
Abstract
Carbon capture and storage (CCS) has a fundamental role in achieving the goals of the Paris Agreement to limit anthropogenic warming to 1.5–2 °C. Most ongoing CCS projects inject CO2 into sedimentary basins and require an impermeable cap rock to prevent the CO2 from migrating to the surface. Alternatively, captured carbon can be stored through injection into reactive rocks (such as mafic or ultramafic lithologies), provoking CO2 mineralization and, thereby, permanently fixing carbon with negligible risk of return to the atmosphere. Although in situ mineralization offers a large potential volume for carbon storage in formations such as basalts and peridotites (both onshore and offshore), its large-scale implementation remains little explored beyond laboratory-based and field-based experiments. In this Review, we discuss the potential of mineral carbonation to address the global CCS challenge and contribute to long-term reductions in atmospheric CO2. Emphasis is placed on the advances in making this technology more cost-effective and in exploring the limits and global applicability of CO2 mineralization.
Key points
-
Carbon capture and storage has a key role in achieving the goals of the Paris Agreement.
-
CO2 storage through mineral carbonation extends the applicability of carbon capture and storage by enabling storage in areas previously not considered feasible.
-
The rapid mineralization of CO2 through injection into reactive rock formations increases storage security.
-
Carbon mineralization in basaltic rocks offers a global storage potential that exceeds anthropogenic emissions.
-
The method can be used for the subsurface storage of CO2, and potentially other environmentally important gases, through water capture, although this approach is water-intensive.
-
Considerable efforts are needed to accelerate the deployment of CO2 storage through mineral carbonation, including more widespread operation in diverse conditions.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 digital issues and online access to articles
$99.00 per year
only $8.25 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout







Similar content being viewed by others
Change history
10 November 2020
An amendment to this paper has been published and can be accessed via a link at the top of the paper.
References
Blunden, J. & Arndt, D. S. State of the climate in 2018. Bull. Am. Meteorol. Soc. 100, Si–S305 (2019).
Intergovernmental Panel on Climate Change. Global warming of 1.5°C (IPCC, 2018).
United Nations Framework Convention on Climate Change. Report of the conference of the Parties on its twenty-first session, held in Paris from 30 November to 13 December 2015 (UNFCCC, 2016).
International Energy Agency. World energy outlook 2017 (IEA, 2017).
Rogelj, J. et al. in Global warming of 1.5°C. Ch. 2 (IPCC, 2018).
Global CCS Institute. The global status of CCS 2018 (Global CCS Institute, 2018).
Benson, S. et al. in IPCC Special Report on Carbon Dioxide Capture and Storage (eds Metz, B. et al.) Ch. 5 (Cambridge Univ. Press, 2005).
Alcalde, J. et al. Estimating geological CO2 storage security to deliver on climate mitigation. Nat. Commun. 9, 2201 (2018).
Seifritz, W. CO2 disposal by means of silicates. Nature 345, 486–490 (1990).
Lackner, K., Wendt, C. H., Butt, D. P., Joyce, E. L. & Sharp, D. H. Carbon dioxide disposal in carbonate minerals. Energy 20, 1153–1170 (1995).
Sigfusson, B. et al. Solving the carbon-dioxide buoyancy challenge: the design and field testing of a dissolved CO2 injection system. Int. J. Greenh. Gas Control 37, 213–219 (2015).
Matter, J. M. et al. Rapid carbon mineralization for permanent disposal of anthropogenic carbon dioxide emissions. Science 352, 1312–1314 (2016). Reports on the results from the rapid mineralization of the CarbFix pilot injection.
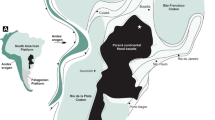
Goldberg, D. S., Kent, D. V. & Olsen, P. E. Potential on-shore and off-shore reservoirs for CO2 sequestration in Central Atlantic magmatic province basalts. Proc. Natl Acad. Sci.USA 107, 1327–1332 (2010).
Goldberg, D. S., Takahashi, T. & Slagle, A. L. Carbon dioxide sequestration in deep-sea basalt. Proc. Natl Acad. Sci. USA 105, 9920–9925 (2008).
