF, which were confirmed to drive RAF dimerization and increase MAPK signaling in vitro76. In preclinical animal models containing in-frame BRAF deletions, the pan-RAF inhibitor LY3009120 led to tumor regression, which was not observed with the BRAFV600E inhibitor vemurafenib. Unfortunately, an early-phase clinical trial of 51 patients with advanced solid cancer treated with LY3009120, including 5 patients with PDAC, revealed no responses83.
Neurotrophic RTK (NTRK) genes drive mitogenic signaling in the central nervous system84,85. Activating NTRK fusions occur in <0.5% of all human cancers, including PDAC, and are also observed in KRAS-wild-type PDAC tumors86,87. In a basket study of patients with advanced solid cancer containing NTRK fusions, 75% of individuals responded to single-agent larotrectinib treatment, with 71% of these responders continuing to respond after 1 year of treatment88. This study led to the regulatory approval of larotrectinib for advanced solid cancers containing NTRK fusions. Of note, this study included only one individual with PDAC, although this individual did experience a PR. Furthermore, an integrated analysis of three ongoing early-phase clinical trials investigating the NTRK inhibitor entrectinib in advanced solid cancers with NTRK fusions reported in three evaluable patients with PDAC two and all three with disease control89. Moreover, the STARTRK-2 study (NCT02568267)90 is enrolling patients with advanced cancer that contain ROS1 fusions, with one individual with PDAC achieving disease stability on entrectinib and remaining on treatment for 7 months (ref. 86). KRAS-wild-type PDAC comprises a minority of cases but features a rich array of druggable non-KRAS driver aberrations in genes such as BRAF and NTRK genes in addition to other fusion events such as RET91, NRG1 (ref.92) and ALK93 (Table 1). Routine tumor sequencing of advanced PDAC tumors is critical in identifying KRAS-wild-type individuals likely to benefit from the targeting of alternative driver events.
Targeting DNA damage repair in PDAC
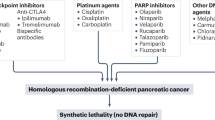
Mutations in DNA damage repair genes result in a deficiency of DNA double-stranded break repair94. In a cohort of 2,818 PDAC tissue samples assayed by NGS, somatic BRCA1, BRCA2 and PALB2 mutations were observed in 1.3%, 3.1% and 0.6% of cases, respectively95. In GEMMs, heterozygous germline Brca2-inactivating truncations promote PDAC formation on a background of oncogenic Kras96. Targeting PARP, a critical enzyme for single-stranded DNA repair, in BRCA1- or BRCA2-deficient preclinical cancer models is an effective therapeutic strategy97,98. The POLO study demonstrated that the PARP inhibitor olaparib has significant activity in patients with platinum-sensitive metastatic PDAC with germline BRCA1 or BRCA2 mutations99. In this study, participants on olaparib experienced a 7.4-month PFS compared to 3.8 months for those on placebo (hazard ratio of 0.53; 95% confidence interval of 0.35–0.82). This study led to the FDA approval of olaparib as a maintenance therapy of platinum-sensitive metastatic PDAC that contain germline BRCA1 or BRCA2 mutations. Importantly, follow-up analysis of the POLO trial demonstrated no statistically significant difference in median OS between the placebo and olaparib arms100. In a single-arm, phase 2 clinical trial, a second PARP inhibitor, rucaparib, also demonstrated clinical activity as a maintenance therapy for patients with platinum-sensitive advanced PDAC and germline or somatic BRCA1, BRCA2 or PALB2 mutations101. Of note, deleterious BRCA1 and BRCA2 mutations are often accompanied by inactivation of the second allele and positively correlate with the BRCA mutation signature102, which is associated with response to platinum-based chemotherapy in PDAC12. In a recent open-label, randomized, multicenter, two-arm phase 2 clinical trial, patients with advanced PDAC and germline mutations in BRCA1, BRCA2 or PALB2 were randomized to cisplatin and gemcitabine with or without the PARP inhibitor veliparib103. The gemcitabine–cisplatin–veliparib (GCV) arm experienced a 74.1% response rate in comparison to the gemcitabine–cisplatin (GC) arm, which demonstrated a response rate of 65.2% (P = 0.55). Moreover, the PFS of the GCV and GC arms was 10.1 months and 9.7 months, respectively (P = 0.73), and the OS was 15.5 months and 16.4 months, respectively (P = 0.60). Although the addition of PARP inhibition did not improve clinical benefits, the combination of gemcitabine and cisplatin without PARP inhibitor is now considered an effective treatment regimen for this subset of patients104. Lastly, a phase 2 randomized trial is investigating the role of adjuvant olaparib in patients with PDAC and germline or somatic BRCA1, BRCA2 or PALB2 mutations (NCT04858334)105.
BRCA1 and BRCA2 mutations positively correlate with increased PD-L1 staining in human PDAC cancer cells95. Retrospective data on BRCA2-mutated breast cancer revealed a superior response to immune checkpoint inhibition compared to BRCA2 wild type, in line with results seen in syngeneic mouse models of breast and colon cancer106, pointing to these as potential candidate biomarkers to stratify patients for ICB. Indeed, an ongoing phase 2 open-label, two-arm clinical trial aims to compare olaparib monotherapy to olaparib plus pembrolizumab as maintenance therapy in patients with metastatic PDAC and germline BRCA1 or BRCA2 mutations (NCT04548752)107. Another PARP inhibitor–PD-1 doublet (niraparib and dostarlimab, respectively) is also being investigated in a phase 2 clinical trial of patients with metastatic PDAC with either somatic or germline BRCA1 or BRCA2 mutations (NCT04493060)108.
The ATM gene is a key regulator of the DNA damage response109 and is mutated in approximately 5% of PDAC cases13. PDAC cells that have mutations in ATM are specifically sensitive to PARP inhibition in combination with inhibition of ATR in vivo, whereas PARP inhibitor monotherapy has limited durable activity110. A DNA replication stress signature that correlates with the PDAC basal-like transcriptional subtype is predictive of response to ATR inhibition in human PDAC organoids111. Multiple early-phase clinical trials testing ATR inhibitors in combination with cytotoxic chemotherapy (NCT04514497)112 or with PARP inhibitors (NCT03682289)113 are currently underway (Table 1).
Translational approaches using transcriptionally defined molecular subtypes
Gene expression profiling studies have significantly improved our understanding of the molecular taxonomy of all major cancers over the past two decades114,115,116,117. The first gene expression-based subtyping study in PDAC revealed the existence of three dominant molecular subtypes of PDAC: quasi-mesenchymal, classical and exocrine-like, with the latter two subtypes displaying a significantly improved survival over quasi-mesenchymal118. The quasi-mesenchymal subtype overexpressed mesenchymal-associated genes, whereas the classical subtype overexpressed cell adhesion- and epithelial-related genes. Subsequently, ‘virtual microdissection’ experiments using expression profiling data revealed the presence of two predominant molecular subtypes of PDAC: basal-like and classical119. The basal-like subtype of PDAC was associated with poor survival and resembled the basal subtype in both breast and bladder cancers. Furthermore, the ICGC demonstrated four distinct molecular subtypes by RNA-sequencing gene expression profiling: pancreatic progenitor, immunogenic, aberrantly differentiated endocrine exocrine (ADEX) and squamous, with the latter resembling the basal-like subtype31. The squamous subtype displayed an increased frequency of TP53 and KDM6A mutations and displayed a significantly poorer prognosis than all other molecular subtypes. The TCGA team further suggested that samples classified as either ADEX or immunogenic had lower purity of neoplastic epithelium and a higher degree of leukocyte infiltration13. After accounting for neoplastic cellularity, all samples were effectively reclassified into either the basal-like/squamous or the classical/progenitor subtypes. More recently, using a cohort of 206 patients with resectable (stages 1 and 2) PDAC and 111 patients with advanced (stages 3 and 4) PDAC, RNA sequencing analysis revealed that 62% of patients with resectable PDAC could be classified as having the classical subtype, whereas in metastatic PDAC, the classical subtype comprised only 46% of individuals120. This study also further subdivided basal-like PDAC into two subgroups: basal-like A and basal-like B. Notably, basal-like A made up only 5% of patients with resectable PDAC but 24% of patients with stage 4 PDAC. Furthermore, the basal-like A subtype was shown to be largely chemoresistant, particularly in advanced disease settings. While bulk RNA-sequencing data suggest that the subtypes of PDAC tumors are dichotomous, single-cell RNA-sequencing analysis has revealed the coexistence of populations of cancer cells displaying classical and basal-like signatures within the same tumor, including the presence of ‘hybrid’ cells bearing features of both subtypes120,121,122. The observed transcriptional subtype heterogeneity in PDAC may have important implications for development of resistance to therapeutic strategies targeting specific subtypes of the disease.
