
Abstract
Anti-PD-1 treatment has shown unprecedented clinical success in the treatment of non-small-cell lung cancer (NSCLC), but the underlying mechanisms remain incompletely understood. Here, we performed temporal single-cell RNA and paired T-cell receptor sequencing on 47 tumor biopsies from 36 patients with NSCLC following PD-1-based therapies. We observed increased levels of precursor exhausted T (Texp) cells in responsive tumors after treatment, characterized by low expression of coinhibitory molecules and high expression of GZMK. By contrast, nonresponsive tumors failed to accumulate Texp cells. Our data suggested that Texp cells were unlikely to be derived from the reinvigoration of terminally exhausted cells; instead, they were accumulated by (1) local expansion and (2) replenishment by peripheral T cells with both new and pre-existing clonotypes, a phenomenon we named clonal revival. Our study provides insights into mechanisms underlying PD-1-based therapies, implicating clonal revival and expansion of Texp cells as steps to improve NSCLC treatment.
Similar content being viewed by others


Main
Immune-checkpoint blockade (ICB) is able to potentiate antitumor immunity by disrupting coinhibitory T-cell signaling and represents a dramatic paradigm shift in cancer treatment1,2,3. However, the remarkable responses to such therapies are limited to a minority of patients with NSCLC4, highlighting the need for additional therapeutic strategies. To facilitate the rational design of next-generation immunotherapies, it is of critical importance to first understand the molecular and immunologic mechanisms underlying ICB-induced tumor rejection. Previous studies reported that PD-1 pathway blockade could reverse the dysfunctional state of terminally exhausted Texp cells during chronic viral infection5,6, presenting a widely presumed mechanism of ICB treatment. However, with concentrated efforts in addressing this question, emerging evidence showed that the epigenetic stability of terminal Tex cells is difficult to alter and could limit this reinvigoration7,8,9. Recent observations have also supported such a notion, showing that newly infiltrated novel clones rather than pre-existing ones play a predominant role in cancer treatment10. Furthermore, multiple studies have linked clinical outcomes to Texp cells, including stem-like cells11,12,13 and transitory cells that subsequently convert to a terminal Tex subset14,15,16,17, but how terminal Tex and Texp cells respond to anti-PD-1 during the treatment of human cancer remains elusive.
Previous lung cancer studies aimed at illuminating the mechanisms of ICB treatment have focused on whole-exome sequencing of bulk tumors18, but such strategies are limited in regard to understanding the cellular basis for immune responses to checkpoint blockade. Single-cell RNA sequencing (scRNA-seq) is a powerful approach to dissect the cellular diversity of complex biological systems, but current scRNA-seq analyses of lung cancer largely focus on-treatment-naïve tumors19, primarily due to the challenge of obtaining high-quality human tumor samples at multiple treatment time points. One recent lung cancer study analyzed neoantigen-specific T cells in post-treatment tumors20, while the temporal changes of such cells following treatment remain elusive. To address these limitations, we utilized scRNA-seq analyses to comprehensively characterize the temporal dynamics of T cells from dissociated tumor biopsies following combination therapies of anti-PD-1 with chemotherapy, allowing us to better explain the fundamental mechanisms of PD-1-based treatment.
Characterization of T cells pre- and post-PD-1 blockade by scRNA-seq
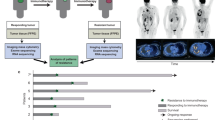
We employed droplet-based 5′ scRNA-seq and paired T-cell receptor (scTCR-seq) sequencing to profile a set of 47 tumor biopsies from 36 patients with NSCLC, spanning 33 treatment-naïve, nine site-matched post-treatment responsive (from eight tumors) and five post-treatment nonresponsive tumor specimens following combination therapies of anti-PD-1 with chemotherapy (Fig. 1a,b, Extended Data Fig. 1 and Supplementary Table 1). After stringent quality-control filtering (Methods) a total of 135,391 T cells remained (Extended Data Fig. 2a,b) of which 56.6% had productive TCR α- and β-chain pairs (Supplementary Table 2). These T cells were further grouped into 43,713 distinct clonotypes by matching both α- and β-chain pairs (Extended Data Fig. 2c), allowing us to track clonal lineages and measure clonal expansion following treatment.

a, Scheme of the overall study design. scRNA-seq and scTCR-seq were applied to cells derived from treatment-naïve and on-treatment tumor biopsies (Supplementary Table 1). b, Representative computed tomography images of a responsive NSCLC tumor (P019) before and after initiation of treatment. c, UMAP plot of 75,735 CD4+ cells from all 47 tumor samples, showing the formation of nine main clusters. Each dot corresponds to a single cell, color coded by cell cluster. d, Proportion of each CD4+ subtype in all CD4 T cells from pretreatment (pre-rx) and post-treatment (post-rx) tumors. Data presented as mean ± s.e.m. P values were determined by Fisher’s exact test, t-test and Dirichlet-multinomial regression model, two-sided (Methods). *P < 0.05, **P < 0.01, ***P < 0.001. e, Volcano plot showing differentially expressed genes for all Tregs between post-treatment responsive and nonresponsive tumors. Each red dot denotes an individual gene with adjusted P < 0.01, two-sided t-test. f, Average gene expression in all Tregs from post-treatment responsive and nonresponsive tumors. Center line indicates the median value, lower and upper hinges represent the 25th and 75th percentiles, respectively, and whiskers denote 1.5× interquartile range. Each dot corresponds to one sample. *P < 0.05, **P < 0.01, ***P < 0.001, one-sided t-test. g, Number of expanded clonotypes (clone size >2) in the Th1-like cell subset between post-treatment responsive and nonresponsive tumors. Data presented as mean ± s.e.m. *P < 0.05, **P < 0.01, ***P < 0.001, one-sided t-test. h, Number of expanded clonotypes (clone size >2) in the Th1-like cell subset. Data presented as mean ± s.e.m. *P < 0.05, **P < 0.01, ***P < 0.001, one-sided Wilcoxon test. Data were summarized from n = 6 pre-rx responsive, n = 6 post-rx responsive, n = 5 pre-rx nonresponsive and n = 5 post-rx nonresponsive tumor samples.
Among CD4+ T cells (Fig. 1c and Extended Data Fig. 2d–g) we found that responsive tumors showed a slight decrease in FOXP3+ regulatory T cells (Tregs) after treatment, whereas post-treatment nonresponsive tumors exhibited a notable increase of such cells compared to responsive tumors (Fig. 1d). We previously observed heterogeneity within Treg cells21, characterized by the bimodal distribution of 4-1BB (TNFRSF9), a known Treg activation marker22. To investigate whether this heterogeneity is associated with response to treatment, we further performed differential expression analysis for Tregs. Compared to post-treatment responsive tumors, Tregs in both pretreatment and nonresponsive tumors consistently upregulated signatures of 4-1BB+ Tregs (Fig. 1e and Extended Data Fig. 2h), including genes associated with immunosuppressive functions (IL1R2, REL and LAYN) (Fig. 1f and Extended Data Fig. 2i). Using gene set enrichment analysis (GSEA), we further demonstrated the enrichment of 4-1BB+ Tregs in nonresponsive and pretreatment tumors (Extended Data Fig. 2j), consistent with a previous study which reported that 4-1BB+ Tregs were correlated with poor prognosis in patients with Cancer Genome Atlas lung adenocarcinoma (LUAD)21. In contrast to Tregs, CXCL13-expressing Th1-like cells exhibited an increase in responsive tumors following treatment (Fig. 1d). Further examination of T-cell clonality revealed a sizable increase in expanded Th1-like cells in responsive tumors compared to nonresponsive (Fig. 1g), which supports the critical role of such cells in response to ICB therapy23. Using previously published basal cell carcinoma (BCC) datasets10, we also observed enrichment of Th1-like cells in post-treatment responsive tumors (Fig. 1h), indicative of improved antitumor immunity accompanying tumor rejection.
To assess infiltrating CD8+ T cells, we first performed immunohistochemistry (IHC) staining for remaining tumor biopsies and observed a significant correlation in the proportion of CD8+ T cells between scRNA-seq and staining data, which indicates that scRNA-seq might reflect, at least in part, the real infiltration level of these cell populations (Fig. 2a and Extended Data Fig. 3a). When comparing CD8+ T-cell populations between treatment responses, we found that responsive tumors exhibited a higher abundance of CD8+ T cells in comparison with nonresponsive tumors (Fig. 2b), consistent with a previous observation that CD8+ T-cell infiltration into tumors could predict survival24,25 and response to immunotherapy26. To gain further insight into the response of CD8+ T cells to PD-1-based therapy, we clustered those 59,656 CD8+ T cells and identified three broad cell groups—terminal Tex, non-exhausted and proliferative CD8+ T cells (Fig. 2c and Extended Data Fig. 3b–e). Terminal Tex cells were characterized by high expression of exhaustion signatures, including PDCD1, CTLA4 and HAVCR2 (Fig. 2d). Emerging evidence demonstrates that terminal Tex cells in tumors are specifically derived from tumor-specific T cells27,28,29,30,31, whereas T cells responsible for acute infections do not give rise to Tex cells32. Thus, a terminal Tex subset could be used as a proxy for a tumor-reactive T-cell compartment31,33. To further support this notion, we analyzed previously reported signature genes of tumor antigen-specific T cells30,31—ENTPD1 and ITGAE—and found that they were specifically expressed in the terminal Tex subset (Extended Data Fig. 3e). In addition, we further examined three previously published tumor-specific CD8+ T-cell datasets31,34,35 and found that these cells consistently highly expressed a set of exhaustion signature genes (Fig. 2e), showing profound similarities with the terminal Tex subset in our data (Fig. 2f). Previous studies have shown that PD-1 pathway blockade could improve the effector function of the terminal Tex subset to a certain extent during chronic viral infection9. To determine whether such a phenomenon could be observed in human lung cancer, we performed GSEA analysis with those reported effector genes following a similar procedure by Pauken et al.9 and, indeed, the terminal Tex subset was significantly enriched for such genes after treatment in responsive tumors (Fig. 2g, Extended Data Fig. 3f and Supplementary Table 3), indicative of their improved effector function following therapy.