Ellis, A. J. The solubility of calcite in carbon dioxide solutions. Am. J. Sci. 257, 354–365 (1959).
Ellis, A. J. The solubility of calcite in sodium chloride solutions at high temperatures. Am. J. Sci. 261, 259–267 (1963).
Saldi, G. D., Jordan, G., Schott, J. & Oelkers, E. H. Magnesite growth rates as a function of temperature and saturation state. Geochim. Cosmochim. Acta 73, 5646–5657 (2009).
Johnson, N. C. et al. Olivine dissolution and carbonation under conditions relevant for in situ carbon storage. Chem. Geol. 373, 93–105 (2014).
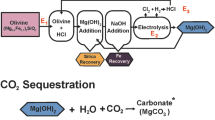
Gadikota, G., Matter, J., Kelemen, P. & Park, A. H. Chemical and morphological changes during olivine carbonation for CO2 storage in the presence of NaCl and NaHCO3. Phys. Chem. Chem. Phys. 16, 4679–4693 (2014).
Turvey, C. C. et al. Hydrotalcites and hydrated Mg-carbonates as carbon sinks in serpentinite mineral wastes from the Woodsreef chrysotile mine, New South Wales, Australia: controls on carbonate mineralogy and efficiency of CO2 air capture in mine tailings. Int. J. Greenh. Gas Control 79, 38–60 (2018).
Rogers, K. L., Neuhoff, P. S., Pedersen, A. K. & Bird, D. K. CO2 metasomatism in a basalt-hosted petroleum reservoir, Nuussuaq, West Greenland. Lithos 92, 55–82 (2006).
Gysi, A. P. & Stefánsson, A. CO2-water–basalt interaction. Low temperature experiments and implications for CO2 sequestration into basalts. Geochim. Cosmochim. Acta 81, 129–152 (2012).
McGrail, B. P. et al. Field validation of supercritical CO2 reactivity with basalts. Environ. Sci. Technol. Lett. 4, 6–10 (2017). Reports the results from the rapid carbonation during the Wallula pilot injection.
Snæbjörnsdóttir, S. Ó. et al. CO2 storage potential of basaltic rocks in Iceland and the oceanic ridges. Energy Procedia 63, 4585–4600 (2014).
Gysi, A. P. & Stefánsson, A. CO2–water–basalt interaction. Numerical simulation of low temperature CO2 sequestration into basalts. Geochim. Cosmochim. Acta 75, 4728–4751 (2011).
Voigt, M., Pearce, C. R., Baldermann, A. & Oelkers, E. H. Stable and radiogenic strontium isotope fractionation during hydrothermal seawater-basalt interaction. Geochim. Cosmochim. Acta 240, 131–151 (2018).
Snæbjörnsdóttir, S. Ó., Gislason, S. R., Galeczka, I. M. & Oelkers, E. H. Reaction path modelling of in-situ mineralisation of CO2 at the CarbFix site at Hellisheidi, SW-Iceland. Geochim. Cosmochim. Acta 220, 348–366 (2018).
Godard, M., Luquot, L., Andreani, M. & Gouze, P. Incipient hydration of mantle lithosphere at ridges: a reactive-percolation experiment. Earth Planet. Sci. Lett. 371, 92–102 (2013).
Kelemen, P. B. & Hirth, G. Reaction-driven cracking during retrograde metamorphism: Olivine hydration and carbonation. Earth Planet. Sci. Lett. 345, 81–89 (2012).
Zhu, W. L. et al. Experimental evidence of reaction-induced fracturing during olivine carbonation. Geophys. Res. Lett. 43, 9535–9543 (2016).
Clark, D. E. et al. The chemistry and potential reactivity of the CO2-H2S charged injected waters at the basaltic CarbFix2 site, Iceland. Energy Procedia 146, 121–128 (2018).
Wolff-Boenisch, D., Gislason, S. R. & Oelkers, E. H. The effect of crystallinity on dissolution rates and CO2 consumption capacity of silicates. Geochim. Cosmochim. Acta 70, 858–870 (2006).