Gemcitabine/nab-paclitaxel (GnP) and modified FOLFIRINOX (mFOLFIRINOX) are both category one recommended regimens for the treatment of advanced PDAC104. Nonetheless, retrospective case series indicated that mFOLFIRINOX may be associated with greater radiographic responses and OS in early123 and advanced stage disease124,125. However, these data may be confounded by the use of mFOLFIRINOX in patients with better performance status. Indeed preliminary evidence from a phase 2 trial (SWOG S1505), in which patients with resectable PDAC were randomized to perioperative chemotherapy with either GnP or mFOLFIRINOX, displayed no significant difference in median OS between the two regimens126. The PRODIGE-24 study demonstrated a median OS of 54 months in individuals who received adjuvant mFOLFIRINOX in comparison to 35 months in individuals who received only adjuvant gemcitabine. The use of gene expression profiling and molecular classification of patients with PDAC may offer insight into the molecular determinants of response to either chemotherapy regimen.
The COMPASS trial in advanced PDAC assessed the response to chemotherapy in classical and basal-like subtypes16. In this trial, 195 participants underwent tumor biopsy followed by RNA sequencing, which classified tumors as either classical (80%) or basal-like (20%)127. There was a 60% rate of radiographic progression in participants with basal-like tumors treated with mFOLFIRINOX in contrast to a 15% rate of progression in classical tumors. Moreover, GATA6 expression was significantly increased in classical subtype tumors and may be an appropriate surrogate marker of chemosensitivity across molecular subtypes. Although this was the first report that gene expression profiling may predict response to chemotherapy, the clinical utility of these findings should be prospectively validated. To that end, The Pancreatic Adenocarcinoma Signature Stratification for Treatment (PASS-01) trial is a phase 2, multicenter clinical trial randomizing patients with metastatic PDAC to either mFOLFIRINOX or GnP treatment (NCT04469556)128. The primary endpoint is PFS and will provide further insight into comparative efficacies of the two regimens. The correlative studies will provide insight into the responses to both chemotherapy regimens in specific molecular subtypes by integrating the analysis of GATA6 expression (classical subtype surrogate marker), cytokeratin 5 and cytokeratin 17 expression (basal-like subtype surrogate markers) and the classical–basal subtype gene expression signatures. This and subsequent randomized phase 3 trials may allow clinicians to select first-line mFOLFIRINOX or GnP chemotherapy regimens using expression profiles or surrogate markers of PDAC molecular subtypes.
Immunotherapeutic approaches to PDAC
Immunotherapies are transforming cancer therapy across tumor types. In this section, we review recent advances and challenges in PDAC immunotherapy and ongoing strategies to reprogram the tumor microenvironment in PDAC.
Immunotherapy trials in PDAC
ICB therapies have revolutionized cancer treatment and clinical prospects129, as monoclonal antibodies targeting PD-1/PD-L1 and CTLA-4 have proven highly effective across numerous solid tumors130,131,132,133,134,135. Although anti-PD-1 antibodies have a tissue-agnostic indication for all metastatic solid tumors with MSI-H status136, this occurs in approximately 1–2% of PDAC137. ICB has otherwise proven ineffective in the treatment of PDAC in several early-phase clinical trials138,139. Most recently, a phase 2 clinical trial tested durvalumab (anti-PD-L1 monoclonal antibody) plus tremelimumab (anti-CTLA-4 monoclonal antibody) versus durvalumab monotherapy in individuals previously treated with chemotherapy for metastatic PDAC140. Both arms displayed favorable toxicity profiles, but the objective response rates were 3.1% and 0%, respectively. These disappointing results highlight the need for translational studies seeking to understand and reverse the recalcitrant nature of the PDAC tumor immune microenvironment.
The immune landscape of PDAC
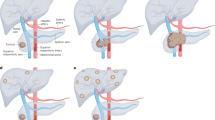
T cell-targeted immunotherapy strategies have been largely unsuccessful in PDAC beyond the ~1% of patients with MSI-H tumors141,142. Nevertheless, new efforts to augment T cell function using vaccines, adoptive cell therapies and novel checkpoint blockade targets and efforts aimed at increasing major histocompatibility (MHC) class I expression on tumor cells could offer new paths forward for enhancing antitumor immunity in PDAC143,144,145. Although CD8+ T cells are present at relatively high frequencies in approximately one-third of PDAC primary tumors, spatial analysis revealed that T cells are rarely located adjacent to the tumor cell nests146,147. Instead, neoplastic cells are surrounded by macrophages and fibroblasts, and close proximity of alternatively activated macrophages correlates with poor prognosis147. Both malignant cells and fibroblasts secrete chemokines and growth factors that attract monocytes and granulocytes to the tumor microenvironment, thereby replenishing a short-lived pool of immunosuppressive myeloid cells. These include CXCL2, CCL2, granulocyte colony-stimulating factor (G-CSF) and granulocyte–macrophage colony-stimulating factor (GM-CSF)148,149. Tumor-associated macrophages, derived either from infiltrating monocytes or from pancreas-resident macrophages, can directly support malignant cells through growth factors such as transforming growth factor-β (TGFβ) and provision of metabolites that interfere with nucleoside analog-based chemotherapies150,151,152. Given the complex immunosuppressive landscape of the PDAC microenvironment143,144,145, we will focus here on immune targeting strategies with three primary aims: (1) removing short-lived immunosuppressive myeloid cells; (2) reprogramming dendritic cells (DCs) to better prime tumor-specific T cell responses, particularly in the context of KRAS vaccines; and (3) targeting macrophage and fibroblast stromal support networks (Fig. 2 and Tables 2 and 3).

a, Schematic representation of two immunotherapy approaches to PDAC: targeting immunosuppressive myeloid cells and reprogramming DCs and macrophages, with both strategies converging on increased CD8+ lymphocyte activity against PDAC cancer cells. Specific receptors amenable to therapeutic intervention on the immunosuppressive myeloid cells granulocytic MDSCs (gMDSCs) and moMDSCs are CXCR2 and CCR2, respectively. FLT3 ligand (FLT3L) promotes the recruitment of cDC1s from the bone marrow, while agonistic anti-CD40 monolonal antibody and CpG promote cDC1 and tumoricidal macrophage activity. Three other denoted molecules (polyinosinic–polycytidylic acid and poly-l-lysine (poly-ICLC), Toll-like receptor 7 (TLR7) agonist and lipid nanoparticle vaccines) exert potential anti-PDAC antibody activity primarily through increased activity of cDC1s. b, The PDAC microenvironment features poor infiltration of cytotoxic T cells and a desmoplastic stroma with an abundance of activated fibroblasts, extracellular matrix proteins and tumor-promoting myeloid cells. Reprogramming of activated fibroblasts through one of several investigational approaches may attenuate the desmoplastic stroma and improve PDAC chemotherapy and ICB sensitivity. IL-1β, interleukin-1β; PIN1, peptidyl-prolyl cis-trans isomerase NIMA-interacting 1; TH1 cell, type 1 helper T cell.
Targeting short-lived immunosuppressive myeloid cells
The importance of the chemokine receptors CXCR2 and CCR2 and their ligands in regulating neutrophil and monocyte cell trafficking to tumors is well established153. Given the short-lived nature of these cells, blocking their influx rapidly leads to a decline in intratumoral moMDSCs and gMDSCs. In mice, transient depletion of neutrophils or selective interference of neutrophil trafficking can lead to reduction in PDAC tumor burden, although such strategies cannot be maintained long term due to the critical importance of neutrophils for host defense154,155,156. Agonism of the pan-myeloid integrin CD11b prevents accumulation of most myeloid cell types in PDAC mouse models and strongly synergizes with PD-1 blockade157. Similarly, mice treated with inhibitors of CCR2 that targeted circulating monocytes showed reduced PDAC tumor burden158. In humans, a landmark clinical trial of FOLFIRINOX combined with a small-molecule inhibitor of CCR2 versus FOLFIRINOX alone in patients with locally advanced PDAC159 demonstrated a retention of monocytes in the bone marrow, correlating with a notable drop in monocytes from circulation and moMDSCs from the tumor microenvironment in CCR2 inhibitor-treated individuals. This was accompanied by impressive reductions in primary tumor burden, leading to downstaging of disease and eligibility for surgery in 39% of the cohort159. Unfortunately, a similar study of a CCR2 inhibitor combined with GnP in metastatic PDAC failed for both safety and efficacy160. One possible interpretation is that blockade of monocyte trafficking may be suited as an adjunct to neoadjuvant chemotherapy or to immunotherapies that engage tumor-specific T cells but could result in therapy-related adverse events when used long term. Currently, an early-phase clinical trial is seeking to assess the response of CCR2 inhibition in combination with the anti-PD-1 monoclonal antibody nivolumab plus chemotherapy (NCT03496662)161. However, because chemotherapy affects hematopoietic cell survival and differentiation, as do concurrent therapies, such as a recombinant G-CSF, how these treatments affect MDSCs should be thoroughly investigated.