a, Correlation of the proportion of CD8+ T cells between scRNA-seq and staining data. P values were determined by two-tailed Spearman’s rank-order correlation test on log-transformed values to linearize the data; n = 7 tumors. b, Proportion of CD8 T cells in all CD45+ cells. Data presented as mean ± s.e.m. *P < 0.05, **P < 0.01, ***P < 0.001, two-sided Wilcoxon test. c, UMAP plot of 59,656 CD8+ T cells from all 47 tumor samples, color coded by the three broad cell groups. d, Violin plots showing the expression levels of selected marker genes in different CD8+ cell groups from c. e, Expression heatmap of terminal exhaustion signatures in tumor-reactive T cells from refs. 35,34,31 (top, bottom left, bottom right, respectively). f, GSEA enrichment of the terminal Tex subset in this study for signatures of previously reported tumor-reactive T cells. P values were determined by one-tailed permutation test by GSEA. NES, normalized enrichment score. Data were summarized from n = 47 tumor samples. g, Enrichment plot for the set of effector genes in the transcriptome of anti-PD-1-treated terminal Tex cells versus matched pretreatment terminal Tex cells by GSEA; n = 8 pre-rx and n = 9 post-rx samples from eight responsive tumors. P values were determined by a one-tailed permutation test by GSEA. h, Proportion of terminal Tex cells in eight responsive tumors following treatment, shown as percentage of total CD8+ cells. P values were determined by two-sided t-test, *P < 0.05, **P < 0.01, ***P < 0.001.
To address whether tumor regression was mainly driven by terminal Tex cells, as shown in other cancer types10,36, we further analyzed changes in terminal Tex cell frequencies and observed two distinct patterns (Fig. 2h). For responsive tumors with low terminal Tex infiltration before treatment, PD-1 blockade preferentially increased the proportions of such cells, suggestive of increased infiltration or expansion of tumor-reactive T cells following treatment, and this was consistent with previous clinical observations that an increase in exhausted-like T cells was associated with anti-PD-1 therapies in human melanoma25 and breast cancer37. By contrast, tumors with high baseline terminal Tex infiltration exhibited the opposite pattern with these cells dramatically decreasing in relative frequencies (Fig. 2h), indicating that, in addition to terminal Tex cells, other cell populations might also play important roles in tumor control. Since multiple mouse studies have demonstrated that anti-PD-1 could also target Texp cells to enhance antitumor immunity11,24,38,39, we next sought to identify these precursors to address fluctuation in terminal Tex cell proportions.
Tex precursors within the tumor
A typical procedure for analysis of Texp cells would be to first identify different cell groups by clustering, followed by examination of clusters with most shared TCRs with the terminal Tex subset. However, our previous analysis40 showed that such clusters may contain TCRs independently connected with terminal Tex and blood effector cells, implying that non-exhausted tumor-reactive T cells and potential bystander cells30,31 may fall into the same cluster due to similar transcriptional phenotypes. To better identify and characterize Texp cells, we applied a TCR-based selection strategy with two primary considerations: (1) the terminal Tex subset could be easily distinguished from the other cells by unsupervised clustering based on the high expression of exhaustion markers (Fig. 2c), and (2) TCR provides a reliable molecular tag to track the lineage of Tex cells. Thus, here we selected only terminal Tex and non-exhausted CD8+ T cells sharing TCRs with terminal Tex cells and applied unsupervised clustering to these cells (Fig. 3a). In addition to terminal and proliferative Tex cells, we identified two non-exhausted cell states with TCR sharing with the terminal Tex subset—GZMK+NR4A2– and GZMK+NR4A2+ T cells, each with its distinct signature genes (Fig. 3b,c and Extended Data Fig. 3g–j). Both clusters showed reduced expression of coinhibitory molecules and increased expression of GZMK (Fig. 3c,d and Extended Data Fig. 4).

a, TCR-based selection and identification of Tex (terminal Tex and Texp) and Tex-irrelevant cells. Terminal Tex cells and those non-exhausted cells sharing the same TCRs with terminal Tex were selected and considered as Tex cells, followed by a second round of unsupervised clustering. b, UMAP plot of Tex cells from samples with Tex cell count >100, showing the four transcriptionally distinct cell states present in n = 20 NSCLC tumor samples. c, Heatmap showing mean relative expression of certain signature genes in the four identified cell populations in b. d, UMAP plots showing the expression levels of certain signature genes in the four cell clusters from b,c. e, GSEA enrichment of each Tex subset in this study for signatures of previously reported Tex cell states. P values were determined by one-tailed permutation test by GSEA. f, RNA velocities visualized on diffusion map of cells from the largest Tex clone from pre- and post-treatment P019 tumor biopsies, color coded by cluster. g, Comparison of average expression of each gene in Tex cells (y axis) versus Tex-irrelevant cells (x axis). Red points indicate significantly upregulated genes in Tex cells with adjusted P < 0.01, one-tailed t-test. Data were summarized from n = 20 samples from b. h, Violin plots showing the expression level of CXCL13 in Tex subsets from n = 33 pretreatment, n = 9 post-treatment responsive and n = 5 post-treatment nonresponsive tumors. i, Exhaustion score and expression of exhaustion signatures plotted along the expression of CXCL13. Data from n = 33 treatment-naïve/pretreatment NSCLC tumors were summarized. P values were determined from Pearson correlation coefficient r, two-tailed linear regression t-test. ***P < 2.2 × 10−16. j, Heatmap showing the proportion of terminal Tex and Texp metacells in all CXCL13+ metacells from the four external datasets. k, Density plot showing the expression of CXCL13 in terminal Tex- and Tex-TCR-sharing peripheral T cells using data from a liver cancer study43.
Previous mouse studies have defined detailed developmental frameworks for Tex cells15,16,38, and we therefore performed further GSEA analysis using reported signature genes of stem, transitory and terminal Tex cells to better characterize Tex subset-specific biology in human lung cancer (Fig. 3e). Assuredly, GZMK+NR4A2–/+ cells shared the highest similarities with intermediate Tex cells defined by Beltra et al.16 and transitory Tex cells by Hudson et al.15. GZMK+NR4A2–/+ cells also showed high similarity with Texprog2 cells defined by Beltra et al., although GZMK+NR4A2– cells had stronger enrichment in comparison with GZMK+NR4A2+ cells. Of note, terminal Tex cells in our study were selectively enriched for terminally differentiated signatures of mouse Tex cells, demonstrating the stability of this cluster. Furthermore, our analysis of resident memory and circulating T-cell signatures41 revealed increased tissue-residency phenotypes from GZMK+NR4A2– to GZMK+NR4A2+ cells, and further to terminal Tex cells, with circulating phenotypes showing the opposite pattern (Fig. 3e). Similar analysis using previously published lung cancer Smart-seq2 data21 further demonstrated precursor phenotypes of GZMK+NR4A2–/+ cells and the stability of the terminal Tex subset (Extended Data Fig. 5a). These analyses provided valuable insights into the stepwise developmental program from GZMK+NR4A2– to GZMK+NR4A2+ cells, and further to terminal Tex cells, and this potential differentiation trajectory was further supported by our RNA velocity42 analysis (Fig. 3f and Extended Data Fig. 5b), although additional functional validation is needed. Together, using a TCR-based selection approach, we showed that GZMK+NR4A2–, GZMK+NR4A2+, terminal and proliferative Tex cells are four transcriptionally distinct Tex populations present in human lung tumors. For clarity, GZMK+NR4A2–/+ subsets are referred to here as Texp1 (GZMK+NR4A2–) and Texp2 (GZMK+NR4A2+) cells.
CXCL13 expression in Tex cells
When comparing Tex (terminal Tex and Texp) cells with those CD8+ T cells sharing no TCR with the terminal Tex subset (Tex-irrelevant cells), we found that CXCL13 was highly and exclusively expressed in terminal Tex and Texp cells (Fig. 3g and Extended Data Fig. 4). CXCL13 produced by Tex cells could mediate immune cell recruitment to the tumor29 and may be one of the drivers of the formation of tertiary lymphoid structures33. However, previous studies of treatment-naïve tumors have reported that CXCL13 was expressed only by the terminal Tex subset21,29,33. To address this, we analyzed the expression of CXCL13 in Texp cells before and after treatment and found that CXCL13 was highly expressed only in Texp cells post- as opposed to pretreatment (Fig. 3h). To further validate this, we collected four published datasets of treatment-naïve T cells from both NSCLC19,21 and other cancer types40,43, in an attempt to survey the expression of CXCL13 in tumor-infiltrating CD8+ T cells. To address the potential bias introduced by dropout events, we applied the MetaCell algorithm44 to identify homogeneous groups of cells—metacells (Methods), analysis of which revealed a significant correlation between exhaustion score and the expression of CXCL13 (Fig. 3i and Extended Data Fig. 5c,d). Furthermore, we found that CXCL13 was highly and exclusively expressed in the terminal Tex subset across all analyzed cancer types, with barely detectable expression in Texp and other CD8+ T cells (Fig. 3j and Extended Data Fig. 5e,f). To further explore the expression of CXCL13, we focused on datasets with matched blood samples and analyzed expression in peripheral Tex-TCR-sharing CD8+ T cells, which were in a non-exhausted state and could readily be distinguished from the terminal Tex subset. Of note, CXCL13 showed only rare expression in peripheral Texp cells (Fig. 3k and Extended Data Fig. 5g), implying that CXCL13 is upregulated only when cells are in the tumor microenvironment. Recent studies have shown that CXCL13 is upregulated by CD8+ T cells in the presence of concurrent TCR stimulation and TGF-β receptor signaling45, further supporting the view that, in treatment-naïve tumors, upregulation of CXCL13 in tumor-reactive T cells is along the exhaustion axis due to chronic antigen stimulation33. Our observations therefore hinted that those post-treatment CXCL13-expressing Texp cells may have been formed by blockade of the transition to terminal Tex cells by PD-1 antibodies, with CXCL13 expression unaffected.