Dessert, C., Dupre, B., Gaillardet, J., Francois, L. M. & Allegre, C. J. Basalt weathering laws and the impact of basalt weathering on the global carbon cycle. Chem. Geol. 202, 257–273 (2003). Assesses the natural weathering potential of basalts worldwide and their contribution to the carbon cycle.
Kappel, E. S. & Ryan, W. B. F. Volcanic episodicity and a non-steady state rift valley along northeast Pacific spreading centers: evidence from Sea MARC I. J. Geophys. Res. 91, 13925–13940 (1986).
Karson, J. A. Geologic structure of the uppermost oceanic crust created at fast-to intermediate-rate spreading centers. Annu. Rev. Earth Planet. Sci. 30, 347–384 (2002).
Alt, J. C. & Teagle, D. A. H. The uptake of carbon during alteration of ocean crust. Geochim. Cosmochim. Acta 63, 1527–1535 (1999).
Coogan, L. A., Parrish, R. & Roberts, N. M. W. Early hydrothermal carbon uptake by the upper oceanic crust: insight from in-situ U-Pb dating. Geology 44, 147–150 (2016).
Wiese, F., Fridriksson, T. & Ármannsson, H. CO 2 Fixation by calcite in high-temperature geothermal systems in Iceland (ÍSOR, 2008).
Marieni, C., Henstock, T. J. & Teagle, D. A. H. Geological storage of CO2 within the oceanic crust by gravitational trapping. Geophys. Res. Lett. 40, 6219–6224 (2013).
Callow, B., Falcon-Suarez, I., Ahmed, S. & Matter, J. M. Assessing the carbon sequestration potential of basalt using X-ray micro-CT and rock mechanics. Int. J. Greenh. Gas Control 70, 146–156 (2018).
McGrail, B. P. et al. Potential for carbon dioxide sequestration in flood basalts. J. Geophys. Res. 111, B12201 (2006).
Kelemen, P. B. & Matter, J. M. In situ carbonation of peridotite for CO2 storage. Proc. Natl Acad. Sci. USA 105, 17295–17300 (2008). Reviews the carbonation of peridotites and their potential for CO 2 storage.
Hansen, L. D., Dipple, G. M., Gordon, T. M. & Kellett, D. A. Carbonated serpentinite (listwanite) at Atlin, British Columbia: a geological analogue to carbon dioxide sequestration. Can. Mineral. 43, 225–239 (2005).
Wilson, S. A. et al. Carbon dioxide fixation within mine wastes of ultramafic-hosted ore deposits: examples from the Clinton Creek and Cassiar chrysotile deposits, Canada. Economic Geol. 104, 95–112 (2009).
Wilson, S. A. et al. Offsetting of CO2 emissions by air capture in mine tailings at the Mount Keith Nickel Mine, Western Australia: Rates, controls and prospects for carbon neutral mining. Int. J. Greenh. Gas Control 25, 121–140 (2014).
Oskierski, H. C., Dlugogorski, B. Z. & Jacobsen, G. Sequestration of atmospheric CO2 in chrysotile mine tailings of the Woodsreef Asbestos Mine, Australia: quantitative mineralogy, isotopic fingerprinting and carbonation rates. Chem. Geol. 358, 156–169 (2013).
Power, I. M. et al. in Geochemistry of Geologic CO 2 Sequestration Vol. 77 Ch. 9 (eds DePaolo, D. J., Cole, D. R., Navrotsky, A. & Bourg, I. C.) 305–360 (Mineralogical Society of America, 2013). Reviews all aspects of carbon mineralization.
Marini, L. Geological Sequestration of Carbon Dioxide: Thermodynamics, Kinetics, and Reaction Path Modeling (Elsevier, 2007). A compilation of the theoretical background of CO 2 storage.
Blum, A. E. & Lasaga, A. C. Role or surface speciation in the low-temperature dissolution of minerals. Nature 331, 431–433 (1988).
Oelkers, E. H. An experimental study of forsterite dissolution rates as a function of temperature and aqueous Mg and Si concentrations. Chem. Geol. 175, 485–494 (2001).
Pokrovsky, O. S. & Schott, J. Kinetics and mechanism of forsterite dissolution at 25°C and pH from 1 to 12. Geochim. Cosmochim. Acta 64, 3313–3325 (2000).
