
Abstract
Diseases that have a complex genetic architecture tend to suffer from considerable amounts of genetic variants that, although playing a role in the disease, have not yet been revealed as such. Two major causes for this phenomenon are genetic variants that do not stack up effects, but interact in complex ways; in addition, as recently suggested, the omnigenic model postulates that variants interact in a holistic manner to establish disease phenotypes. Here we present DiseaseCapsule, as a capsule-network-based approach that explicitly addresses to capture the hierarchical structure of the underlying genome data, and has the potential to fully capture the non-linear relationships between variants and disease. DiseaseCapsule is the first such approach to operate in a whole-genome manner when predicting disease occurrence from individual genotype profiles. In experiments, we evaluated DiseaseCapsule on amyotrophic lateral sclerosis (ALS) and Parkinson’s disease, with a particular emphasis on ALS, which is known to have a complex genetic architecture and is affected by 40% missing heritability. On ALS, DiseaseCapsule achieves 86.9% accuracy on hold-out test data in predicting disease occurrence, thereby outperforming all other approaches by large margins. Also, DiseaseCapsule required sufficiently less training data for reaching optimal performance. Last but not least, the systematic exploitation of the network architecture yielded 922 genes of particular interest, and 644 ‘non-additive’ genes that are crucial factors in DiseaseCapsule, but remain masked within linear schemes.
Similar content being viewed by others

Main
Amyotrophic lateral sclerosis (ALS) is a rare primary neurodegenerative syndrome characterized by human motor system degeneration. So far, ALS is still not curable, but symptomatic treatment can significantly improve life quality and survival of the affected1. As the diagnosis of ALS often comes at a considerable delay2, most patients miss the advantageous opportunities of early intervention3; for example, recent studies show that NAD+ replenishment can improve clinical features of patients with ALS, indicating an encouraging potential novel treatment for ALS4,5. These explain why efficient methods and tools for predicting the prevalence and occurrence of ALS have life-saving potential.
Various studies6,7,8 have demonstrated that ALS is a complex disorder that has an encompassing genetic background9 and its heritability amounts to 50% (ref. 10). However, the genetic variants delivered by genome-wide association studies (GWAS) have been amounting to only 10% of the heritability11 of ALS. It is therefore reasonable to assume that the missing heritability of ALS amounts to approximately 40%.
The striking amount of missing heritability, or even the idea that genotype–phenotype relationships are based on the omnigenic12,13 and not the polygenic model—where only the polygenic model renders the concept of missing heritability a truly reasonable concept—is supposed to be among the major reasons for the poor prognosis of ALS. In summary, the major methodological challenges are as follows: (1) the association signals of complex diseases can spread across most of the genome instead of involving just a few core pathways12,14, which means that the omnigenic model applies; (2) when following the polygenic model, the corresponding linear models that underlie the GWAS analysis techniques cannot detect non-additive genetic effects such as epistasis15,16. Several studies have modelled gene–gene interactions17,18,19. However, only a few of them have been directly applied to predicting the prevalence of complex genetic diseases.
Methods that aim to exploit non-additive relationships have left behind various open questions. When being based on statistical hypothesis testing, they tend to suffer from a lack of power. On the other hand, approaches that are based on potentially omnigenic machine learning models are relatively rare, and so far have left ample room for improvements.
Deep learning, as a predominant machine learning approach, has established the state of the art in many areas. Extensions of the universal approximation theorem20 provide a theoretical basis for the insight that not only the width of the layers but also the depth of the network is crucial for reaching superior levels of prediction accuracy21,22. The intuitive idea is to detect and arrange patterns in a hierarchical way, which leads to elevated levels of resolution when mapping the data23.
Convolutional neural networks (CNNs) reflect network architectures that are particularly suitable to implement the idea of hierarchies of patterns 24. While CNNs such as AlexNet22, VGG25, ResNet26 and DenseNet27 indeed achieved the breakthrough successes in deep learning, major criticisms with regard to interpretability (‘deep black boxes’)28 and the enormous demand for training data for reaching superior performance29 had been remaining. However, in clinical applications, data can be expensive, and the inability to explain lets one remain with ethical concerns30,31.