Accumulation of Texp cells in responsive tumors after treatment
To further test the above hypothesis and address the decrease in terminal Tex cells (Fig. 2h), we next investigated how the abundance of Texp cells changed during treatment. Out of 14 post-treatment tumor samples, 13 were collected from either primary lung tumors (n = 7) or lymph node metastases (n = 6). We therefore first evaluated whether Tex subsets were comparable between different types of biopsy site and found that these populations could be consistently captured by both unsupervised clustering and supervised cell-type classification (Extended Data Fig. 6), demonstrating their stability. To enable precise evaluation of Tex subset proportions, we next analyzed each dominant high-confidence Tex clone (Methods) in each tumor. Focusing on responsive tumors, we observed that most Tex cells from pretreatment tumors were in a terminally exhausted status whereas post-treatment Tex cells were enriched for Texp cells (Fig. 4a and Extended Data Fig. 7), and such patterns were consistently observed across six out of the eight responsive tumors (P010, P013, P019, P030, P033 and P035; Fig. 4b,c and Extended Data Fig. 7). Of note, one of the responsive tumors, P029, was not included in this analysis due to a low Tex cell count (<30). Further differential expression analysis for each dominant Tex clone in responsive tumors revealed that post-treatment Tex cells consistently expressed lower levels of exhaustion genes in comparison with pretreatment (Extended Data Fig. 8), supporting an increase in Texp cells in response to therapy. By performing multicolored IHC staining on remaining biopsy materials from four treatment-naïve and three on-treatment responsive tumors (Methods), we confirmed the existence of CXCL13-expressing Texp cells (CD8A+CXCL13+TIM3–) and terminal Tex cells (CD8A+CXCL13+TIM3+; Extended Data Fig. 9a and Supplementary Fig. 1). In addition, CXCL13-expressing Texp cells showed higher abundance after treatment (Extended Data Fig. 9a–d), consistent with our scRNA-seq data.

a, Gene expression heatmap for cells from the largest Tex clone from pre- and post-treatment P019 tumor biopsies. Rows represent selected differentially expressed genes and columns represent individual cells. b, Comparison of the frequency of Texp cells in all Tex cells (terminal Tex and Texp) from pretreatment (n = 19), post-treatment nonresponsive (n = 4) and post-treatment responsive (n = 8) tumors. Samples from pretreatment P001, P013 and P033 were removed due to low cell counts. Each dot represents one tumor biopsy sample while the center line indicates the median value. The lower and upper hinges represent the 25th and 75th percentiles, respectively, and whiskers denote 1.5× interquartile range. *P < 0.05, **P < 0.01, ***P < 0.001, two-sided t-test. c, Proportion of Texp cells in the top three qualified dominant Tex clones from pretreatment (n = 29 clones), post-treatment responsive (n = 14 clones) and post-treatment nonresponsive (n = 11 clones) tumors. Tex clones with ten or fewer cells not shown. The center line indicates the median value, the lower and upper hinges represent the 25th and 75th percentiles, respectively, and whiskers denote 1.5× interquartile range. *P < 0.05, **P < 0.01, ***P < 0.001, two-sided t-test. d, Volcano plot showing differentially expressed genes for all Tex cells from n = 9 post-treatment responsive and n = 5 post-treatment nonresponsive tumors. Each red dot denotes an individual gene with adjusted P < 0.01, two-sided t-test. e, Comparison of clonal expansion levels between Tex (terminal Tex and Texp) and Tex-irrelevant cells from pre- (n = 8) and post-treatment (n = 9) responsive tumors. Data are presented as mean + s.e.m. *P < 0.05, **P < 0.01, ***P < 0.001, two-sided t-test. f, Clone size in each Tex-irrelevant cell cluster across the eight responsive tumors, separated by treatment status. Center line indicates the median value, lower and upper hinges represent the 25th and 75th percentiles, respectively, and whiskers denote 1.5× interquartile range, two-sided t-test. g, Metacell-based in silico FACS for CD8+ T cells from n = 8 responsive NSCLC, n = 6 melanoma, n = 6 BCC and n = 2 SCC tumors. The x axis represents the expression of CXCL13 and the y axis the exhaustion score. For NSCLC tumors, data were summarized for eight pretreatment and nine site-matched post-treatment samples from eight responsive tumors. h, Metacell-based in silico FACS for CD8+ T cells from 14 post-treatment nonresponsive melanoma tumors.
We next focused our analysis on post-treatment nonresponsive tumors as well as the remaining treatment-naïve tumors. Like pretreatment responsive samples, these tumors failed to accumulate Texp cells with the terminal Tex subset accounting for the major proportion (Fig. 4b,c and Extended Data Fig. 9e–g). Accordingly, Tex cells in post-treatment nonresponsive tumors exhibited overall higher expression levels of exhaustion signatures compared to responsive tumors (Fig. 4d), demonstrating the importance of Texp cells in the treatment of cancer. We next compared Tex-irrelevant cells with Tex cells in an attempt to further understand how the former respond to treatment. By defining clonal expansion with the previously developed STARTRAC index40, we observed that Tex cells exhibited a significantly higher level of expansion compared to Tex-irrelevant cells (Fig. 4e), supporting the tumor specificity of Tex clones. In addition, closer examination of Tex-irrelevant cell clusters revealed that these subsets did not show consistently higher clonal expansion following treatment (Fig. 4f and Extended Data Fig. 10a,b), suggesting that Tex subsets were possibly the major cell types responding to PD-1-based therapies as previously reported29,38,39,46, although future studies are required for validation.
To address whether the effect of Texp cells is present only during the treatment of lung cancer, we analyzed three existing datasets of pre- and post-ICB-treatment CD8+ T cells from responsive BCC, squamous cell carcinoma (SCC)10 and melanoma tumors12. Because the TCR-based identification of Texp cells requires the capture of large clones both pre and post treatment, which might be not suited to these datasets, we then performed metacell-based in silico fluorescent activated cell sorting (FACS; Methods) to distinguish CXCL13-expressing Texp cells from other cells. Of note, this approach readily identified terminal Tex and CXCL13+ Texp cells in our data (Fig. 4g) and yielded results similar to TCR-selection-based clustering (Fig. 4b,c). Further analyses of BCC and SCC revealed increased terminal Tex cells following PD-1 blockade (Fig. 4g), indicative of increased numbers of tumor-reactive cells within the tumor, but CXCL13+ Texp cells were not observed in these two cancer types potentially due to a greater degree of immunosuppression in the tumor microenvironment (TME). We next examined CXCL13+ Texp cells in melanoma. Similar to our data, these cells were enriched in responsive tumors after treatment (Fig. 4g and Extended Data Fig. 10c), with barely detectable abundance in nonresponsive tumors (Fig. 4h). Collectively, our analyses suggest that (1) increase in Texp cells may also exist in melanoma after treatment, and (2) Tex cells may respond differently to ICB treatment in different cancer types, although further studies are required to investigate this.
Local expansion of pre-existing Texp cells
An important question in regard to the increase in Texp cells found in our data is where these cells were derived from. Three possible modes could explain the formation of Texp cells: (1) reprogramming of the terminal Tex subset into Texp cells by treatment; (2) local expansion of pre-existing Texp cells; and (3) replenishment with peripheral T cells10,47. Regarding the first of these, a previous mouse lymphocytic choriomeningitis virus (LCMV) study by Pauken et al. demonstrated that terminal Tex cells could not acquire a memory T-cell phenotype due to their epigenetic stability9 and, in agreement with this report, our above-mentioned RNA velocity analysis revealed a clear one-directional flow from Texp to terminal Tex cells (Fig. 3f), supporting the irreversible differentiation into terminally exhausted T cells14,15, although a direct test for human tumor-derived terminal Tex cells is further required.
We next evaluated whether pre-existing Texp cells could expand in response to treatment. Despite the low abundance of Texp cells, they were indeed identified within pretreatment tumors (Fig. 3b) and this was further supported by closer examination of individual Tex clones (Extended Data Fig. 7). Focusing on responsive tumors, we found that a sizable fraction of post-treatment Tex clonotypes could be detected in pretreatment tumors (Fig. 5a), corresponding to 62.2% of post-treatment Tex cells (Fig. 5b). In addition, such re-emerged (pre-existing) clonotypes were significantly enriched for large size (Fig. 5c), indicating their predominant role in tumor antigen recognition and further antitumor activities. To further validate the second mode, we then focused our analysis on pretreatment proliferative Tex cells to investigate whether this cluster contains certain Texp cells. Here we used metacell-based in silico FACS to distinguish Texp from terminal Tex cells. As a test case, we first show that such an approach could effectively identify progenitor (Tcf1+) and terminal Tex cells in mouse LCMV models38, with an accuracy of 98.1% (Extended Data Fig. 10d). When applying this approach to our data we identified proliferative Texp cells within pretreatment tumors (Fig. 5d), demonstrating that the expansion of pre-existing Texp cells could occur and is an important mode for the accumulation of Texp cells in response to treatment.