Rosso, J. J. & Rimstidt, J. D. A high resolution study of forsterite dissolution rates. Geochim. Cosmochim. Acta 64, 797–811 (2000).
Golubev, S. V., Pokovsky, O. S. & Schott, J. Experimental determination of the effect of dissolved CO2 on the dissolution kinetics of Mg and Ca silicates at 25 °C. Chem. Geol. 217, 227–238 (2005).
Olsen, A. A. & Rimstidt, J. D. Oxalate-promoted forsterite dissolution at low pH. Geochim. Cosmochim. Acta 72, 1758–1766 (2008).
Brantley, S. L. & Chen, Y. in Chemical Weathering Rates of Silicate Minerals Vol. 85 (eds White, A. F. & Brantley S. L.) 1767–1783 (Mineralogical Society of America, 1995).
Carroll, S. A. & Knauss, K. G. Dependence of labradorite dissolution kinetics on CO2(aq), Al(aq), and temperature. Chem. Geol. 217, 213–225 (2005).
Metz, V., Amram, K. & Ganor, J. Stoichiometry of smectite dissolution reaction. Geochim. Cosmochim. Acta 69, 1755–1772 (2005).
Oelkers, E. H. & Cole, D. R. Carbon dioxide sequestration: a solution to a global problem. Elements 4, 305–310 (2008).
Kampman, N., Bickle, M., Becker, J., Assayag, N. & Chapman, H. Feldspar dissolution kinetics and Gibbs free energy dependence in a CO2-enriched groundwater system, Green River, Utah. Earth Planet. Sci. Lett. 284, 473–488 (2009).
Pham, V. T. H., Lu, P., Aagaard, P., Zhu, C. & Hellevang, H. On the potential of CO2–water–rock interactions for CO2 storage using a modified kinetic model. Int. J. Greenh. Gas Control 5, 1002–1015 (2010).
Munz, I. A. et al. Mechanisms and rates of plagioclase carbonation reactions. Geochim. Cosmochim. Acta 77, 27–51 (2012).
Hellevang, H., Pham, V. T. H. & Aagaard, P. Kinetic modelling of CO2–water–rock interactions. Int. J. Greenh. Gas Control 15, 3–15 (2013).
Gudbrandsson, S., Wolff-Boenisch, D., Gislason, S. R. & Oelkers, E. H. Experimental determination of plagioclase dissolution rates as a function of its composition and pH at 22°C. Geochim. Cosmochim. Acta 139, 154–172 (2014).
Gislason, S. R. & Oelkers, E. H. Mechanism, rates, and consequences of basaltic glass dissolution: II. An experimental study of the dissolution rates of basaltic glass as a function of pH and temperature. Geochim. Cosmochim. Acta 67, 3817–3832 (2003).
Oelkers, E. H. & Gislason, S. R. The mechanism, rates and consequences of basaltic glass dissolution: I. An experimental study of the dissolution rates of basaltic glass as a function of aqueous Al, Si and oxalic acid concentration at 25°C and pH = 3 and 11. Geochim. Cosmochim. Acta 65, 3671–3681 (2001).
Wolff-Boenisch, D., Gislason, S. R., Oelkers, E. H. & Putnis, C. V. The dissolution rates of natural glasses as a function of their composition at pH 4 and 10.6, and temperatures from 25 to 74°C. Geochim. Cosmochim. Acta 68, 4843–4858 (2004).
Rimstidt, J. D., Brantley, S. L. & Olsen, A. A. Systematic review of forsterite dissolution rate data. Geochim. Cosmochim. Acta 99, 159–178 (2012).
Giammar, D. E., Bruant, R. G. & Peters, C. A. Forsterite dissolution and magnesite precipitation at conditions relevant for deep saline aquifer storage and sequestration of carbon dioxide. Chem. Geol. 217, 257–276 (2005).
Power, I. M., Kenward, P. A., Dipple, G. M. & Raudsepp, M. Room temperature magnesite precipitation. Cryst. Growth Des. 17, 5652–5659 (2017).
Hänchen, M., Prigobbe, V., Baciocchi, R. & Mazzotti, M. Precipitation in the Mg-carbonate system — effects of temperature and CO2 pressure. Chem. Eng. Sci. 63, 1012–1028 (2008).