Reprogramming DCs
Given the limited efficacy of strategies aimed at reinvigorating exhausted T cells in pancreatic cancer, whether the central issue in patients with PDAC is endogenous T cell exhaustion or a lack of T cell priming remains an open question162. Studies in mice have revealed the importance of cross-presenting type 1 conventional dendritic cells (cDC1s) in priming tumor-specific CD8+ T cell responses and the dearth of these cells in pancreatic cancer163,164. Indeed, a direct comparison of mouse models of PDAC versus lung adenocarcinoma showed a distinct lack of CD103+ cDC1s in PDAC. FLT3L treatment increases intratumoral cDC1s and restores sensitivity to CD40 agonist antibody and radiation therapy in PDAC mouse models164. Ongoing early-phase clinical trials will soon determine the tolerability and efficacy of CD40 agonist antibody in combination with FLT3L in PDAC and other solid tumors (NCT04536077 and NCT03329950)165,166.
DCs phagocytose tumor cell fragments and present antigenic peptides on MHC class I and class II. Following activation, DCs express costimulatory ligands and upregulate CCR7 to travel to draining lymph nodes and prime naive CD8+ and CD4+ T cells129. This process is dependent on the presence of innate immune adjuvants, including damage-associated molecules, such as ATP or HMGB1, released from dying tumor cells157. Although tumor cell death can activate DCs, phagocytosis of tumor cell fragments also induces regulatory programs in DCs that hinder their subsequent engagement with T cells167. This regulatory program can be overcome by exposure of DCs to microbial products that engage TLR signaling, such as the viral nucleic acid mimetic pIpC or CpG DNA, and these innate immune adjuvants are being incorporated into vaccine strategies168. Other approaches include targeted agents that affect DNA replication and repair pathways, which activate the STING pathway, leading to type I interferon production, which in turn promotes DC activation169.
Therapeutic cancer vaccines have historically been unsuccessful, although recent developments have reignited interest in this topic (Table 2). Mutant KRAS epitopes are presented on multiple human HLA alleles and can elicit antitumor T cell responses in patients with cancer, suggesting that direct T cell recognition of mutant KRAS is possible in at least a subset of individuals52,53. Analysis of blood from healthy donors with diverse HLA haplotypes revealed that mutant KRAS-specific T cells could be readily expanded from the naive repertoire170. Given the early emergence of KRAS mutations in preneoplastic lesions, KRAS-mutant-specific T cells may acquire tolerance early in the oncogenic process. However, the near-ubiquitous presence of mutant KRAS in PDAC and its known role as a key oncogenic driver makes KRAS an ideal target for personalized vaccine approaches. Moreover, the development of better computational tools for candidate neoantigen prioritization and the emergence of improved vaccine delivery platforms will spur the development of efficacious personalized vaccines for PDAC171,172.
Targeting macrophages
Macrophages are highly abundant in PDAC, and they participate in tissue repair, support epithelial cell growth and have tumor-promoting properties158. Nevertheless, macrophage depletion strategies in solid tumors have limited efficacy due to compensatory increases in other myeloid cell populations157. Therapies aimed at reprogramming macrophages to phagocytose and kill live tumor cells may provide an interesting twist to this approach. Tumoricidal macrophages were first identified in PDAC treated with agonistic anti-CD40, an antibody that activates the phagocytic program in macrophages173. Anti-CD40 antibody seemed to operate independently of T cells, although it was later found to activate DCs and enhance T cell priming in addition to its effects on macrophages174,175,176. A recent phase 2 trial testing anti-CD40 antibody, anti-PD-1 antibody and GnP revealed no benefit of inclusion of anti-CD40 antibody, suggesting that the T cell-priming role of anti-CD40 antibody in human PDAC may not be its central mechanism of action177. Determining how to better exploit the macrophage-potentiating role of anti-CD40 in combination therapy for PDAC may prove fruitful. Tumoricidal macrophages were also reported in mice treated with the NF-κB modulator LCL-161, which induced lymphotoxin production from T cells that reprogrammed macrophages to phagocytose and kill live pancreatic tumor cells178,179, indicating potential alternate pathways for tumoricidal macrophage induction.
The receptor–ligand interactions that govern macrophage uptake of live tumor cells are still largely unknown. The ‘don’t eat me’ ligand CD47 on tumor cells can engage SIRPα on macrophages and prevent phagocytosis, although blockade of CD47 alone has little effect in most solid tumors without provision of a prophagocytic signal180,181,182,183. Activation of macrophages with the TLR9 ligand CpG can induce phagocytosis even of tumor cells expressing CD47 (ref. 184). Antibodies of IgG1 subclasses engage Fc receptors on macrophages and can strongly induce phagocytosis185. These translational approaches inducing tumoricidal macrophages are of great interest, and only clinical testing will inform of their actual efficacy in patients with PDAC.
Targeting cancer-associated fibroblasts
Activated fibroblasts are a major source of myeloid cell-recruiting chemokines. Although broad targeting of PDAC cancer-associated fibroblasts has been unsuccessful186,187, increased understanding of the heterogeneity of the fibroblast compartment has led to a resurgence in interest in targeting subsets of fibroblasts or their secreted products188,189,190,191.
Antibodies that target TGFβ result in pleiotropic phenotypes, including decreased intratumoral fibroblasts, relieved CD8+ T cell suppression and reduced myeloid cell infiltration192,193,194. An anti-TGFβ monoclonal antibody (NIS793) in combination with GnP and PD-1 blockade is currently being evaluated in a phase 2 clinical trial (NCT04390763)195. The angiotensin II receptor antagonist losartan acts partly via decreased TGFβ levels and is also being tested in combination with PD-1 blockade and chemoradiation in resectable PDAC (NCT03563248)196,197. Fibroblast quiescence may also be achieved using vitamin D157, and two phase 2 clinical trials of paricalcitrol with GnP (NCT03520790)198 and with PD-1 blockade plus GnP/cisplatin (NCT02754726)199 in patients with metastatic PDAC are underway.
PIN1 mediates the phosphorylation and activation of >60 oncoproteins and inactivation of >30 tumor suppressor genes, many of which are downstream of oncogenic KRAS200. A high-throughput small-molecule screen identified all-trans retinoic acid (ATRA) to directly bind, inhibit and degrade PIN1 in vivo201. A selective inhibitor of PIN1, sulfopin, has also demonstrated efficacy in orthotopic mouse models202. ATRA plus arsenic trioxide or sulfopin treatment in PDAC models promotes a quiescent cancer-associated fibroblast state and thereby diminishes the PDAC desmoplastic response; additionally, PIN1 inhibition leads to decreased degradation of the gemcitabine plasma membrane transporter (ENT1), enhancing chemosensitivity, and it may synergize with immunotherapy202,203. Finally, chemotherapy plus ATRA showed promise in a phase 1 clinical trial in metastatic PDAC204, indicating that PIN1-based stromal reprogramming strategies are worth further investigation.
IL-1β is actively produced by macrophages and granulocytes in PDAC and has pleiotropic effects on the tumor microenvironment, including supporting inflammatory cancer-associated fibroblasts, which produce IL-6 and have been shown to support an immunosuppressive microenvironment and tumor cell survival189,205. IL-1β may also be produced by PDAC tumor cells, and blockade of IL-1β in preclinical models synergizes with PD-1 blockade206. The IL-1β-blocking antibody canakinumab and PD-1-blocking antibody spartalizumab are currently being evaluated in combination with GnP in a phase 1 clinical trial (NCT04581343)207. Canakinumab limits the incidence of lung adenocarcinoma208, and the role of innate inflammatory cytokines in tumor-promoting inflammation is recognized209. Whether blockade of IL-1β will be effective in established tumors remains to be determined.