Capsule networks (CapsNets)32,33 were presented as a remedy for addressing such critical issues. The major motivation for CapsNets was to classify distorted or entangled patterns in images correctly. The improved modelling of spatial hierarchies, as empowered by the ‘viewpoint invariance property’, led to major improvements with respect to the accuracy of the predictions. Beyond that, the ‘viewpoint invariance property’ is the likely reason for the reduced requirements in terms of training data that one observed in comparison with CNNs. Further, although not primarily intended, CapsNets also enabled a human-mind-friendly interpretation of results.
Therefore, CapsNets have shown to have the potential to resolve two issues of primary concern in biomedical applications. Recent studies indicate that the potential of CapsNets to learn complex hierarchical structures can indeed be leveraged also for biomedical data34,35. These applications of CapsNets add to the earlier (innumerous) applications36,37,38,39,40 of ordinary CNNs or machine learning models in biology or medicine.
There also have been recent applications of deep learning when predicting phenotype from genotype profiles, such as the detection of epistasis41,42, or the prediction of ALS from genotype profiles of the promoter regions from four chromosomes43. Last but not least, an approach was presented that models non-linearities within the range of LD (linkage disequilibrium) blocks, while joining effects of LD blocks in a linear manner44, which prevents recognition of non-linear interactions across LD blocks.
The omnigenic model12 requires the modelling of gene–gene interactions across the entire genome, in a way that allows genes to interact in non-additive ways for establishing their joint effects. From this point of view, none of the approaches presented so far in the literature builds on the omnigenic model as its foundation.
Our approach will be the first approach to model whole-genome wide, non-additive interactions between genes. For that, it takes a route that one can consider the opposite of the protocol presented in ref. 44: we summarize local (gene-range) effects of variants linearly, and then combine the local effects in non-additive ways globally (across the entire genome). In this Article, we present a deep neural network-based approach that caters to the omnigenic model as a conceptual basis. More specifically, from a methodical point of view, we present the first approach that employs capsule networks, as an advanced deep neural network class of functions, to map genotypes onto phenotypes.
We refer the interested reader to Supplementary Note 1 for full details on arguments referring to deep learning and capsule networks raised in the introduction.
Results
We present DiseaseCapsule as a framework that can handle genome-scale variant input and reveal non-additive interactions across genes. Please see Supplementary Note 2 for an overview of the approach and a summarizing account of the results; here, for the sake of reproducibility, we will report results in sufficient detail. For all methodical details, see Methods.
Workflow
DiseaseCapsule implements a two-step approach to successfully deal with genome-scale variant screens. The first step consists of a novel protocol to perform dimensionality reduction. This protocol enables to capture millions of polymorphic loci in a way that enables subsequent application of non-additive methods. The second step then is the application of capsule networks, as a fundamentally non-additive model, to the reduced data. These two steps are preceded by basic quality control (QC) and batch effect removal, as well as a basic step to filter out variants that do not matter (regardless of the underlying approach). For an illustration, see Fig. 1. In the following, we describe the essence of the two steps, and refer the reader to Methods for full methodical details. In the following, the data on which DiseaseCapsule and competing methods are validated refers to 10,405 DNA-array-based, whole-genome genotype samples from the Dutch cohort of Project MinE8,11,45.

In the tables for each gene, S1, S2, ..., Si represent sample IDs, and PC1, PC2, ..., PCk represent the 1, 2, ..., k-th principal component of each Gene-PCA, respectively. The number of PC is 8 or 4 or 1, which depends on the length of the input, that is, the number of SNPs.
For the first step, the challenge is the fact that the application of principal component analysis (PCA) across the polymorphic loci of the entire genome—which is the standard protocol to reduce the dimensionality of the data—annihilates the effects of subsequent application of genome-wide, non-linear models in so far as non-linear interactions between global, linear combinations of variants remain meaning less for the analyst. Our solution is to apply linear techniques, such as PCA, only for small, biologically well-defined functional units of the genome (that is, genes). As linearization happens only within small regions of the genome, non-linear interactions across such small regions can still be detected. Beyond this theoretical reasoning, our experiments confirm these ideas by revealing the idea of only local PCA as the considerably stronger approach (Table 1 and Supplementary Tables 1 and 2). For more details about descriptions of the challenge and the solution, see Supplementary Note 3.