Qafoku, O. et al. Formation of submicron magnesite during reaction of natural forsterite in H2O-saturated supercritical CO2. Geochim. Cosmochim. Acta 134, 197–209 (2014).
Maroto-Valer, M. M., Fauth, D. J., Kuchta, M. E., Zhang, Y. & Andrésen, J. M. Activation of magnesium rich minerals as carbonation feedstock materials for CO2 sequestration. Fuel Process. Technol. 86, 1627–1645 (2005).
Gislason, S. R., Veblen, D. R. & Livi, K. J. T. Experimental meteoric water-basalt interactions: characterization and interpretation of alteration products. Geochim. Cosmochim. Acta. 57, 1459–1471 (1993).
Xiong, W. et al. CO2 mineral trapping in fractured basalt. Int. J. Greenh. Gas Control 66, 204–217 (2017).
Harrison, A. L., Tutolo, B. M. & DePaolo, D. J. The role of reactive transport modelling in geologic carbon storage. Elements 15, 93–98 (2019).
Aradóttir, E. S. P., Sonnenthal, E. L., Björnsson, G. & Jónsson, H. Multidimensional reactive transport modeling of CO2 mineral sequestration in basalts at the Hellisheidi geothermal field, Iceland. Int. J. Greenh. Gas Control 9, 24–40 (2012).
Snæbjörnsdóttir, S. Ó. et al. The chemistry and saturation states of subsurface fluids during the in situ mineralisation of CO2 and H2S at the CarbFix site in SW-Iceland. Int. J. Greenh. Gas Control 58, 87–102 (2017).
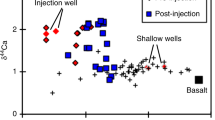
Pogge von Strandmann, P. A. E. et al. Rapid CO2 mineralisation into calcite at the CarbFix storage site quantified using calcium isotopes. Nat. Commun. 10, 1983 (2019).
Trias, R. et al. High reactivity of deep biota under anthropogenic CO2 injection into basalt. Nat. Commun. 8, 1063 (2017).
Gunnarsson, I. et al. The rapid and cost-effective capture and subsurface mineral storage of carbon and sulfur at the CarbFix2 site. Int. J. Greenh. Gas Control 79, 117–126 (2018).
Sigfússon, B. et al. Reducing emissions of carbon dioxide and hydrogen sulphide at Hellisheidi power plant in 2014–2017 and the role of CarbFix in achieving the 2040 Iceland climate goals. Energy Procedia 146, 135–145 (2018).
McGrail, B. P., Spane, F. A., Amonette, J. E., Thompson, C. R. & Brown, C. F. Injection and monitoring at the Wallula basalt pilot project. Energy Procedia 63, 2939–2948 (2014).
McGrail, B. P. et al. Wallula Basalt Pilot demonstration project: post-injection results and conclusions. Energy Procedia 114, 5783–5790 (2017).
The National Academies of Sciences, Engineering, and Medicine. Negative emissions technologies and reliable sequestration: a research agenda (National Academies Press, 2018). A comprehensive review on carbon mineralization prepared by the US National Academy of Sciences.
Zakharova, N. V., Goldberg, D. S., Sullivan, E. C., Herron, M. M. & Grau, J. A. Petrophysical and geochemical properties of Columbia River flood basalt: implications for carbon sequestration. Geochem. Geophys. Geosyst. 13, Q11001 (2012).
Weingarten, M., Ge, S., Godt, J. W., Bekins, B. A. & Rubinstein, J. L. High-rate injection is associated with the increase in US mid-continent seismicity. Science 348, 1336–1340 (2015).
Thorsteinsson, H. & Gunnarsson, G. Induced seismicity – stakeholder engagement in Iceland. GRC Trans. 38, 879–882 (2014).
Juncu, D. et al. Injection-induced surface deformation and seismicity at the Hellisheidi geothermal field, Iceland. J. Volcanol. Geotherm. Res. https://doi.org/10.1016/j.jvolgeores.2018.03.019 (2018).