a, Venn diagram showing Tex clonotype sharing between pre- and post-treatment responsive tumors. b, Left, pie chart of all post-treatment Tex clonotypes, colored by whether the clonotype was observed in the same tumor before treatment. Right, pie chart of all Tex cells corresponding to Tex clonotypes on the left. Data were summarized from n = 8 responsive tumors. c, Boxplot showing clone size of pretreatment-specific (n = 98), post-treatment-specific (n = 159) and re-emerged (n = 152) clonotypes from n = 8 responsive tumors. Center line indicates the median, lower and upper hinges represent the 25th and 75th percentiles, respectively, and whiskers denote 1.5× interquartile range. *P < 0.05, **P < 0.01, ***P < 0.001, two-sided t-test. d, Metacell-based in silico FACS for pretreatment proliferative Tex cells. The x axis represents the exhaustion score and the y axis the proliferation score (Methods).
Clonal revival of tumor-specific T cells
In addition to clones present both pre and post treatment (Fig. 3a–c), we explored the phenomenon of the increase in Texp cells by further analyzing new clones that were absent before treatment, and we found that these new clones also exhibited a higher proportion of Texp cells relative to clones specific to baseline controls in responsive tumors (Extended Data Fig. 10e–j). It has been suggested previously that new Tex clones are derived from sites outside tumors10,47,48, prompting us to think that those re-emerged clones in our study, like new clones, might also be replenished with recently recruited fresh cells in addition to the local expansion mode.
To further investigate whether Texp cells could be replenished from the periphery, we performed bulk TCR-seq on matched pre- and post-treatment blood samples for four responsive samples (Methods), and found that those clonotypes expanded in tumor tissue could also be detected in blood (Fig. 6a). Furthermore, we extended our analysis by matching all shared clonotypes in tumors with those in blood and observed a general and significant correlation in clone size (Fig. 6b), consistent with the previous notion that peripheral T cells could be supplied as a pool of intratumoral T cells47. To further demonstrate this notion, we analyzed the clone abundance of new intratumoral Tex clonotypes that appeared after treatment in peripheral blood. Of note, most new Tex clonotypes (87%) were detected only in post-treatment blood (Fig. 6c–f), supporting the activation of novel clones with new tumor antigen specificities following treatment10. Interestingly, a similar pattern was also observed for those pre-existing intratumoral Tex clonotypes after treatment (Fig. 6c–f), with 63% of pre-existing Tex clonotypes detected only in post-treatment blood and many of such clonotypes being of large size in tumors before treatment (Fig. 6g). To address whether this phenomenon resulted from potential sequencing bias, we analyzed the published BCC dataset and further confirmed the enrichment of re-emerged Tex clonotypes in post-treatment blood (Extended Data Fig. 10k–m). It is likely that the treatment enhanced the activation and expansion of tumor-reactive clones, resulting in increased abundance of such clones in peripheral blood.

a, Venn diagram showing TCR clonotype overlap between peripheral and intratumoral T cells. b, Correlated clone sizes in blood and tumor. Scatter plots of the clone size of each clonotype in tumor (x axis) and blood (y axis), with data summarized from P010, P013, P019 and P030. P values were determined by two-tailed linear regression t-test, on log-transformed clone sizes to linearize the data. c, Scatter plot of the clone size of each detected Tex clonotype in blood. Tex clonotypes were divided into pretreatment-specific, post-treatment re-emerged (pre-existing) and new clonotypes appearing after treatment. d, Abundance of each detected new (n = 23) and pre-existing (n = 63) Tex clonotype (clone size >1) in blood before and after treatment, with random jitter added to distinguish points. Black lines indicate median. *P < 0.05, **P < 0.01, ***P < 0.001, two-sided Wilcoxon test. n = 4 patients (P010, P013, P019 and P030). e, Abundance of Tex clonotypes in blood before and after treatment. Tex clonotypes were divided into pretreatment-specific, post-treatment re-emerged (pre-existing) and new clonotypes appearing after treatment. f, Pie charts showing the expansion patterns of Tex clonotypes in blood, defined by whether these clonotypes could be exclusively detected in pre- or post-treatment blood by bulk TCR-seq; n = 4 patients. g, Scatter plots of intratumoral Tex clone sizes before and after treatment for P010 and P019 in this study. Each dot denotes one Tex clonotype, color coded by whether these clonotypes could be exclusively detected in pre- or post-treatment blood by bulk TCR-seq. h, UMAP plot of all 8,433 cells from post-treatment P019 blood, color coded by cell clusters. i, UMAP plot of CXCL13 expression level in post-treatment intratumoral Tex cells from P019. j, UMAP plot of T cells from a post-treatment blood sample (P019), color coded by whether they shared TCR sequences with intratumoral Tex cells. A total of 14 peripheral Texp cells were identified. k, Average CXCL13 expression in post-treatment peripheral and intratumoral Texp cells. Distribution of average CXCL13 expression in intratumoral Texp cells was estimated with n = 1,000 permutation tests (n = 14 random cells for each test). l, Contribution of new and re-emerged (pre-existing) Tex clones. Data from NSCLC, BCC and SCC are summarized, showing the ratio of the number of re-emerged Tex clonotypes (top) and cells (bottom) to the number of new Tex clonotypes (top) and cells (bottom) after treatment across patients, as a function of the number of pretreatment Tex clonotypes (top) and cells (bottom). P values were determined by two-tailed linear regression t-test on log-transformed values to linearize data. m. Comparison of the number of Tex clonotypes (y axis) detected in n = 5 BCC and n = 8 NSCLC pretreatment tumors. Center line indicates median, lower and upper hinges represent the 25th and 75th percentiles, respectively, and whiskers denote 1.5× interquartile range. Two-sided t-test.
Despite sharing of TCRs between peripheral and intratumoral T cells, the direction of Texp cell migration is unclear. To further address this, we performed paired scRNA-seq and scTCR-seq on remaining frozen post-treatment blood samples for P019 (Fig. 6h and Extended Data Fig. 10n), the sampling time point of which was the same as that for the post-treatment tumor biopsy. These intratumoral Texp cells have been shown to highly express the CXCL13 gene (Fig. 6i) and, if it was intratumoral Texp cells leaking into the blood, we should be able to detect the expression of CXCL13 in post-treatment peripheral Texp cells. Using the TCR-based lineage tracking approach, we identified certain peripheral CD8+ T cells (n = 14) sharing identical TCRs with intratumoral Tex cells (Fig. 6j), but CXCL13 was expressed neither in these peripheral Texp nor in other CD8+ T cells (Extended Data Fig. 10n). To address whether this observation was due to dropout events, we performed permutation analysis for post-treatment intratumoral Texp cells to estimate the distribution of average CXCL13 expression, and this analysis further demonstrated the difference in CXCL13 expression between peripheral and intratumoral Texp cells (Fig. 6k). Our data, along with previous reports47,49,50, support T-cell migration to the tumor although more direct evidence is needed to confirm this migration direction.
As our analysis indicates, in addition to clonal replacement with new Tex clones10, cells sharing the same clonotypes with pre-existing Tex clones could be reactivated and further enter the tumor following treatment (Fig. 6b–d). To further understand the contribution of new and re-emerged (pre-existing) Tex clones during cancer treatment, we combined our data with published BCC and SCC datasets and matched the number of Tex clones in tumors before treatment with the proportion of re-emerged Tex clones after treatment. It is evident that the frequency of re-emerged Tex clones increased monotonically with the number of pretreatment Tex clones (Fig. 6l). This analysis suggested that most BCC tumors have low baseline Tex infiltration (Fig. 6m) and thus PD-1 blockade would enhance the expansion and infiltration of new Tex clones, presumably by boosting the processing of new tumor neoantigens. By contrast, lung cancer samples tended to be ‘hot tumors’, with the pool of intratumoral Tex clones before treatment already including substantial amounts of tumor-specific clonotypes. Collectively, our data indicated that both new and pre-existing Tex clones could be important in the treatment of cancer, and we termed the phenomenon of their replenishment with peripheral T cells following treatment ‘clonal revival’ (Fig. 7), serving as a confirmation and extension of a previous model of clonal replacement.

Sketch map showing the process of clonal revival and expansion of Texp cells. Intratumoral Texp cells were accumulated by the expansion of pre-existing Texp cells and replenishment with peripheral T cells with both new and pre-existing clonotypes (clonal revival). Further action of PD-1 antibodies on these Texp cells would block their transition to terminal Tex cells. MHC, major histocompatibility complex.
Discussion
In this study we leveraged scRNA-seq to understand the mechanisms underlying combination therapies of anti-PD-1 with chemotherapy. Using TCR-based lineage tracking, we identified two Texp cell states present in human lung cancer associated with high expression of GZMK and low expression of exhaustion signatures. These Texp cells increased substantially in responsive tumors following treatment and are likely to be the major cell type responding to PD-1-based therapies, consistent with previous studies29,38,39,46. Our data suggested that these Texp cells were unlikely to have been derived from the reinvigoration of the terminal Tex subset; instead, they were accumulated by both clonal revival and expansion of pre-existing Texp cells, providing insights into the mechanisms of ICB treatment.
Despite the high baseline Tex infiltration cells in certain NSCLC tumors, many pre-existing Tex clonotypes in tumors were detected only in post-treatment blood (Fig. 6c–f). A possible explanation is that, with increased immunosuppressive activity accompanying tumor progression, certain tumor-reactive clones activated in the early cancer stages gradually diminished in blood over time. With PD-1-based treatment such clones are activated and expanded, resulting in increased abundance in blood (Fig. 6d). In addition, clonal revival of both new and pre-existing Tex clones suggests that newly recruited Texp cells, either from the periphery or other sources such as lymphoid organs11, may be another key factor in successful treatment in addition to local expansion, although the respective contribution of these two modes to antitumor immunity requires further investigation.