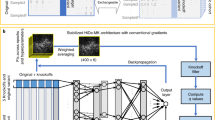
For the second step, see Fig. 2 for an illustration and a brief description of the architecture of DiseaseCapsule; for full details, see Methods.

The input is the concatenation of the compressed features from all Gene-PCA models, where each feature corresponds to one Gene-PCA. The number of Gene-PCAs is 75,584, so the dimensionality of the input is 75,584 × 1. DiseaseCapsule consists of three layers: a fully connected layer (FC), a primary capsule layer (PrimaryCaps) and a phenotype capsule layer (PhenoCaps). The FC layer consists of 150 neurons followed by ReLU as activation function. The PrimaryCaps is composed of 32 primary capsules. Each of them involves four different convolutional filters (kernel size 5 × 1, stride 2, no padding). PhenoCaps consists of two 16-dimensional vectors. Each phenotype capsule receives input from all 32 primary capsules. The output is a binary classification label (Healthy or ALS).
Predicting ALS: Gene-PCA + DiseaseCapsule yields optimal performance
For the following, see Table 1. We evaluated the performance of DiseaseCapsule with various state-of-the-art approaches, including predominant machine learning approaches and polygenic risk scores (PRS). To further evaluate the contribution of the novel gene-scale dimensionality reduction protocol, we combined each method with standard genome-scale PCA (‘All-PCA’) on the one hand, and the novel, local PCA-based protocol (‘Gene-PCA’) on the other hand. We discuss results briefly in the following; for full details, see Supplementary Note 4.
The first observation is that all neural network-based (so fully non-additive) models profit from using ‘Gene-PCA’ instead of ‘All-PCA’: the accuracy of DiseaseCapsule, multilayer perceptron (MLP) and CNN increases from 81.9 to 86.9, from 72.5 to 84.2 and from 53.8 to 74.5, respectively. All other approaches are run on ‘Gene-PCA’ or ‘All-PCA’ at near-identical performance. This points out that Gene-PCA predominantly caters to neural network models. From a conceptual point of view, this was to be anticipated, because Gene-PCA preserves the potential to detect non-linearities across genes, whereas ordinary PCA does not. Since applying local linearization before a linear model-based analysis still results in an approach that is linear overall, as the concatenation of two linear functions, linear methods cannot profit from Gene-PCA; still they fail to pick up non-linearities.
In an overall account, DiseaseCapsule achieves a prediction accuracy of 86.9, which establishes the top performance, rivalled only by MLP, as an alternative non-additive approach (84.2). The third-best performance is achieved by DiseaseCapsule on ‘All-PCA’ (81.9), closely followed by PRS at a GWAS threshold of 5 × 10−2 (81.8). All other performance rates drop below 79. This means in particular that, in a relative comparison with PRS, as a standard prediction technique, DiseaseCapsule leaves 28% fewer individuals misclassified, which establishes considerable, relevant progress, both from the point of view of clinical applications and from the point of view of predictive power in general. It also establishes a first quantification of the contribution of non-additive constellations of variants/genes to identifying ALS (for further analyses, see Supplementary Note 4).
DiseaseCapsule also clearly reveals itself as the most balanced and strongest approach overall: DiseaseCapsule achieves precision and recall of 85.2 and 89.4, respectively, which combines into an F1 score of 87.2, which is unrivalled by the other approaches.
In addition, results show that, compared with other classifiers, DiseaseCapsule is less sensitive to batch-induced or cohort-specific confounding effects (Supplementary Table 3) and needs less data for training (Supplementary Table 4). For a fully detailed discussion of the corresponding experiments, see Supplementary Notes 5 and 6.