Zoback, M. D. & Gorelick, S. M. Earthquake triggering and large-scale geologic storage of carbon dioxide. Proc. Natl Acad. Sci. USA 109, 10164–10168 (2012).
Aradóttir, E. S. & Hjálmarsson, E. CarbFix – public engagement and transparency. Energy Procedia 146, 115–120 (2018).
Eiken O. in Geophysics and Geosequestration (eds Davis, T. L., Landrø, M. & Wilson, M.) 209–234 (Cambridge Univ. Press, 2019).
Goldberg, D. S., Lackner, K. S., Han, P., Slagle, A. L. & Wang, T. Co-location of air capture, subseafloor CO2 sequestration, and energy production on the Kerguelen plateau. Environ. Sci. Technol. 47, 7521–7529 (2013).
World Health Organization. Guidelines for drinking-water quality, 4th edition (WHO, 2011).
Flaathen, T. K., Gislason, S. R., Oelkers, E. H. & Sveinbjörnsdóttir, Á. E. Chemical evolution of the Mt. Hekla, Iceland, groundwaters: a natural analogue for CO2 sequestration in basaltic rocks. Appl. Geochem. 24, 463–474 (2009).
Olsson, J., Stipp, S. L. S., Makovicky, E. & Gislason, S. R. Metal scavenging by calcium carbonate at the Eyjafjallajökull volcano: a carbon capture and storage analogue. Chem. Geol. 384, 135–148 (2014).
Goldberg, D. & Lackner, K. Creating negative emissions at remote CO2 sequestration sites. Greenh. Gas Sci. Technol. 5, 238–240 (2015).
Keith, D. W., Holmes, G., St. Angelo, D. & Heidel, K. A process for capturing CO2 from the atmosphere. Joule 2, 1573–1594 (2018).
Gutknecht, V., Snæbjörnsdóttir, S. Ó., Sigfússon, B., Aradóttir, E. S. & Charles, L. Creating a carbon dioxide removal solution by combining rapid mineralization of CO2 with direct air capture. Energy Procedia 146, 129–134 (2018).
Goldberg, D. et al. Geological storage of CO2 in sub-seafloor basalt: the CarbonSAFE pre-feasibility study offshore Washington State and British Columbia. Energy Procedia 146, 158–165 (2018).
Wolff-Boenisch, D. On the buffer capacity of CO2-charged seawater used for carbonation and subsequent mineral sequestration. Energy Procedia 4, 3738–3745 (2011).
Luhmann, A. J. et al. Whole rock basalt alteration from CO2-rich brine during flow-through experiments at 150°C and 150 bar. Chem. Geol. 453, 92–110 (2017).
Schaef, H. T., McGrail, B. P. & Owen, A. T. Carbonate mineralisation of volcanic province basalts. Int. J. Greenh. Gas Control 4, 249–261 (2010).
Aradóttir, E. S. P., Sonnenthal, E. L. & Jónsson, H. Development and evaluation of a thermodynamic dataset for phases of interest in CO2 mineral sequestration in basaltic rocks. Chem. Geol. 304–305, 26–38 (2012).
Husebye, J., Brunsvold, A. L., Roussanaly, S. & Zhang, X. Techno economic evaluation of amine based CO2 capture: impact of CO2 concentration and steam supply. Energy Procedia 23, 381–390 (2012).
Webb, R. M. & Gerrard, M. B. Overcoming impediments to offshore carbon dioxide storage: legal issues in the U.S. and Canada. Environ. Law Rep. 49, ELR10634 (2019).
European Commission. Report from the Commission to the European Parliament and the Council. Report on the functioning of the European carbon market (European Commission, 2019).
Folger, P. Carbon capture and sequestration (CCS) in the United States (Congressional Research Service, 2018).
Gaillardet, J., Dupré, B., Louvat, P. & Allégre, C. J. Global silicate weathering and CO2 consumption rates deduced from the chemistry of the large rivers. Chem. Geol. 159, 3–30 (1999).
Sanna, A., Uibu, M., Carmanna, G., Kuusik, R. & Maroto-Valer, M. M. A review of mineral carbonation technologies to sequester CO2. Chem. Soc. Rev. 43, 8049–8080 (2014). Reviews the potential of in situ and ex situ storage through mineral carbonation.