Our analysis revealed that ICB treatment in different cancer types may have varied effects on Tex cell subsets. BCC and SCC tumors exhibited a notable expansion of terminal Tex cells in response to ICB and this expansion was also observed in a recent breast cancer study37, whereas lung cancer and melanoma tumors tended to accumulate Texp cells, with terminal Tex cells dramatically decreased following treatment. This difference was possibly due to a variable degree of immunosuppression in different cancer types, or to the concurrent chemotherapy used in these patients with lung cancer. Future therapeutic strategies to enhance the accumulation of Texp cells may further improve the clinical response of these cancers. In this study, we considered terminal Tex cells as a proxy for a tumor-reactive T-cell compartment and performed subsequent analysis based on these Tex clonotypes, although future in vitro experiments would be needed to demonstrate tumor specificity. Given the challenge in collecting consecutive clinical tumor biopsies from patients with NSCLC before and during anti-PD-1 treatment, limited samples were used in this study and thus future work based on larger cohorts of anti-PD-1-treated patients is needed to generalize our findings. In addition, our results are mainly based on transcriptomic data and further functional experiments are required for validation.
Methods
This study complies with all relevant ethical regulations and was approved by the Ethics Committee of Peking University. Written informed consent was provided by all participants.
Human specimens
Thirty-six patients pathologically diagnosed with NSCLC were included in this study. None of the patients was treated with checkpoint blockade inhibitors, radiotherapy or chemotherapy before the first biopsy. Patients with LUAD receiving anti-PD-1 therapy were treated with 200 mg of pembrolizumab, carboplatin (area under the curve (AUC) = 6) and 500 mg m–2 pemetrexed every 3 weeks, and those with lung SCC were treated with 200 mg of pembrolizumab, carboplatin (AUC = 6) and 260 mg m–2 albumin-bound paclitaxel every 3 weeks. Clinical response was assessed for each target lesion based on RECIST v.1.1 (ref. 51). For P001, lymph node metastasis denoted a responsive tumor with the primary lung tumor refractory to treatment. For P013, liver metastasis was responsive and lymph node metastasis newly appeared following treatment (nonresponsive). The clinical metadata for collected tumor biopsies are summarized in Supplementary Table 1 and Extended Data Fig. 1.
scRNA-seq data generation
Fresh tumor biopsies were cut into approximately 1–2-mm3 pieces in RPMI-1640 medium (Gibco) with 10% fetal bovine serum (FBS, Gibco) and enzymatically digested with gentleMACS (Miltenyi) for 60 min on a rotor at 37 °C, according to the manufacturer’s instructions. Dissociated cells were subsequently passed through a 100-μm SmartStrainer and centrifuged at 400g for 5 min. After removal of supernatant the pelleted cells were suspended in red blood cell lysis buffer (TIANDZ) and incubated on ice for 1–2 min to lyse red blood cells. After washing twice with 1× PBS (Gibco), cell pellets were resuspended in sorting buffer (PBS supplemented with 1% FBS). The concentration of single-cell suspensions was adjusted to ~500–1,200 cells μl–1. In total, ~10,000–18,000 cells were used for 10X Chromium Single cell 5′ and human variable, diversity and joining (VDJ) library construction (10X Genomics), according to the manufacturer’s instructions. All subsequent steps were performed following the standard manufacturer protocols. Purified libraries were analyzed using an Illumina Hiseq X Ten sequencer with 150-base pair (bp) paired-end reads.
scRNA-seq data processing and clustering
The scRNA-seq data generated were aligned and quantified using the Cell Ranger Single-Cell toolkit (v.3.0.2) against the GRCh38 human reference genome. Preliminary filtered data generated from Cell Ranger were used for downstream analysis. Further quality control was applied to filter out low-quality cells with either (1) <600 expressed genes or (2) >25,000 or <600 UMIs. We applied the expression entropy (S-E) model to raw count data for selection of highly variable genes with the ROGUE package52 and utilized the library size-correction method to normalize raw counts with the NormalizeData function in Seurat53. The top 3,000 highly variable genes were used for principal component analysis (PCA). To remove batch effects between patients we performed the BBKNN pipeline54 to obtain the batch-corrected space, and further used the Leiden clustering approach implemented in scanpy55 to identify each cell cluster.
Separation and identification of T-cell subtypes
Clusters of T and natural killer (NK) cells were identified by unsupervised clustering following the strategy described above, characterized by high expression of CD3D, CD3E and NKG7. A second round of unsupervised clustering was further performed to identify NK, CD4+ and CD8+ T cells based on the expression of signature genes including CD8A, CD8B, CD4, IL7R and NKG7 (Extended Data Fig. 2a,b).
scTCR-seq data processing
The TCR sequence data from 10X Genomics were processed using Cell Ranger software (v.3.1) with the manufacturer-supplied human VDJ reference genome. For each sample, the output file filtered_contig_annotations.csv, containing TCR α- and β-chain CDR3 nucleotide sequences, was used for downstream analysis. Only those assembled chains that were productive, highly confident, full length, with a valid cell barcode and an unambiguous chain type (for example, alpha) assignment were retained. If a cell had two or more qualified chains of the same type, only that chain with the highest UMI count was qualified and retained. For each patient, cells with an identical α/β-chain pair were considered as having originated from the same clonotype and were therefore identified as clonal cells.
Bulk TCR-seq data processing and analysis
To identify the expansion patterns of Tex clonotypes in peripheral blood, we performed bulk TCR-seq using pre- and post-treatment blood samples from P010, P013, P019 and P030. Total RNA of each sample was extracted from peripheral blood mononuclear cells (PBMCs) using TriZol Reagent (Thermo Fisher Scientific) and the RNeasy Mini Kit (Qiagen). Total RNA of each sample was checked by 1.2% agarose gel and then quantified on an Agilent 2100 Bioanalyzer (Agilent Technologies) and Nanodrop (Thermo Fisher Scientific). Total RNA (100 ng–1 µg) with RNA integrity number >7 was used as input for sequencing library preparation, performed according to procedures developed by GENEWIZ. Reverse transcription was performed according to the manufacturer’s instructions using reverse transcriptase. First-strand complementary DNA was then amplified using primers containing partial Illumina adapters for heavy and light chains. PCR product was amplified using primers containing Illumina adapters and indices with six to eight cycles (annealing temperature, 60 °C). PCR products were cleaned using the AxyPrep Mag PCR Clean-up Kit (Axygen) and then examined using an Agilent 2100 Bioanalyzer and a Qubit 2.0 Fluorometer (Invitrogen). Libraries with different indices were then multiplexed and loaded on an Illumina MiSeq instrument according to the manufacturer’s instructions. Sequencing was carried out using a 2 × 300-bp paired-end configuration; image analysis and base calling were conducted using MiSeq control software on the MiSeq instrument.
Sequences were processed and analyzed using the MiXCR method. Bulk TCR-seq clonotypes were linked to intratumoral Tex clonotypes by matching the TCR β-chain to any TCR β-chain from a clonotype in the scTCR-seq data.
Proportion analysis
To identify changes in the proportion of different cell clusters between treatment conditions, we used three complementary types of statistical test: the Dirichlet-multinomial regression model, Fisher’s exact test and t-test. The Dirichlet-multinomial regression model was used to test for differences in cell composition between treatment conditions by accounting for dependencies in the proportion of different cell clusters; Fisher’s exact test was used to reflect the enrichment of each cell cluster in each treatment condition, and the t-test was used to identify changes in proportion by accounting for the sample from which each cell was collected. Specially, only the t-test was used for analysis of the proportion of Tex subsets because only two cell groups were involved.
RNA velocity analysis
The spliced and unspliced UMIs for each gene in each cell were counted using the python package velocyto; subsequent analyses were performed by scanpy and scvelo56. Specifically, count matrices were normalized by library size, then a k nearest-neighbor graph (k = 30) was built using the top 30 principal components derived from PCA performed on the logarithmized spliced matrix. For each cell, the moments (means and uncentered variances) of normalized spliced/unspliced counts were computed using the 30 nearest neighbors from the function scv.pp.moments. These moments facilitated RNA velocity estimation implemented in the function scv.tl.velocity, with mode set to ‘dynamical’. Based on estimated velocities, a velocity graph representing transition probabilities was constructed using the function scvelo.tl.velocity_graph. The velocity graph was then used to embed RNA velocities in the uniform manifold approximation and projection (UMAP) map in the form of streamlines, with the function scv.pl.velocity_embedding_stream.
Identification of high-confidence Tex clones and Tex proportion analysis
During analysis of Tex clones we found that a small number did not express any exhaustion markers either before or after treatment, suggesting that some ‘non-self’ cells fell into the terminal Tex subset. This is unavoidable because unsupervised clustering may not be 100% accurate and could lead to inaccurate estimation of cell proportions, especially for samples with low cell counts. CXCL13 has been shown to be exclusively expressed by terminal Tex cells in treatment-naïve tumors21,29,33, and its expression has now also been observed in post-treatment Texp but not in Tex-irrelevant cells (Extended Data Fig. 4). Thus we included only high-confidence Tex clones in subsequent analysis, with two additional filters: (1) clone size at least five cells and (2) cells from a certain clone expressing CXCL13—that is, normalized average CXCL13 expression of a certain clone >0.1.
To gain a more accurate assessment of Tex subset proportions, we performed unsupervised clustering for Tex cells from responsive primary lung tumors and lymph node metastases (Extended Data Fig. 6), respectively, and further calculated the frequency of each Tex subset. The identities of Tex cells from the remaining samples were determined by supervised cell-type classification using SciBet57, with cells from Extended Data Fig. 6 as the reference set.
Identification of markers
To characterize each cluster we applied both the FindMarkers procedure in Seurat, which identified markers using log fold changes (FC) of mean expression, and E-test57, a supervised gene selection method that identifies cell-type-specific genes with high accuracy and speed.