Future directions
Over the past decade, we have gained a deep understanding of recurrent driver mutations in PDAC by in-depth analyses of human samples and GEMMs. These tools have delineated specific vulnerabilities in PDAC and have improved our understanding of the tumor microenvironment. Future translational studies using PDAC GEMMs will need to account for the real-world presence of multiple driver genes. As we have seen with the recent successful approvals of PARP inhibitors and PD-1 blockade for molecularly defined subclasses of PDAC, precision-based and immunotherapy-based preclinical and clinical pipelines open new therapeutic avenues. The ongoing developments in KRAS-specific inhibitors is especially encouraging for PDAC. As encountered in other major malignancies, once these therapies reach the clinic, we will need to address how they will be given in concert with current standard of care chemotherapy. In addition, understanding the mechanisms of chemotherapy resistance in PDAC is essential to control this systemic illness. Lastly, as PDAC treatment approaches hopefully advance, attention will also need to focus on measures aimed at improving the quality of life of patients, such as cancer-associated cachexia. We predict that the next decade will feature an abundance of precision oncology approaches to this recalcitrant cancer, which will benefit an ever-larger group of patients.
References
Siegel, R. A. et al. Cancer Statistics 2022. CA Cancer J. Clin. 72, 7–33 (2022).
Conroy, T. et al. FOLFIRINOX or gemcitabine as adjuvant therapy for pancreatic cancer. N. Engl. J. Med. 379, 2395–2406 (2018).
Gobbi, P. G. et al. The prognostic role of time to diagnosis and presenting symptoms in patients with pancreatic cancer. Cancer Epidemiol. 37, 186–190 (2013).
Sohal, D. P. S. et al. Metastatic pancreatic cancer: ASCO clinical practice guideline update. J. Clin. Oncol. 36, 2545–2556 (2018).
Conroy, T. et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 364, 1817–1825 (2011).
Von Hoff, D. D. et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 369, 1691–1703 (2013).
Van Cutsem, E. et al. Randomized phase III trial of pegvorhyaluronidase α with nab-paclitaxel plus gemcitabine for patients with hyaluronan-high metastatic pancreatic adenocarcinoma. J. Clin. Oncol. 38, 3185–3194 (2020).
Tempero, M. et al. Ibrutinib in combination with nab-paclitaxel and gemcitabine for first-line treatment of patients with metastatic pancreatic adenocarcinoma: phase III RESOLVE study. Ann. Oncol. 32, 600–608 (2021).
Hecht, J. R. et al. Randomized phase III study of FOLFOX alone and with pegilodecakin as second-line therapy in patients with metastatic pancreatic cancer (SEQUOIA). J. Clin. Oncol. 39, 1108–1118 (2021).
Biankin, A. V. et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 491, 399–405 (2012).
Witkiewicz, A. K. et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat. Commun. 6, 6744 (2015).
Waddell, N. et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 518, 495–501 (2015).
The Cancer Genome Atlas Research Network. Integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell 32, 185–203 (2017).
Pishvaian, M. J. et al. Overall survival in patients with pancreatic cancer receiving matched therapies following molecular profiling: a retrospective analysis of the Know Your Tumor registry trial. Lancet Oncol. 21, 508–518 (2020).
Lowery, M. A. et al. Real-time genomic profiling of pancreatic ductal adenocarcinoma: potential actionability and correlation with clinical phenotype. Clin. Cancer Res. 23, 6094–6100 (2017).
Aung, K. L. et al. Genomics-driven precision medicine for advanced pancreatic cancer: early results from the COMPASS trial. Clin. Cancer Res. 24, 1344–1354 (2018).
Aguirre, A. J. et al. Real-time genomic characterization of advanced pancreatic cancer to enable precision medicine. Cancer Discov. 8, 1096–1111 (2018).
Zehir, A. et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 23, 703–713 (2017).
Waters, A. M. & Der, C. J. KRAS: the critical driver and therapeutic target for pancreatic cancer. Cold Spring Harb. Perspect. Med. 8, a031435 (2018).
Bamford, S. et al. The COSMIC (catalogue of somatic mutations in cancer) database and website. Br. J. Cancer 91, 355–358 (2004).
Hingorani, S. R. et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 4, 437–450 (2003).
Aguirre, A. J. et al. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 17, 3112–3126 (2003).
Ying, H. et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 149, 656–670 (2012).
Ying, H. et al. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 30, 355–385 (2016).
Hingorani, S. R. et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 7, 469–483 (2005).
Poulin, E. J. et al. Tissue-specific oncogenic activity of KRASA146T. Cancer Discov. 9, 738–755 (2019).
Cook, J. H. et al. The origins and genetic interactions of KRAS mutations are allele- and tissue-specific. Nat. Commun. 12, 1808 (2021).
Gultawatvichai, P. I., Tomaszewicz, K., Bathini, V. G. & Hutchinson, L. Prevalence of KRAS mutation subtypes and MSI status in pancreatic cancer. Ann. Oncol. 29, VIII673 (2018).
Qian, Z. R. et al. Association of alterations in main driver genes with outcomes of patients with resected pancreatic ductal adenocarcinoma. JAMA Oncol. 4, e173420 (2018).
McCormick, F. Targeting KRAS directly. Annu. Rev. Cancer Biol. 2, 81–90 (2018).
Bailey, P. et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 531, 47–52 (2016).
Janes, M. R. et al. Targeting KRAS mutant cancers with a covalent G12C-specific inhibitor. Cell 172, 578–589 (2018).
Canon, J. et al. The clinical KRASG12C inhibitor AMG 510 drives anti-tumour immunity. Nature 575, 217–223 (2019).
Lanman, B. A. et al. Discovery of a covalent inhibitor of KRASG12C (AMG 510) for the treatment of solid tumors. J. Med. Chem. 63, 52–65 (2020).
Skoulidis, F. et al. Sotorasib for lung cancers with KRAS p.G12C mutation. N. Engl. J. Med. 384, 2371–2381 (2021).
NCT03600883: A phase 1/2, study evaluating the safety, rolerability, PK, and efficacy of AMG 510 in subjects with solid tumors with a specific KRAS mutation (CodeBreak 100). https://clinicaltrials.gov/ct2/show/NCT03600883 (2018).
Hong, D. S. et al. CodeBreak 100: phase I study of AMG 510, a novel KRASG12C inhibitor, in patients (pts) with advanced solid tumors other than non-small cell lung cancer (NSCLC) and colorectal cancer (CRC). J. Clin. Oncol. 38, 3511 (2020).
Hallin, J. et al. The KRASG12C inhibitor MRTX849 provides insight toward therapeutic susceptibility of KRAS-mutant cancers in mouse models and patients. Cancer Discov. 10, 54–71 (2020).
Bekaii-Saab, T. S. et al. KRYSTAL-1: updated activity and safety of adagrasib (MRTX849) in patients (pts) with unresectable or metastatic pancreatic cancer (PDAC) and other gastrointestinal (GI) tumors harboring a KRASG12C mutation. J. Clin. Oncol. 40, 519 (2022).
Wang-Gillam, A. et al. NAPOLI-1 phase 3 study of liposomal irinotecan in metastatic pancreatic cancer: final overall survival analysis and characteristics of long-term survivors. Eur. J. Cancer 108, 78–87 (2019).
NCT03785249: Phase 1/2 study of MRTX849 in patients with cancer having a KRASG12C mutation KRYSTAL-1. https://clinicaltrials.gov/ct2/show/NCT03785249 (2018).
NCT04330664: Adagrasib in combination with TNO155 in patients with cancer (KRYSTAL 2). https://clinicaltrials.gov/ct2/show/NCT04330664 (2018).
James G. Christensen. Discovery and characterization of MRTX1133, a selective non-covalent inhibitor of KRASG12D. AACR-NCI-EORTC Virtual International Conference on Molecular Targets and Cancer Therapeutics. Plenary Session 5: Drugging Difficult Targets. San Diego, California (7 October 2021).
Mirati Therapeutics. Mirati Therapeutics reports investigational adagrasib (MRTX849) preliminary data demonstrating tolerability and durable anti-tumor activity as well as initial MRTX1133 preclinical data. https://ir.mirati.com/press-releases/press-release-details/2020/Mirati-Therapeutics-Reports-Investigational-Adagrasib-MRTX849-Preliminary-Data-Demonstrating-Tolerability-and-Durable-Anti-Tumor-Activity-as-well-as-Initial-MRTX1133-Preclinical-Data/default.aspx (2020).
Wang, X. et al. Identification of MRTX1133, a noncovalent, potent, and selective KRASG12D inhibitor. J. Med. Chem. 65, 3123–3133 (2022).
Schulze C. J. et al. Tri-complex inhibitors of the oncogenic, GTP-bound form of KRASG12C overcome RTK-mediated escape mechanisms and drive tumor regressions in vivo. Mol. Cancer Ther. 18, abstr. PR10 (2020).