Mervine, E. M. et al. Potential for offsetting diamond mine carbon emissions through mineral carbonation of processed kimberlite: an assessment of De Beers mine sites in South Africa and Canada. Mineral. Petrol. 112, 755–765 (2018).
Harrison, A. L., Power, I. M. & Dipple, G. M. Accelerated carbonation of brucite in mine tailings for carbon sequestration. Environ. Sci. Technol. 47, 126–134 (2012).
Power, I. M. et al. Strategizing carbon-neutral mines: a case for pilot projects. Minerals 4, 399–436 (2014).
Hartmann, J. & Moosdorf, N. The new global lithological map database GLiM: a representation of rock properties at the Earth surface. Geochem. Geophys. Geosyst. 13, Q12004 (2012).
Whittaker, J. M. et al. Long-term interaction between mid-ocean ridges and mantle plumes. Nat. Geosci. 8, 479–483 (2015).
Müller, R. D. et al. Ocean basin evolution and global-scale plate reorganization events since Pangea breakup. Annu. Rev. Earth Planet. Sci. 44, 107–138 (2016).
Johansson, L., Zahirovic, S. & Müller, R. D. The interplay between the eruption and weathering of Large Igneous Provinces and the deep-time carbon cycle. Geophys. Res. Lett. 45, 5380–5389 (2018).
Bales, R. C. & Morgan, J. J. Dissolution kinetics of chrysotile at pH 7 to 10. Geochim. Cosmochim. Acta 49, 2281–2288 (1985).
Gudbrandsson, S., Wolff-Boenisch, D., Gislason, S. R. & Oelkers, E. H. An experimental study of crystalline basalt dissolution from 2 ≤ pH ≤ 11 and temperatures from 5 to 75°C. Geochim. Cosmochim. Acta 75, 5496–5509 (2011).
Critelli, T. et al. Dissolution rates of actinolite and chlorite from a whole-rock experimental study of metabasalt dissolution from 2 ≤ pH ≤ 12 at 25°C. Chem. Geol. 390, 100–108 (2014).
Gebald, C., Repond, N. & Wurzbacher, J. A. Steam assisted vacuum desorption process for carbon dioxide capture. US Patent 2017/0203249 (2015).
Jackson, S. & Brodal, E. A comparison of the energy consumption for CO2 compression process alternatives. IOP Conf. Ser. 167, 012031 (2018).
Acknowledgements
S.Ó.S., B.S., C.M., S.R.G. and E.H.O. work on the CarbFix project funded by the European Union’s Horizon 2020 research and innovation programme under grant agreement 764760 (CarbFix2), 818169 (GECO) and 764810 (S4CE). D.G. works on the Solid Carbon project funded by the Pacific Institute for Climate Solutions. The authors thank E.S. Aradóttir Pind, R.B. Bragadóttir, K. Helgason, M. Voigt and R. Þrastarson for discussions and assistance in developing Fig. 2.
Author information
Authors and Affiliations
Contributions
The authors contributed equally to all aspects of the article.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests
Additional information
Peer review information
Nature Reviews Earth & Environment thanks G. Gadikota and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Snæbjörnsdóttir, S.Ó., Sigfússon, B., Marieni, C. et al. Carbon dioxide storage through mineral carbonation. Nat Rev Earth Environ 1, 90–102 (2020). https://doi.org/10.1038/s43017-019-0011-8
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s43017-019-0011-8
This article is cited by
-
Accelerating the climate transition through scientist-led CO2 management pilot projects
Nature Chemical Engineering (2024)
-
Unraveling the rapid CO2 mineralization experiment using the Paraná flood basalts of South America
Scientific Reports (2024)
-
Simulation of CO2 Storage Using a Parameterization Method for Essential Trapping Physics: FluidFlower Benchmark Study
Transport in Porous Media (2024)
-
Advancement of Carotenogenesis of Astaxanthin from Haematococcus pluvialis: Recent Insight and Way Forward
Molecular Biotechnology (2024)
-
Replacement reactions for carbon geosequestration may be faster in calcium olivine vs magnesium olivine
Communications Earth & Environment (2023)