Validating DiseaseCapsule on PD
We also validated the predictive performance of DiseaseCapsule in Parkinson’s disease (PD) data46,47,48,49, following the exact same protocol as for ALS. In a summary of results, in analogy to ALS, Gene-PCA + DiseaseCapsule outperforms all other approaches in terms of accuracy (62.0%), recall (68.1%) and F1 score (64.2%) in PD (Supplementary Table 5). For more details, see Supplementary Note 7. The loss of accuracy in comparison with ALS, observed for all methods, can be attributed to the relatively small amounts of polymorphic loci inspected for the PD cohorts, which has a clear impact on the expressiveness of all models.
Increasing number of genes improves classification
For the classification performance of DiseaseCapsule when varying the number of genes, see Supplementary Fig. 1, and for full details, see Supplementary Note 8. It is immediately evident that increasing the number of genes improves results in all aspects. This arguably supports the hypothesis that the omnigenic model12 is in effect. It remains to design a strategy through which to select the genes that are most relevant for classification; most likely, such genes have key roles in establishing or preventing the disease.
Determining genes decisive for classification
For explanations in the following, see Extended Data Fig. 1 and see ‘The architecture and parameters of DiseaseCapsule’ and ‘Model interpretation’ subsections in Methods. While i indexes primary capsules, j indexes higher-level (‘phenotype’) capsules. In the DiseaseCapsule network, the vectors of the two phenotype capsules ‘ALS’ and ‘Healthy’ (sj in Extended Data Fig. 1) consist of linear combinations of output vectors provided by the primary capsules (uj∣i in Extended Data Fig. 1). The linear weights cij that connect the sj with the uj∣i are referred to as coupling coefficients. Unlike ordinary parameters of the network (for example, Wij in Extended Data Fig. 1), the cij are not learned (using backpropagation), but determined through the dynamic routing procedure, as a novel and characteristic component of capsule networks. The dynamic routing algorithm induces situations that favour only few—often even only one—large cij over several equally large cij for each i. This means that each primary capsule predominantly ‘routes’ its output to only few or even only one higher-level capsule.
Primary capsules that have great coupling coefficients in connection with the ‘ALS’ capsule are likely to code for constellations of genes that drive the disease. Vice versa, primary capsules sharing links with the ‘Healthy’ output capsule that are equipped with large coupling coefficients code for genes whose activation (or de-activation) distinguishes healthy individuals from the ones affected with ALS.
Primary capsules that predominantly route their output to one of the phenotype capsules, ‘ALS’ or ‘Healthy’, are crucial factors for the classification process. We therefore investigated how primary capsules related to the ‘ALS’ output capsule (which predominantly fires when ALS is to be predicted) on the one hand, and the ‘Healthy’ output capsule (which predominantly fires when the individual is to be classified as not being affected by ALS) on the other hand.
To highlight primary capsules that share exceptionally large coupling coefficients with one of the two phenotype capsules, we ran all training and all test samples through the network, amounting to two separate runs, one for the training and one for the test data. The intuition is to demonstrate that, despite not having been part of the training, effects reproduce on data that had not been seen before. We collected the resulting coupling coefficients; we recall that coupling coefficients are computed individually for each sample by way of the ‘dynamic routing’ protocol during the forward pass32. If coupling coefficients were determined as part of backpropagation during training, coupling coefficients would be equal for all individuals. We averaged the resulting coupling coefficients across all samples, for each of the 2 × 32 possible combinations of PrimaryCaps and PhenoCaps. As above-mentioned, we did this for both training and test data. The corresponding averaged coupling coefficients are displayed in the two heat maps in Fig. 3, where Fig. 3a is for the training data and Fig. 3b is for the test data. For further details on the experiments and the corresponding visualization process, see also Methods.

a, Only plot for training individuals. b, Only plot for test individuals. The x axis and y axis represent the 32 primary capsule groups and 2 phenotype capsules, respectively. All 18,279 genes involved in DiseaseCapsule model are retained when predicting on test samples.
The most striking effect is that primary capsule 5 establishes the strongest link to the ‘ALS’ output capsule, both for training and test data. Far lesser so, but still apparent, primary capsule 28 activates the ‘ALS’ capsule. The agreement between training and test data demonstrates that effects do not only get manifested on data that were used to establish the parameters of the network (namely, the training data). In summary, activation of primary capsule 5 is the by far predominant effect from which to determine whether an individual is affected with ALS.