Gustafson, W. C. et al. Direct targeting of RAS in pancreatic ductal adenocarcinoma with RMC-6236, a first-in-class, RAS-selective, orally bioavailable, tri-complex RASMULTI(ON) inhibitor. J. Clin. Oncol. 40, 591–591 (2022).
Freedman, T. S. et al. A Ras-induced conformational switch in the Ras activator Son of Sevenless. Proc. Natl Acad. Sci. USA 103, 16692–16697 (2006).
Hofmann, M. H. et al. BI-3406, a potent and selective SOS1–KRAS interaction inhibitor, is effective in KRAS-driven cancers through combined MEK inhibition. Cancer Discov. 11, 142–157 (2021).
NCT04111458: A study to test different doses of BI 1701963 alone and combined with trametinib in patients with different types of advanced cancer (solid tumours with KRAS mutation). https://clinicaltrials.gov/ct2/show/NCT04111458 (2019).
Baden, L. R. et al. Efficacy and safety of the mRNA-1273 SARS-CoV-2 vaccine. N. Engl. J. Med. 384, 403–416 (2021).
Tran, E. et al. T-cell transfer therapy targeting mutant KRAS in cancer. N. Engl. J. Med. 375, 2255–2262 (2016).
Wang, Q. J. et al. Identification of T-cell receptors targeting KRAS-mutated human tumors. Cancer Immunol. Res. 4, 204–214. (2016).
NCT03948763: A study of mRNA-5671/V941 as monotherapy and in combination with pembrolizumab (V941-001). https://clinicaltrials.gov/ct2/show/NCT03948763 (2019).
Kamerkar, S. et al. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature 546, 498–503 (2017).
NCT03608631: iExosomes in treating participants with metastatic pancreas cancer with KRASG12D mutation. https://clinicaltrials.gov/ct2/show/NCT03608631 (2018).
Xue, J. Y. et al. Rapid non-uniform adaptation to conformation-specific KRASG12C inhibition. Nature 577, 421–425 (2020).
Gandara, D. et al. A phase 1b study evaluating the combination of sotorasib, a KRASG12C inhibitor, and afatinib, a pan-ErbB tyrosine kinase inhibitor, in advanced KRAS p.G12C mutated non-small cell lung cancer (NSCLC). Mol. Cancer Ther. 20, abstr. P05-02 (2021.
NCT04185883: Sotorasib activity in subjects with advanced solid tumors with KRAS p.G12C mutation (CodeBreak 101). https://clinicaltrials.gov/ct2/show/NCT04185883 (2019).
Awad, M. M. et al. Acquired resistance to KRASG12C inhibition in cancer. N. Engl. J. Med. 384, 2382–2393 (2021).
Tanaka, N. et al. Clinical acquired resistance to KRASG12C inhibition through a novel KRAS switch-II pocket mutation and polyclonal alterations converging on RAS–MAPK reactivation. Cancer Discov. 11, 1913–1922 (2021).
Kopetz, S. et al. Encorafenib, binimetinib, and cetuximab in BRAFV600E-mutated colorectal cancer. N. Engl. J. Med. 381, 1632–1643 (2019).
Kinsey, C. G. et al. Protective autophagy elicited by RAF→MEK→ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat. Med. 25, 620–627 (2019).
Bryant, K. L. et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat. Med. 25, 628–640 (2019).
NCT03825289: Trametinib and hydroxychloroquine in treating patients with pancreatic cancer (THREAD). https://clinicaltrials.gov/ct2/show/NCT03825289 (2019).
NCT04132505: Binimetinib and hydroxychloroquine in treating patients with KRAS mutant metastatic pancreatic cancer. https://clinicaltrials.gov/ct2/show/NCT04132505 (2019).
NCT04386057: LY3214996 ± HCQ in pancreatic cancer. https://clinicaltrials.gov/ct2/show/NCT04386057 (2020).
Collins, M. A. et al. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J. Clin. Invest. 122, 639–653 (2012).
Viale, A. et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 514, 628–632 (2014).
Genovese, G. et al. Synthetic vulnerabilities of mesenchymal subpopulations in pancreatic cancer. Nature 542, 362–366 (2017).
Zheng, X. et al. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 527, 525–530 (2015).
Kapoor, A. et al. Yap1 activation enables bypass of oncogenic Kras addiction in pancreatic cancer. Cell 158, 185–197 (2014).
McIntyre, C. A. et al. Alterations in driver genes are predictive of survival in patients with resected pancreatic ductal adenocarcinoma. Cancer 126, 3939–3949 (2020).
Singhi, A. D. et al. Real-time targeted genome profile analysis of pancreatic ductal adenocarcinomas identifies genetic alterations that might be targeted with existing drugs or used as biomarkers. Gastroenterology 156, 2242–2253 (2019).
Guan, M. et al. Molecular and clinical characterization of BRAF mutations in pancreatic ductal adenocarcinomas (PDACs). J. Clin. Oncol. 36, 214 (2018).
Chen, S. H. et al. Oncogenic BRAF deletions that function as homodimers and are sensitive to inhibition by RAF dimer inhibitor LY3009120. Cancer Discov. 6, 300–315 (2016).
Collisson, E. A. et al. A central role for RAF→MEK→ERK signaling in the genesis of pancreatic ductal adenocarcinoma. Cancer Discov. 2, 685–693 (2012).
Kawaguchi, K. et al. MEK inhibitors cobimetinib and trametinib, regressed a gemcitabine-resistant pancreatic-cancer patient-derived orthotopic xenograft (PDOX). Oncotarget 8, 47490–47496 (2017).
Infante, J. R. et al. A randomised, double-blind, placebo-controlled trial of trametinib, an oral MEK inhibitor, in combination with gemcitabine for patients with untreated metastatic adenocarcinoma of the pancreas. Eur. J. Cancer 50, 2072–2081 (2014).
NCT04390243: Binimetinib and encorafenib for the treatment of pancreatic cancer in patients with a somatic BRAFV600E mutation. https://clinicaltrials.gov/ct2/show/NCT04390243 (2020).
Robert, C. et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N. Engl. J. Med. 372, 30–39 (2015).
Shi, H. et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 4, 80–93 (2014).
Sullivan, R. J. et al. A phase I study of LY3009120, a pan-RAF inhibitor, in patients with advanced or metastatic cancer. Mol. Cancer Ther. 19, 460–467 (2020).
Kaplan, D. R. et al. The trk proto-oncogene product: a signal transducing receptor for nerve growth factor. Science 252, 554–558 (1991).
Kaplan, D. R., Martin-Zanca, D. & Parada, L. F. Tyrosine phosphorylation and tyrosine kinase activity of the trk proto-oncogene product induced by NGF. Nature 350, 158–160 (1991).
Pishvaian, M. J. et al. Entrectinib in TRK and ROS1 fusion-positive metastatic pancreatic cancer. JCO Precis. Oncol. 2, 1–7 (2018).
Okamura, R. et al. Analysis of NTRK alterations in pan-cancer adult and pediatric malignancies: implications for NTRK-targeted therapeutics. JCO Precis. Oncol. 2018, PO.18.00183 (2018).
Drilon, A. et al. Efficacy of larotrectinib in TRK fusion-positive cancers in adults and children. N. Engl. J. Med. 378, 731–739 (2018).
Doebele, R. C. et al. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: integrated analysis of three phase 1–2 trials. Lancet Oncol. 21, 271–282 (2020).
NCT02568267: Basket study of entrectinib (RXDX-101) for the treatment of patients with solid tumors harboring NTRK 1/2/3 (Trk A/B/C), ROS1, or ALK gene rearrangements (fusions) (STARTRK-2). https://clinicaltrials.gov/ct2/show/NCT02568267 (2015).
Subbiah, V. et al. Clinical activity of the RET inhibitor pralsetinib (BLU-667) in patients with RET fusion-positive solid tumors. J. Clin. Oncol. 39, 467 (2021).
Heining, C. et al. NRG1 fusions in KRAS wild-type pancreatic cancer. Cancer Discov. 8, 1087–1095 (2018).
Singhi, A. D. et al. Identification of targetable ALK rearrangements in pancreatic ductal adenocarcinoma. J. Natl Compr. Canc. Netw. 15, 555–562 (2017).
Stoppa-Lyonnet, D. The biological effects and clinical implications of BRCA mutations: where do we go from here? Eur. J. Hum. Genet. 24, S3–S9 (2016).