Multicolored IHC
To validate the existence of terminal Tex and CXCL13+ Texp cells, we performed multiplexed IHC (mIHC) staining on the remaining biopsy materials from four treatment-naïve (P001, P010, P013 and P029) and three on-treatment responsive tumors (P001, P010 and P029). Specimens were provided by the Chinese PLA General Hospital. The specimens were collected within 30 min of biopsy sampling and fixed in formalin for 48 h. Dehydration and embedding in paraffin were performed following routine methods. Paraffin blocks were cut into 5-μm slices and adhered to glass slides. Sections were then placed in 70 °C paraffin oven for 1 h before deparaffinization in xylene and successive rehydration in 100%, 90% and 70% alcohol. Antigen was retrieved by citric acid buffer (pH 6.0) in a 95 °C water bath for 20 min. Endogenous peroxidase was inactivated by incubation in 3% H2O2 for 15 min. Following preincubation with 10% normal goat serum to block nonspecific sites for 30 min, sections were incubated with primary antibodies in a humidified chamber at 4 °C overnight. The antibodies used in the validation of terminal Tex and Texp cells were rabbit anti-BCA1 (no. Ab246518), rabbit anti-TIM3 (no. CST45208) and mouse anti-CD8A (no. CST70306). After washing sections twice with PBS for 5 min, antigenic binding sites were visualized using the GT Vision TMII Detection System (Mo&Rb) according to the manufacturer’s protocol.
Differential expression analysis
To identify differentially expressed genes between two groups of clusters, we used t.test in Seurat to evaluate the significance of each gene with multiple hypothesis correction using the Benjamini–Hochberg procedure. Genes with adjusted P < 0.01 were considered differentially expressed. In addition, log2(FC) was calculated by subtracting log2-transformed mean counts in each group.
Analysis of BCC, SCC and melanoma datasets
Datadets for scRNA-seq and scTCR-seq (only for BCC and SCC) were obtained from the Gene Expression Omnibus (GEO), nos. GSE123813 and GSE120575. Bulk TCR-seq sequences and template counts of BCC were obtained from the ImmuneACCESS Web portal by Adaptive Biotechnologies (https://clients.adaptivebiotech.com/pub/yost-2019-natmed). For the melanoma dataset, only patients treated with PD-1 antibodies or the combination of PD-1 and CTLA-4 antibodies were included in this study.
Metacell analysis
To address the potential bias introduced by dropout events we used the MetaCell algorithm44 to compute cell-to-cell similarity and identify homogeneous groups of cells—metacells. We applied the MetaCell package to raw count data with default parameters for both UMI- and full-length-based datasets, and further performed correlation, expression and in silico FACS analyses using those metacells. The expression of each gene in a given metacell is defined as the average expression of that gene in corresponding individual cells. Terminal Tex metacells were identified based on exhaustion score.
Calculation of exhaustion and proliferation scores
Because our calculation of exhaustion score was based on metacells, which addresses the potential bias introduced by dropout events, we used only four well-defined exhaustion markers (HAVCR2, ENTPD1, LAYN and LAG3) to improve the accuracy of terminal Tex cell identification. Exhaustion score was defined as the sum of the expression of these four genes. In addition, proliferation score was defined as the sum of the expression of STMN1 and TUBB (top two highly expressed genes in proliferative cells).
The power to detect TCR overlap between biopsies and new clonotype analysis
In our study we analyzed new Tex clonotypes that appeared after treatment, to (1) indicate the direction of T-cell migration (Extended Data Fig. 10e) and (2) investigate the contribution of new and re-emerged (pre-existing) Tex clones during cancer treatment (Fig. 6l). An important question here is whether the new Tex clonotypes detected by scTCR-seq were really absent before treatment or were missed in the first biopsy. To evaluate the power to detect TCR overlap, and the credibility of new Tex clonotypes, here we analyzed previously published BCC datasets with matched scTCR-seq and bulk TCR-seq data. Both the BCC datasets and our studies used the 10X Genomics Chromium approach to profile TCR sequences of individual T cells, and the number of detected clonotypes in our study is at a level similar to that in the BCC study (Extended Data Fig. 10f). Thus, the BCC datasets could be considered a suitable resource for this analysis. Here we considered bulk TCR-seq data as ground truth. By direct comparison of the number of clonotypes detected by these two platforms for each BCC sample, we found that coverage of scTCR-seq was one-sixth that of bulk TCR-seq (Extended Data Fig. 10f). In addition, within the 83 new Tex clonotypes (clone size at least three) detected by scTCR-seq, 42.2% (n = 35) were missed at the first biopsy (Extended Data Fig. 10g) although the remaining new Tex clonotypes (n = 48) were also validated by bulk TCR-seq data. Because both our study and the BCC study performed bulk TCR-seq on matched blood samples, we next investigated whether such data could help improve the accuracy of new Tex clonotype identification. Of note, in the two BCC samples (bcc.su001 and bcc.su008) with matched PBMC data, we observed that the false-positive rate of new Tex clonotypes could be reduced to 17% by filtering out those clonotypes detected in pretreatment blood (Extended Data Fig. 10h). We therefore applied the same filtering strategy to our data and found that these new Tex clones also exhibit a higher proportion of Texp cells relative to clones specific to baseline controls in responsive tumors (Extended Data Fig. 10i), showing that ICB treatment could also modulate certain new Texp cells to enhance antitumor immunity.
In addition, Yost et al.10 previously demonstrated that new Tex clonotypes were preferentially observed in BCC tumors following PD-1 blockade. However, we observed a different dynamic change in lung cancer with those pre-existing Tex clonotypes accounting for the major proportion after treatment (Fig. 5a,b). Thus, we also included the new Tex clonotype in Fig. 6l to address this difference. To address the potential concern that the relatively low coverage of scTCR-seq may represent an intrinsic challenge in this analysis, we matched the number of new Tex clonotypes detected by scTCR-seq and bulk TCR-seq using the BCC datasets and observed a significant correlation between these two approaches (Extended Data Fig. 10j), showing that the analysis in Fig. 6l is reliable.
Statistics and reproducibility
No statistical method was used to predetermine sample size, but our sample sizes are similar to those reported in previous publications10,20. No data were excluded from the analyses. The experiments were not randomized. The Investigators were not blinded to allocation during experiments and outcome assessment. Data collection and analysis were not performed blind to the conditions of the experiments.
Statistical analysis was performed in R v.3.6.1. Three complementary types of statistical test—Dirichlet-multinomial regression model, Fisher’s exact test and t-test—were used to identify changes in the proportion of different cell clusters between treatment conditions and responses. Comparisons of numerical variables between treatment conditions and responses were carried out using either the Wilcoxon rank-sum test or t-test. When using the latter, normality and equal variances were tested and data distribution was found to be normal.
Single-cell sequencing for each tumor biopsy was performed in one experimental run. This is typical for human studies because cell counts from a given biopsy sample are usually low and cells cannot be analyzed more than once. The mIHC experiments for each tumor sample were performed in one experimental run, with approximately ten fields of view captured to verify reproducibility. In addition, at least three tumor samples from each group were collected to verify reproducibility (Extended Data Fig. 9a–d). Further information on research design is available in the Nature Research reporting summary linked to this article.
Reporting summary
Further information on research design is available in the Nature Research reporting summary linked to this article.
Data availability
All processed scRNA-seq, scTCR-seq and bulk TCR-seq data that support the findings of this study have been deposited with GEO under accession code GSE179994. An interactive web server for analysis and visualization of scRNA-seq data is available at http://nsclcpd1.cancer-pku.cn/. Previously published scRNA-seq, scTCR-seq and bulk TCR-seq data reanalyzed here are available under accession code nos. GSE123814 (ref. 10), GSE120575 (ref. 12), GSE99254 (ref. 21), GSE108989 (ref. 40), GSE98638 (ref. 43), GSE113590 (ref. 31), GSE141878 (ref. 35), GSE24536 (ref. 34) and E-MTAB-6149 (ref. 19). Source data are provided with this paper. All other data supporting the findings of this study are available from the corresponding author on reasonable request.
Code availability
No new algorithms were developed for this manuscript. All code generated for analysis is available from the corresponding authors upon reasonable request.
References
Sharma, P. & Allison, J. P. The future of immune checkpoint therapy. Science 348, 56–61 (2015).
Hellmann, M. D. et al. Nivolumab plus ipilimumab as first-line treatment for advanced non-small-cell lung cancer (CheckMate 012): results of an open-label, phase 1, multicohort study. Lancet Oncol. 18, 31–41 (2017).
Brahmer, J. R. et al. Safety and activity of anti–PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 366, 2455–2465 (2012).
Topalian, S. L. et al. Safety, activity, and immune correlates of anti–PD-1 antibody in cancer. N. Engl. J. Med. 366, 2443–2454 (2012).
Wherry, E. J. & Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 15, 486–499 (2015).
Barber, D. L. et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 439, 682–687 (2006).
Scott, A. C. et al. TOX is a critical regulator of tumour-specific T cell differentiation. Nature 571, 270–274 (2019).
Khan, O. et al. TOX transcriptionally and epigenetically programs CD8+ T cell exhaustion. Nature 571, 211–218 (2019).
Pauken, K. E. et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 354, 1160–1165 (2016).
Yost, K. E. et al. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat. Med. 25, 1251–1259 (2019).
Im, S. J. et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 537, 417–421 (2016).
Sade-Feldman, M. et al. Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell 175, 998–1013 (2018).
Brummelman, J. et al. High-dimensional single cell analysis identifies stem-like cytotoxic CD8+ T cells infiltrating human tumors. J. Exp. Med. 215, 2520–2535 (2018).
Chen, Z. et al. TCF-1-centered transcriptional network drives an effector versus exhausted CD8 T cell-fate decision. Immunity 51, 840–855 (2019).
Hudson, W. H. et al. Proliferating transitory T Cells with an effector-like transcriptional signature emerge from PD-1+ stem-like CD8+ T cells during chronic infection. Immunity 51, 1043–1058 (2019).
Beltra, J.-C. et al. Developmental relationships of four exhausted CD8+ T cell subsets reveals underlying transcriptional and epigenetic landscape control mechanisms. Immunity 52, 825–841 (2020).