Seeber, A. et al. Molecular characteristics of BRCA1/2 and PALB2 mutations in pancreatic ductal adenocarcinoma. ESMO Open 5, e000942 (2020).
Skoulidis, F. et al. Germline Brca2 heterozygosity promotes KrasG12D-driven carcinogenesis in a murine model of familial pancreatic cancer. Cancer Cell 18, 499–509 (2010).
Farmer, H. et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434, 917–921 (2005).
Patel, A. G., Sarkaria, J. N. & Kaufmann, S. H. Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP) inhibitor lethality in homologous recombination-deficient cells. Proc. Natl Acad. Sci. USA 108, 3406–3411 (2011).
Golan, T. et al. Maintenance olaparib for germline BRCA-mutated metastatic pancreatic cancer. N. Engl. J. Med. 381, 317–327 (2019).
Golan, T. et al. Overall survival from the phase 3 POLO trial: maintenance olaparib for germline BRCA-mutated metastatic pancreatic cancer. J. Clin. Oncol. 39, 378 (2021).
Reiss, K. A. et al. Phase II study of maintenance rucaparib in patients with platinum-sensitive advanced pancreatic cancer and a pathogenic germline or somatic variant in BRCA1, BRCA2, or PALB2. J. Clin. Oncol. 39, 2497–2505 (2021).
Alexandrov, L. B. et al. Signatures of mutational processes in human cancer. Nature 500, 415–421 (2013).
O’Reilly, E. M. et al. Randomized, multicenter, phase II trial of gemcitabine and cisplatin with or without veliparib in patients with pancreas adenocarcinoma and a germline BRCA/PALB2 mutation. J. Clin. Oncol. 38, 1378–1388 (2020).
Tempero, M. A. et al. Pancreatic adenocarcinoma, version 1.2019. J. Natl Compr. Canc. Netw. 17, 202–210 (2019).
NCT04858334: A randomized study of olaparib or placebo in patients with surgically removed pancreatic cancer who have a BRCA1, BRCA2 or PALB2 mutation, the APOLLO trial. https://clinicaltrials.gov/ct2/show/NCT04858334 (2021).
Samstein, R. M. et al. Mutations in BRCA1 and BRCA2 differentially affect the tumor microenvironment and response to checkpoint blockade immunotherapy. Nat. Cancer 1, 1188–1203 (2021).
NCT04548752: Testing the addition of pembrolizumab, an immunotherapy cancer drug to olaparib alone as therapy for patients with pancreatic cancer that has spread with inherited BRCA mutations. https://clinicaltrials.gov/ct2/show/NCT04548752 (2020).
NCT04493060: Niraparib and dostarlimab for the treatment of germline or somatic BRCA1/2 and PALB2 mutated metastatic pancreatic cancer. https://clinicaltrials.gov/ct2/show/NCT04493060 (2020).
Blackford, A. N. & Jackson, S. P. ATM, ATR, and DNA-PK: the trinity at the heart of the DNA damage response. Mol. Cell 66, 801–817 (2017).
Gout, J. et al. Synergistic targeting and resistance to PARP inhibition in DNA damage repair-deficient pancreatic cancer. Gut 70, 743–760 (2021).
Dreyer, S. B. et al. Targeting DNA damage response and replication stress in pancreatic cancer. Gastroenterology 160, 362–377 (2021).
NCT04514497: Testing the addition of an anti-cancer drug, BAY 1895344, to usual chemotherapy for advanced stage solid tumors, with a specific focus on patients with small cell lung cancer, poorly differentiated neuroendocrine cancer, and pancreatic cancer. https://clinicaltrials.gov/ct2/show/NCT04514497 (2020).
NCT03682289: Phase II trial of AZD6738 alone and in combination with olaparib. https://clinicaltrials.gov/ct2/show/NCT03682289 (2018).
The Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 487, 330–337 (2012).
The Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 511, 543–550 (2014).
The Cancer Genome Atlas Research Network. The molecular taxonomy of primary prostate cancer. Cell 163, 1011–1025 (2015).
The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70 (2012).
Collisson, E. A. et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 17, 500–503 (2011).
Moffitt, R. A. et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 47, 1168–1178 (2015).
Chan-Seng-Yue, M. et al. Transcription phenotypes of pancreatic cancer are driven by genomic events during tumor evolution. Nat. Genet. 52, 231–240 (2020).
Lee, J. J. et al. Elucidation of tumor-stromal heterogeneity and the ligand-receptor interactome by single cell transcriptomics in real-world pancreatic cancer biopsies. Clin. Cancer Res. 27, 5912–5921 (2021).
Raghavan, S. et al. Microenvironment drives cell state, plasticity, and drug response in pancreatic cancer. Cell 184, 6119–6137 (2021).
Perri, G. et al. Response and survival associated with first-line FOLFIRINOX vs gemcitabine and nab-paclitaxel chemotherapy for localized pancreatic ductal adenocarcinoma. JAMA Surg. 155, 832–839 (2020).
Lee, J. C. et al. Comparison of FOLFIRINOX and gemcitabine plus nab-paclitaxel for treatment of metastatic pancreatic cancer: using Korean Pancreatic Cancer (K-PaC) Registry. Am. J. Clin. Oncol. 43, 654–659 (2020).
Chan, K. K. W. et al. Real-world outcomes of FOLFIRINOX vs gemcitabine and nab-paclitaxel in advanced pancreatic cancer: a population-based propensity score-weighted analysis. Cancer Med. 9, 160–169 (2020).
Sohal, D. et al. SWOG S1505: results of perioperative chemotherapy (peri-op CTx) with mfolfirinox versus gemcitabine/nab-paclitaxel (Gem/nabP) for resectable pancreatic ductal adenocarcinoma (PDA). J. Clin. Oncol. 38, 4504 (2020).
O’Kane, G. M. et al. GATA6 expression distinguishes classical and basal-like subtypes in advanced pancreatic cancer. Clin. Cancer Res. 26, 4901–4910 (2020).
NCT04469556: Pancreatic adenocarcinoma signature stratification for treatment (PASS-01). https://clinicaltrials.gov/ct2/show/NCT04469556 (2020).
Dougan, M., Dranoff, G. & Dougan, S. K. Cancer immunotherapy: beyond checkpoint blockade. Annu. Rev. Cancer Biol. 3, 55–75 (2019).
Rini, B. I. et al. Pembrolizumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N. Engl. J. Med. 380, 1116–1127 (2019).
Motzer, R. J. et al. Nivolumab plus ipilimumab versus sunitinib in advanced renal-cell carcinoma. N. Engl. J. Med. 378, 1277–1290 (2018).
Schmid, P. et al. Pembrolizumab for early triple-negative breast cancer. N. Engl. J. Med. 382, 810–821 (2020).
Gandhi, L. et al. Pembrolizumab plus chemotherapy in metastatic non-small-cell lung cancer. N. Engl. J. Med. 378, 2078–2092 (2018).
Reck, M. et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N. Engl. J. Med. 375, 1823–1833 (2016).
Larkin, J. et al. Five-year survival with combined nivolumab and ipilimumab in advanced melanoma. N. Engl. J. Med. 381, 1535–1546 (2019).
Marcus, L. et al. FDA approval summary: pembrolizumab for the treatment of microsatellite instability-high solid tumors. Clin. Cancer Res. 25, 3753–3758 (2019).
Luchini, C. et al. Comprehensive characterisation of pancreatic ductal adenocarcinoma with microsatellite instability: histology, molecular pathology and clinical implications. Gut 70, 148–156 (2021).
Royal, R. E. et al. Phase 2 trial of single agent ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J. Immunother. 33, 828–833 (2010).
Wainberg, Z. A. et al. Open-label, phase I study of nivolumab combined with nab-paclitaxel plus gemcitabine in advanced pancreatic cancer. Clin. Cancer Res. 26, 4814–4822 (2020).
O’Reilly, E. M. et al. Durvalumab with or without tremelimumab for patients with metastatic pancreatic ductal adenocarcinoma: a phase 2 randomized clinical trial. JAMA Oncol. 5, 1431–1438 (2019).
Le, D. T. et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 357, 409–413 (2017).
Balachandran, V. P., Beatty, G. L. & Dougan, S. K. Broadening the impact of immunotherapy to pancreatic cancer: challenges and opportunities. Gastroenterology 156, 2056–2072 (2019).
Ho, W. J., Jaffee, E. M. & Zheng, L. The tumour microenvironment in pancreatic cancer—clinical challenges and opportunities. Nat. Rev. Clin. Oncol. 17, 527–540 (2020).
Bear, A. S., Vonderheide, R. H. & O’Hara, M. H. Challenges and opportunities for pancreatic cancer immunotherapy. Cancer Cell 38, 788–802 (2020).
Schmiechen, Z. C. & Stromnes, I. M. Mechanisms governing immunotherapy resistance in pancreatic ductal adenocarcinoma. Front. Immunol. 11, 613815 (2020).
Liudahl, S. M. et al. Leukocyte heterogeneity in pancreatic ductal adenocarcinoma: phenotypic and spatial features associated with clinical outcome. Cancer Discov. 11, 2014–2031 (2021).
Vayrynen, S. A. et al. Composition, spatial characteristics, and prognostic significance of myeloid cell infiltration in pancreatic cancer. Clin. Cancer Res. 27, 1069–1081 (2021).
Vonderheide, R. H. & Bear, A. S. Tumor-derived myeloid cell chemoattractants and T cell exclusion in pancreatic cancer. Front. Immunol. 11, 605619 (2020).
Hosein, A. N., Brekken, R. A. & Maitra A. Pancreatic cancer stroma: an update on therapeutic targeting strategies. Nat. Rev. Gastroenterol. Hepatol. 17, 487–505 (2020).
Hou, P. et al. Tumor microenvironment remodeling enables bypass of oncogenic KRAS dependency in pancreatic cancer. Cancer Discov. 10, 1058–1077 (2020).
Zhu, Y. et al. Tissue-resident macrophages in pancreatic ductal adenocarcinoma originate from embryonic hematopoiesis and promote tumor progression. Immunity 47, 323–338 (2017).
Kraut, H., Lipps, H. J. & Prescott, D. M. The genome of hypotrichous ciliates. Int. Rev. Cytol. 99, 1–28 (1986).
Nywening, T. M. et al. Targeting both tumour-associated CXCR2+ neutrophils and CCR2+ macrophages disrupts myeloid recruitment and improves chemotherapeutic responses in pancreatic ductal adenocarcinoma. Gut 67, 1112–1123 (2018).
Stromnes, I. M. et al. Targeted depletion of an MDSC subset unmasks pancreatic ductal adenocarcinoma to adaptive immunity. Gut 63, 1769–1781 (2014).
Chao, T., Furth, E. E. & Vonderheide, R. H. CXCR2-dependent accumulation of tumor-associated neutrophils regulates T-cell immunity in pancreatic ductal adenocarcinoma. Cancer Immunol. Res. 4, 968–982 (2016).
Steele, C. W. et al. CXCR2 inhibition profoundly suppresses metastases and augments immunotherapy in pancreatic ductal adenocarcinoma. Cancer Cell 29, 832–845 (2016).
Twyman-Saint Victor, C. et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 520, 373–377 (2015).
Mitchem, J. B. et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 73, 1128–1141 (2013).
Nywening, T. M. et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: a single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. 17, 651–662 (2016).
Noel, M. et al. Phase 1b study of a small molecule antagonist of human chemokine (C–C motif) receptor 2 (PF-04136309) in combination with nab-paclitaxel/gemcitabine in first-line treatment of metastatic pancreatic ductal adenocarcinoma. Invest. New Drugs 38, 800–811 (2020).
NCT03496662: BMS-813160 with nivolumab and gemcitabine and nab-paclitaxel in borderline resectable and locally advanced pancreatic ductal adenocarcinoma (PDAC). https://clinicaltrials.gov/ct2/show/NCT03496662 (2018).
Vonderheide, R. H. The immune revolution: a case for priming, not checkpoint. Cancer Cell 33, 563–569 (2018).
Lin, J. H. et al. Type 1 conventional dendritic cells are systemically dysregulated early in pancreatic carcinogenesis. J. Exp. Med. 217, e20190673 (2020).
Hegde, S. et al. Dendritic cell paucity leads to dysfunctional immune surveillance in pancreatic cancer. Cancer Cell 37, 289–307 (2020).
NCT04536077: Immunologic effects of CDX-301 and CDX-1140 in resectable pancreatic cancer patients. https://clinicaltrials.gov/ct2/show/NCT04536077 (2020).
NCT03329950: A study of CDX-1140 (CD40) as monotherapy or in combination in patients with advanced malignancies. https://clinicaltrials.gov/ct2/show/NCT03329950 (2017).
Maier, B. et al. A conserved dendritic-cell regulatory program limits antitumour immunity. Nature 580, 257–262 (2020).
Salmon, H. et al. Expansion and activation of CD103+ dendritic cell progenitors at the tumor site enhances tumor responses to therapeutic PD-L1 and BRAF inhibition. Immunity 44, 924–938 (2016).
Zhang, Q. et al. Inhibition of ATM increases interferon signaling and sensitizes pancreatic cancer to immune checkpoint blockade therapy. Cancer Res. 79, 3940–3951 (2019).
Bear, A. S. et al. Biochemical and functional characterization of mutant KRAS epitopes validates this oncoprotein for immunological targeting. Nat. Commun. 12, 4365 (2021).
Balachandran, V. P. et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 551, 512–516 (2017).
Liu, H. et al. Structure-based programming of lymph-node targeting in molecular vaccines. Nature 507, 519–522 (2014).
Beatty, G. L. et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science 331, 1612–1616 (2011).
Morrison, A. H. et al. Sufficiency of CD40 activation and immune checkpoint blockade for T cell priming and tumor immunity. Proc. Natl Acad. Sci. USA 117, 8022–8031 (2020).
Byrne, K. T. & Vonderheide, R. H. CD40 stimulation obviates innate sensors and drives T cell immunity in cancer. Cell Rep. 15, 2719–2732 (2016).
Long, K. B. et al. IFNγ and CCL2 cooperate to redirect tumor-infiltrating monocytes to degrade fibrosis and enhance chemotherapy efficacy in pancreatic carcinoma. Cancer Discov. 6, 400–413 (2016).
O’Hara, M. H. et al. Gemcitabine (Gem) and nab-paclitaxel (NP) ± nivolumab (nivo) ± CD40 agonistic monoclonal antibody APX005M (sotigalimab), in patients (Pts) with untreated metastatic pancreatic adenocarcinoma (mPDAC): phase (Ph) 2 final results. J. Clin. Oncol. 39, 2021 (2021).
Dougan, S. K. & Dougan, M. Regulation of innate and adaptive antitumor immunity by IAP antagonists. Immunotherapy 10, 787–796 (2018).
& Roehle, K. et al. cIAP1/2 antagonism eliminates MHC class I-negative tumors through T cell-dependent reprogramming of mononuclear phagocytes. Sci. Transl. Med. 13, eabf5058 (2021).
Weiskopf, K. et al. Engineered SIRPα variants as immunotherapeutic adjuvants to anticancer antibodies. Science 341, 88–91 (2013).
Feng, M. et al. Phagocytosis checkpoints as new targets for cancer immunotherapy. Nat. Rev. Cancer. 19, 568–586 (2019).
Chao, M. P. et al. Anti-CD47 antibody synergizes with rituximab to promote phagocytosis and eradicate non-Hodgkin lymphoma. Cell 142, 699–713 (2010).
Sockolosky, J. T. et al. Durable antitumor responses to CD47 blockade require adaptive immune stimulation. Proc. Natl Acad. Sci. USA 113, E2646–E2654 (2016).
Liu, M. et al. Metabolic rewiring of macrophages by CpG potentiates clearance of cancer cells and overcomes tumor-expressed CD47-mediated ‘don’t-eat-me’ signal. Nat. Immunol. 20, 265–275 (2019).
Wang, T. T. & Ravetch, J. V. Functional diversification of IgGs through Fc glycosylation. J. Clin. Invest. 129, 3492–3498 (2019).
Ozdemir, B. C. et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 25, 719–734 (2014).
Rhim, A. D. et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 25, 735–747 (2014).
Elyada, E. et al. Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Discov. 9, 1102–1123 (2019).
Biffi, G. et al. IL1-induced JAK/STAT signaling is antagonized by TGFβ to shape CAF heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discov. 9, 282–301 (2019).
Buechler, M. B. et al. Cross-tissue organization of the fibroblast lineage. Nature 593, 575–579 (2021).
Chen, Y. et al. Type I collagen deletion in αSMA+ myofibroblasts augments immune suppression and accelerates progression of pancreatic cancer. Cancer Cell 39, 548–565 (2021).
Grauel, A. L. et al. TGFβ-blockade uncovers stromal plasticity in tumors by revealing the existence of a subset of interferon-licensed fibroblasts. Nat. Commun. 11, 6315 (2020).
Mariathasan, S. et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 554, 544–548 (2018).