Zander, R. et al. CD4+ T cell help is required for the formation of a cytolytic CD8+ T cell subset that protects against chronic infection and cancer. Immunity 51, 1028–1042 (2019).
Rizvi, N. A. et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348, 124–128 (2015).
Lambrechts, D. et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat. Med. 24, 1277–1289 (2018).
Caushi, J. X. et al. Transcriptional programs of neoantigen-specific TIL in anti-PD-1-treated lung cancers. Nature 596, 126–132 (2021).
Guo, X. et al. Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat. Med. 24, 978–985 (2018).
Bacher, P. et al. Regulatory T cell specificity directs tolerance versus allergy against aeroantigens in humans. Cell 167, 1067–1078 (2016).
Jiao, S. et al. Differences in tumor microenvironment dictate T helper lineage polarization and response to immune checkpoint therapy. Cell 179, 1177–1190 (2019).
Jansen, C. S. et al. An intra-tumoral niche maintains and differentiates stem-like CD8 T cells. Nature 576, 465–470 (2019).
Galon, J. et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 313, 1960–1964 (2006).
Herbst, R. S. et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 515, 563–567 (2014).
Ahmadzadeh, M. et al. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood 114, 1537–1544 (2009).
Matsuzaki, J. et al. Tumor-infiltrating NY-ESO-1–specific CD8+ T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Proc. Natl Acad. Sci. USA 107, 7875–7880 (2010).
Thommen, D. S. et al. A transcriptionally and functionally distinct PD-1+ CD8+ T cell pool with predictive potential in non-small-cell lung cancer treated with PD-1 blockade. Nat. Med. 24, 994–1004 (2018).
Duhen, T. et al. Co-expression of CD39 and CD103 identifies tumor-reactive CD8 T cells in human solid tumors. Nat. Commun. 9, 2724 (2018).
Simoni, Y. et al. Bystander CD8+ T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature 557, 575–579 (2018).
Angelosanto, J. M., Blackburn, S. D., Crawford, A. & Wherry, E. J. Progressive loss of memory T cell potential and commitment to exhaustion during chronic viral infection. J. Virol. 86, 8161–8170 (2012).
van der Leun, A. M., Thommen, D. S. & Schumacher, T. N. CD8+ T cell states in human cancer: insights from single-cell analysis. Nat. Rev. Cancer 20, 218–232 (2020).
Baitsch, L. et al. Exhaustion of tumor-specific CD8+ T cells in metastases from melanoma patients. J. Clin. Invest. 121, 2350–2360 (2011).
Yang, R. et al. Distinct epigenetic features of tumor-reactive CD8+ T cells in colorectal cancer patients revealed by genome-wide DNA methylation analysis. Genome Biol. 21, 2 (2019).
Wei, S. C. et al. Distinct cellular mechanisms underlie anti-CTLA-4 and anti-PD-1 checkpoint blockade. Cell 170, 1120–1133 (2017).
Bassez, A. et al. A single-cell map of intratumoral changes during anti-PD1 treatment of patients with breast cancer. Nat. Med. 27, 820–832 (2021).
Miller, B. C. et al. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol. 20, 326–336 (2019).
Siddiqui, I. et al. Intratumoral Tcf1+PD-1+CD8+ T cells with stem-like properties promote tumor control in response to vaccination and checkpoint blockade immunotherapy. Immunity 50, 195–211 (2019).
Zhang, L. et al. Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature 564, 268–272 (2018).
Milner, J. J. et al. Runx3 programs CD8+ T cell residency in non-lymphoid tissues and tumours. Nature 552, 253–257 (2017).
La Manno, G. et al. RNA velocity of single cells. Nature 560, 494–498 (2018).
Zheng, C. et al. Landscape of infiltrating T cells in liver cancer revealed by single-cell sequencing. Cell 169, 1342–1356 (2017).
Baran, Y. et al. MetaCell: analysis of single-cell RNA-seq data using K-nn graph partitions. Genome Biol. 20, 206 (2019).
Workel, H. H. et al. A transcriptionally distinct CXCL13+CD103+CD8+ T-cell population is associated with B-cell recruitment and neoantigen load in human cancer. Cancer Immunol. Res. 7, 784–796 (2019).
Huang, A. C. et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 545, 60–65 (2017).
Dammeijer, F. et al. The PD-1/PD-L1-checkpoint restrains T cell immunity in tumor-draining lymph nodes. Cancer Cell 38, 685–700 (2020).
Wu, T. D. et al. Peripheral T cell expansion predicts tumour infiltration and clinical response. Nature 579, 274–278 (2020).
Chen, D. S. & Mellman, I. Oncology meets immunology: the cancer-immunity cycle. Immunity 39, 1–10 (2013).
Muthuswamy, R. et al. NF-κB hyperactivation in tumor tissues allows tumor-selective reprogramming of the chemokine microenvironment to enhance the recruitment of cytolytic T effector cells. Cancer Res. 72, 3735–3743 (2012).
Eisenhauer, E. A. et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur. J. Cancer 45, 228–247 (2009).
Liu, B. et al. An entropy-based metric for assessing the purity of single cell populations. Nat. Commun. 11, 3155 (2020).
Stuart, T. et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902 (2019).
Polański, K. et al. BBKNN: fast batch alignment of single cell transcriptomes. Bioinformatics 36, 964–965 (2020).
Wolf, F. A., Angerer, P. & Theis, F. J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, 15 (2018).
Bergen, V., Lange, M., Peidli, S., Wolf, F. A. & Theis, F. J. Generalizing RNA velocity to transient cell states through dynamical modeling. Nat. Biotechnol. 38, 1408–1414 (2020).
Li, C. et al. SciBet as a portable and fast single cell type identifier. Nat. Commun. 11, 1818 (2020).
Acknowledgements
This project was supported by the National Natural Science Foundation of China (nos. 81988101, 91742203, 91942307 and 31991171) and Beijing Advanced Innovation Center for Genomics. Part of the analysis was performed at the High Performance Computing and National Center for Protein Sciences at Peking University. We thank members of the Zhang laboratory for discussions and W. He for assistance with data analysis.
Author information
Authors and Affiliations
Contributions
Z.Z. and W.H. conceived and designed this study. B.L. conceived and performed bioinformatic data analysis and interpreted the results. K.F., S.Z., Y.H. and Z.X. collected clinical tumor biopsies. X.H. and R.G. performed experiments. Y.Z. assisted with data analysis. E.C. provided valuable discussions. B.L. and Z.Z. wrote the manuscript, with all authors contributing to writing and providing feedback.
Corresponding authors
Ethics declarations
Competing interests
E.C. is an employee of Boehringer-Ingelheim. Z.Z. is a founder of Analytical Biosciences and is a consultant for InnoCare Pharma and ArsenalBio. All financial interests are unrelated to this study. The other authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Sample statistics.
a, Summary of clinical metadata for patients with post-treatment tumour samples, showing the time from the first biopsy sampling, biopsy sites and responses to the treatment. b,c, Representative computed tomography (CT) images of responsive (b) and non-responsive (c) NSCLC tumours before and after initiation of treatment, corresponding to Fig. 1b.
Extended Data Fig. 2 Characterization of CD4+ T cell subtypes.
a, UMAP plot of N = 150,849 T and NK cells isolated from all sequenced cells, with colour-coded by the four inferred cell types; N = 47 tumour samples. b, UMAP plot of expression levels of selected genes in different clusters of T and NK cells from a. c, Histogram showing the distribution of clone sizes for N = 75,735 CD4+ T cells and 59,656 CD8+ T cells from N = 47 samples. d,e, UMAP of N = 75,735 CD4+ T cells from 47 samples colored by patient identity (d) as well as treatment status and response (e). f, Expression heatmap of cell-type-specific genes in the 9 CD4+ clusters. Rows represent different clusters and columns represent signature genes. g, UMAP plot showing expression levels of cell-type-specific markers in CD4+ T cells from d. h, Volcano plot showing differentially expressed genes for all Tregs from N = 8 pre- and 9 post-treatment samples from 8 responsive tumours. Each red dot denotes an individual gene with adjusted P value < 0.01, two-sided t-test. i, Average gene expression in all Tregs from pre-treatment (n = 33) and post-treatment responsive tumour samples (n = 9). Each dot corresponds to one sample. The center line indicates the median value. The lower and upper hinges represent the 25th and 75th percentiles, respectively, and whiskers denote 1.5 times the interquartile range. *P < 0.05; **P < 0.01; ***P < 0.001; one-sided t-test. j, Enrichment plot for 4-1BB+ Treg signatures derived from Guo et al., and the P value was determined by a one-tailed permutation test using the GSEA package. NES, normalized enrichment score.
Extended Data Fig. 3 Characterization of CD8+ T cell subtypes.
a, CD8A IHC staining for 7 tumour biopsies. Original magnification, x20. b-d, UMAP of CD8+ T cells from N = 47 samples colored by patient identity (b), treatment status and response (c, d). e, UMAP plot showing expression levels of certain naïve markers, memory signatures, effector molecules and transcription factors in CD8+ T cells. f, Expression of effector-associated genes in terminal Tex cells from site-matched pre- and post-treatment responsive tumours. g, UMAP of CD8+ T cells colored by cluster origin. h, Expression heatmap of cell-type-specific genes in the 6 CD8+ clusters. Rows represent different clusters and columns represent signature genes. i, Distribution of Tex and Tex-irrelevant cells in CD8+ T cell clusters from N = 8 responsive tumours, corresponding to c. j, Proportion of each CD8 subtype in all CD8 T cells. Data are presented as mean values ± SEM. P value was determined by t-test and Dirichlet-multinomial regression model, two-sided (Methods). *P < 0.05; **P < 0.01; ***P < 0.001.