Huang, H. et al. Targeting TGFβR2-mutant tumors exposes vulnerabilities to stromal TGFβ blockade in pancreatic cancer. EMBO Mol. Med. 11, e10515 (2019).
NCT04390763: Study of efficacy and safety of NIS793 (with and without spartalizumab) in combination with SOC chemotherapy in first-line metastatic pancreatic ductal adenocarcinoma (mPDAC). https://clinicaltrials.gov/ct2/show/NCT04390763 (2020).
NCT03563248: Losartan and nivolumab in combination with FOLFIRINOX and SBRT in localized pancreatic cancer. https://clinicaltrials.gov/ct2/show/NCT03563248 (2018).
Murphy, J. E. et al. Total neoadjuvant therapy with FOLFIRINOX in combination with losartan followed by chemoradiotherapy for locally advanced pancreatic cancer: a phase 2 clinical trial. JAMA Oncol. 5, 1020–1027 (2019).
NCT03520790: Paricalcitol plus gemcitabine and nab-paclitaxel in metastatic pancreatic cancer. https://clinicaltrials.gov/ct2/show/NCT03520790 (2018).
NCT02754726: Combination therapy for patients with untreated metastatic pancreatic ductal adenocarcinoma. https://clinicaltrials.gov/ct2/show/NCT02754726 (2016).
Zhou, X. Z. & Lu, K. P. The isomerase PIN1 controls numerous cancer-driving pathways and is a unique drug target. Nat. Rev. Cancer 16, 463–478. (2016).
Wei, S. et al. Active Pin1 is a key target of all-trans retinoic acid in acute promyelocytic leukemia and breast cancer. Nat. Med. 21, 457–466 (2015).
Dubiella, C. et al. Sulfopin is a covalent inhibitor of Pin1 that blocks Myc-driven tumors in vivo. Nat. Chem. Biol. 17, 954–963 (2021).
Koikawa, K. et al. Targeting Pin1 renders pancreatic cancer eradicable by synergizing with immunochemotherapy. Cell 184, 4753–4771 (2021).
Kocher, H. M. et al. Phase I clinical trial repurposing all-trans retinoic acid as a stromal targeting agent for pancreatic cancer. Nat. Commun. 11, 4841 (2020).
Steele, N. G. et al. Inhibition of hedgehog signaling alters fibroblast composition in pancreatic cancer. Clin. Cancer Res. 27, 2023–2037. (2021).
Das, S. et al. Tumor cell-derived IL1β promotes desmoplasia and immune suppression in pancreatic cancer. Cancer Res. 80, 1088–1101 (2020).
NCT04581343: A phase 1B study of canakinumab, spartalizumab, nab-paclitaxel, and gemcitabine in metastatic PC patients (PanCAN-SR1). https://clinicaltrials.gov/ct2/show/NCT04581343 (2020).
Ridker, P. M. et al. Effect of interleukin-1β inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet 390, 1833–1842 (2017).
Dougan, M. et al. A dual role for the immune response in a mouse model of inflammation-associated lung cancer. J. Clin. Invest. 121, 2436–2446. (2011).
Le, D. T. et al. PD-1 blockade in tumors with mismatch-repair deficiency. N. Engl. J. Med. 372, 2509–2520 (2015).
Marabelle, A. et al. Efficacy of pembrolizumab in patients with noncolorectal high microsatellite instability/mismatch repair-deficient cancer: results from the phase II KEYNOTE-158 study. J. Clin. Oncol. 38, 1–10 (2020).
O’Reilly, E. M. & Hechtman, J. F. Tumour response to TRK inhibition in a patient with pancreatic adenocarcinoma harbouring an NTRK gene fusion. Ann. Oncol. 30, viii36–viii40 (2019).
Jones, M. R. et al. NRG1 gene fusions are recurrent, clinically actionable gene rearrangements in KRAS wild-type pancreatic ductal adenocarcinoma. Clin. Cancer Res. 25, 4674–4681 (2019).
Geuijen, C. A. W. et al. Unbiased combinatorial screening identifies a bispecific IgG1 that potently inhibits HER3 signaling via HER2-guided ligand blockade. Cancer Cell 33, 922–936 (2018).
NCT02912949: A study of zenocutuzumab (MCLA-128) in patients with solid tumors harboring an NRG1 fusion. https://clinicaltrials.gov/ct2/show/NCT02912949 (2016).
Chou, A. et al. Clinical and molecular characterization of HER2 amplified-pancreatic cancer. Genome Med. 5, 78 (2013).
Tsurutani, J. et al. Targeting HER2 with trastuzumab deruxtecan: a dose-expansion, phase I study in multiple advanced solid tumors. Cancer Discov. 10, 688–701 (2020).
NCT04853017: A study of ELI-002 in subjects with KRAS mutated pancreatic ductal adenocarcinoma (PDAC) and other solid tumors (AMPLIFY-201). https://clinicaltrials.gov/ct2/show/NCT04853017 (2021).
NCT05013216: Mutant Kirsten Rat Sarcoma (KRAS)—targeted long peptide vaccine for patients at high risk of developing pancreatic cancer. https://clinicaltrials.gov/ct2/show/NCT05013216 (2021).
NCT04117087: Pooled mutant KRAS-targeted long peptide vaccine combined with nivolumab and ipilimumab for patients with resected MMR-p colorectal and pancreatic cancer. https://clinicaltrials.gov/ct2/show/NCT04117087 (2019).
NCT03956056: Neoantigen peptide vaccine strategy in pancreatic cancer patients following surgical resection and adjuvant chemotherapy. https://clinicaltrials.gov/ct2/show/NCT03956056 (2019).
NCT04161755: Study of personalized tumor vaccines (PCVs) and a PD-L1 blocker in patients with pancreatic cancer that can be treated with surgery. https://clinicaltrials.gov/ct2/show/NCT04161755 (2019).
NCT02600949: Personalized peptide vaccine in treating patients with advanced pancreatic cancer or colorectal cancer. https://clinicaltrials.gov/ct2/show/NCT02600949 (2015).
Sanford, D. E. et al. Inflammatory monocyte mobilization decreases patient survival in pancreatic cancer: a role for targeting the CCL2/CCR2 axis. Clin. Cancer Res. 19, 3404–3415 (2013).
NCT04477343: A study to evaluate the safety and tolerability of SX-682 in combination with nivolumab as a maintenance therapy in patients with metastatic pancreatic ductal adenocarcinoma. https://clinicaltrials.gov/ct2/show/NCT04477343 (2020).
Sherman, M. H. et al. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell 159, 80–93 (2014).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
A.N.H. has no disclosures. S.K.D. receives grant funding from Novartis, BMS, Genocea and Eli Lilly and is a cofounder and SAB member of Kojin Therapeutics. A.J.A. has consulted for Oncorus, Inc., Arrakis Therapeutics, Syros Pharmaceuticals, Boehringer Ingelheim, T-knife Therapeutics, AstraZeneca, Mirati Therapeutics and Merck & Co., Inc., and has research funding from Mirati Therapeutics, Syros Pharmaceuticals, Revolution Medicines, Novartis, Bristol Myers Squibb, Deerfield, Inc., and Novo Ventures that is unrelated to this work. A.M. receives royalties for a pancreatic cancer biomarker test from Cosmos Wisdom Biotechnology, is listed as an inventor on a patent that has been licensed by Johns Hopkins University to ThriveEarlier Detection and serves as a consultant for Freenome and Tezcat Biotechnology.
Peer review
Peer review information
Nature Cancer thanks Vinod Balachandran, and the other, anonymous reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Hosein, A.N., Dougan, S.K., Aguirre, A.J. et al. Translational advances in pancreatic ductal adenocarcinoma therapy. Nat Cancer 3, 272–286 (2022). https://doi.org/10.1038/s43018-022-00349-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s43018-022-00349-2
This article is cited by
-
Preoperative chemotherapy, radiotherapy and surgical decision-making in patients with borderline resectable and locally advanced pancreatic cancer
Nature Reviews Gastroenterology & Hepatology (2024)
-
Clearance of dead cells by efferocytosis promotes liver metastasis in pancreatic cancer
Nature Cancer (2024)
-
Disruption of the pro-oncogenic c-RAF–PDE8A complex represents a differentiated approach to treating KRAS–c-RAF dependent PDAC
Scientific Reports (2024)
-
KRAS degradation averts PDAC chemoresistance
Nature Cancer (2024)
-
Establishment, characterization, and biobanking of 36 pancreatic cancer organoids: prediction of metastasis in resectable pancreatic cancer
Cellular Oncology (2024)