Extended Data Fig. 4 Gene expression in Tex and Tex-irrelevant cells.
a,b, UMAP of Tex (a) and Tex-irrelevant cells (b) with colour coded by cell cluster. c, UMAP plots showing the expression levels of certain signature genes in Tex and Tex-irrelevant cells.
Extended Data Fig. 5 SciBet classification, RNA velocity and CXCL13 expression analysis.
a, Cell type classification for Tex subsets in this study, with two clusters─CD8_C6-LAYN (terminal Tex) and CD8_C4-GZMK (Tem-like) from Guo et al. as the reference set. b, RNA velocities are visualized on the diffusion map of cells from Tex clones > 300 cells from P019, P010 and P030. c, Metacell-based in-silico FACS for all CD8+ cells from 33 treatment-naive tumours in this study. d, Exhaustion score and the expression of exhaustion signatures are plotted along the expression of CXCL13 for CD8+ T cells from the four published datasets. Data were summarized from N = 12,040 cells from 5 patients for E-MTAB-6149, 3,628 cells from 12 patients for GSE108989, 3,325cells from 14 patients for GSE99254, and 1,579 cells from 6 patients for GSE98638. P values were determined from the Pearson correlation coefficient r, two-tailed linear regression t-test. ***P < 2.2×10−16. e, Metacell-based in-silico FACS for all CD8+ cells in treatment-naive tumours from the four published datasets, corresponding to d. f, Comparison of exhaustion score and the expression of CXCL13 between terminal Tex and non-terminal Tex cells using the metacell-based approach for the four published datasets. The center line indicates the median value. The lower and upper hinges represent the 25th and 75th percentiles, respectively, and whiskers denote 1.5 times the interquartile range. *P < 0.05; **P < 0.01; ***P < 0.001; two-sided t-test. g, Density plot showing the expression of CXCL13 in terminal Tex and Tex-TCR-sharing peripheral T cells using data from the lung and colon cancer studies. Data were summarized from N = 799 terminal Tex cells and 35 peripheral T cells from 12 patients for GSE108989, and 476 terminal Tex cells and 35 peripheral cells from 14 patients for GSE99254.
Extended Data Fig. 6 Tex cell subsets from LN metastases and non-LN tumours.
a, UMAP of Tex cells from N = 3 responsive LN metastases, with colored by cell clusters; N = 6,982 cells. b, UMAP plot showing expression levels of cell-type-specific markers in Tex subsets, corresponding to a. c, UMAP of Tex cells from N = 5 tumours, with colour coded by cell clusters; N = 1,476 cells. d, UMAP plot showing expression levels of cell-type-specific markers in Tex subsets, corresponding to c. e, Cell type classification for Tex cells from LN metastases, with cells from non-LN metastases as the reference set.
Extended Data Fig. 7 Cell states of individual dominant Tex clones from responsive tumours.
Gene expression heatmap for cells from dominant high-confidence Tex clones from pre- and post-treatment tumour biopsies. Rows represent certain differentially expressed genes (adjusted P value < 0.01, two-tailed t-test) and columns represent individual cells. All the N = 8 responsive tumours were included, with representative clones (with largest sizes) from each sample shown here.
Extended Data Fig. 8 Differential expression analysis of individual dominant Tex clones from responsive tumours.
Differentially expressed genes for the most representative individual dominant Tex clones from pre- and post-treatment tumour biopsies. Red points indicate significantly up- or down-regulated genes with adjusted P value < 0.01, two-tailed t-test. Data are representative of N = 5 responsive tumours.
Extended Data Fig. 9 mIHC staining and Tex subset proportions.
a, Three representative fields of view from P010 (pre & post) and P029 (post) samples, stained by mIHC. Original magnification, x20. Data are representative of n = 3 tumour samples, with data from all the 7 samples summarized in panels b-d. b, Distribution of cell densities across pre-treatment (n = 4) and post-treatment (n = 3) samples, showing that the cell density in pre-treatment samples was at a similar level to that in post-treatment ones. For each sample, 10–16 fields of view (690 µm×520 µm) were collected. c, Comparison of the number and proportion of CD8A+BCA1+TIM3− T cells per 690 µm×520 µm field in pre-treatment (n = 4) and post-treatment (n = 3) responsive tumour samples, with random jitter added to distinguish points. The center line indicates the median value. The lower and upper hinges represent the 25th and 75th percentiles, respectively, and whiskers denote 1.5 times the interquartile range. Each dot denotes a field of view. Two-sided t-test. d, Barplot showing the number and proportion of CD8A+BCA1+TIM3− T cells in each sample. The pre-treatment P029 sample was removed in the proportion analysis because only two CD8A+BCA1+ cells were detected in all ten fields of view. Data are presented as mean values ± SEM. P values were determined by a two-tailed t-test. *P < 0.05; **P < 0.01; ***P < 0.001. e,f, Gene expression heatmap for cells from dominant high-confidence Tex clones from post-treatment non-responsive and the remaining 25 treatment-naive tumour biopsies. Rows represent certain differentially expressed genes and columns represent individual cells. g, Comparison of the proportion of Texp cells in all Tex cells between pre-treatment LN metastases and non-LN biopsy sites. The center line indicates the median value. The lower and upper hinges represent the 25th and 75th percentiles, respectively, and whiskers denote 1.5 times the interquartile range.
Extended Data Fig. 10 Analyses of Tex-irrelevant cell, Tex subset proportion and peripheral Tex clonotypes.
a, UMAP of N = 11,476 Tex-irrelevant cells from 8 responsive tumours, with colour coded by cluster origin. b, Expression heatmap of cell-type-specific genes in the 5 Tex-irrelevant cell clusters. Rows represent different clusters and columns represent signature genes, corresponding to a. c, Frequency of terminal Tex, CXCL13+ Texp and other metacells as the proportion of all CD8+ metacells in each patient with melanoma. d, Metacell-based in-silico FACS for N = 9,197 Tex cells from a mouse LCMV model. e, Comparison of the proportion of Texp cells in all Tex cells between pre-treatment- (n = 7) and post-treatment-specific (n = 36) Tex clones. Data were summarized from N = 4 responsive tumours (P010, P013, P019 and P030). *P < 0.05; **P < 0.01; ***P < 0.001; two-sided t-test. f, Boxplot showing the number of detected TCR clonotypes (y axis) from NSCLC scTCR-seq (N = 25 tumours), BCC scTCR-seq (N = 22 tumours) and matched bulk TCR-seq (N = 22 tumours) data. The center line indicates the median value. The lower and upper hinges represent the 25th and 75th percentiles, respectively, and whiskers denote 1.5 times the interquartile range. g, Barplot showing the proportion of true new Tex clonotypes (validated by bulk TCR-seq) and false positive new clonotypes (only detected by scTCR-seq) for each BCC tumour. h, Barplot showing the proportion of true and false positive new Tex clonotypes after filtering out clonotypes detected in pre-treatment blood (bulk TCR-seq). i, Comparison of the proportion of Texp cells between pre-treatment- (n = 7) and post-treatment-specific (n = 32) Tex clones in this study after filtering out clonotypes detected in pre-treatment blood, corresponding to e; two-sided t-test. *P < 0.05; **P < 0.01; ***P < 0.001. j, Correlation of number of detected new Tex clonotypes between scRNA-seq and bulk TCR-seq using n = 11 BCC samples. P value was determined by a two-tailed linear regression t-test. k, Scatter plots of intra-tumoural Tex clone abundance before and after treatment by bulk TCR-seq for patient su001 from BCC data. P value was determined by a two-tailed linear regression t-test. l, Abundance of Tex clonotypes in blood before and after treatment. Tex clonotypes were divided into pre-treatment-specific, post-treatment re-emerged and new clonotypes appeared after treatment based on k. m, Pie charts showing the expansion patterns of Tex clonotypes in blood. n, UMAP plot of expression levels of selected marker genes in different clusters from the post-treatment P019 blood sample.
Supplementary information
Supplementary Information
Supplementary Fig. 1.
Supplementary Tables
Supplementary Tables 1–3.
Source data
Source Data Fig. 1
Statistical source data.
Source Data Fig. 2
Statistical source data.
Source Data Fig. 3
Statistical source data.
Source Data Fig. 4
Statistical source data.
Source Data Fig. 5
Statistical source data.
Source Data Fig. 6
Statistical source data.
Source Data Extended Data Fig. 2
Statistical source data.
Source Data Extended Data Fig. 3
Statistical source data.
Source Data Extended Data Fig. 4
Statistical source data.
Source Data Extended Data Fig. 5
Statistical source data.
Source Data Extended Data Fig. 6
Statistical source data.
Source Data Extended Data Fig. 7
Statistical source data.
Source Data Extended Data Fig. 8
Statistical source data.
Source Data Extended Data Fig. 9
Statistical source data.
Source Data Extended Data Fig. 10
Statistical source data.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, B., Hu, X., Feng, K. et al. Temporal single-cell tracing reveals clonal revival and expansion of precursor exhausted T cells during anti-PD-1 therapy in lung cancer. Nat Cancer 3, 108–121 (2022). https://doi.org/10.1038/s43018-021-00292-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s43018-021-00292-8
This article is cited by
-
Spatial features of specific CD103+CD8+ tissue-resident memory T cell subsets define the prognosis in patients with non-small cell lung cancer
Journal of Translational Medicine (2024)
-
Sex disparities revealed by single-cell and bulk sequencing and their impacts on the efficacy of immunotherapy in esophageal cancer
Biology of Sex Differences (2024)
-
Semi-supervised integration of single-cell transcriptomics data
Nature Communications (2024)
-
Modeling T cell temporal response to cancer immunotherapy rationalizes development of combinatorial treatment protocols
Nature Cancer (2024)
-
Regulatory T cell-mediated immunosuppression orchestrated by cancer: towards an immuno-genomic paradigm for precision medicine
Nature Reviews Clinical Oncology (2024)