Correspondingly, we developed an algorithmic protocol according to which to determine core genes that markedly contribute to the activation of primary capsule 5; for details, see Methods. Using this protocol, we obtained 922 core genes that contribute to the activation of primary capsule 5 (Fig. 4a). This means that these 922 genes are important for DiseaseCapsule to identify the occurrence of ALS. To validate the predictive power of these 922 genes, we masked all other genes in the test data (that is, we set entries in the input vector to zero if referring to genes not among the 922 selected ones), and ran the modified test data through the (trained) DiseaseCapsule model. As a result, using these 922 genes alone—which as we recall are crucial for activating primary capsule 5—a test accuracy of 76% was achieved. Note that random selections of 922 genes yielded 70% accuracy on average, with a standard deviation of 1%; for corresponding results, see Fig. 4b. So, the 76% achieved by the genes selected through our protocol is significantly greater (P < 2.2 × 10−16).

a, The distribution of coupling coefficients between primary capsule 5 and phenotype capsule ALS for all genes. The red dashed line indicates the 95th percentile. A total of 922 genes whose coupling coefficients are above the 95th percentile are selected as core genes decisive for classification. The vertical coordinates adopt scientific notation (×106). b, Test accuracy distribution of using 922 randomly chosen genes as input for DiseaseCapsule model (repeat 1,000 times), while the other genes are masked (set as zero). The red dashed line indicates the test accuracy of using 922 core genes as input. c, Heat map for average coupling coefficient matrices (test data) using 922 core genes as input.
To further corroborate the predictive power of the 922 selected genes, we computed the average of the coupling coefficients across all test individuals when using only the 922 preferentially selected core genes, see Fig. 4c. The level of activation of primary capsule 5 (0.0342) nearly matches the one when using all genes (0.0399).
It therefore made sense to examine and classify these 922 genes further; for the resulting list, see Supplementary Table 6. In summary, when examining these 922 genes, we found some overlapping genes with the Amyotrophic Lateral Sclerosis online Database (ALSoD) and other studies 44. Additionally, the collection of enriched Gene Ontology terms and pathways have been shown to significantly relate with human nervous system related diseases, which do include ALS in most cases50,51,52,53,54,55. For full details, see Supplementary Note 9.
644 ‘non-additive’ genes
We designed a simple target function that, for a selection of genes, measures the difference between the genes supporting DiseaseCapsule and the genes supporting a common logistic regression scheme, in terms of accurate classification. This difference is quantified by the difference in accuracy that one achieves when running DiseaseCapsule using these genes alone, on the one hand, and when running the logistic regression scheme using these genes alone, on the other hand. Employing a genetic algorithm, we determined a subset of 644 genes (Supplementary Table 7) that yields a maximum of that target function. So, running methods on these 644 genes maximizes the difference between the accuracy achieved in the non-additive scheme (DiseaseCapsule) and the accuracy achieved in the additive scheme (regression). For full details, see Methods. In other words, these 644 genes remain useless when being used in a linear scheme, but decisively contribute to classification in DiseaseCapsule, as a non-additive scheme; more than that, these 644 genes reflect a selection that is optimal in that respect.
To reinforce that these 644 ‘non-additive’ genes are responsible for predominantly non-additive effects, we ran both DiseaseCapsule (Gene-PCA + DiseaseCapsule in Table 1) and logistic regression (Gene-PCA + logistic regression, which proved the strongest additive protocol; Table 1), on both training (which led to establishing parameters for both DiseaseCapsule and logistic regression) and test data (hitherto unseen by either approach). The differences in accuracy are striking, on both training (difference 0.227) and test (difference 0.162) data.
The difference between training and test data may be due to hidden biases. To prevent such biases, and ensure that these 644 genes nearly exclusively yield non-additive effects—which can only be picked up by DiseaseCapsule—we retrained both DiseaseCapsule and logistic regression on these 644 genes alone. This gives both approaches the same fair chance: try to get the most out of the 644 genes they were provided with.
For the corresponding results, see Table 2. Retraining both models yields accuracies of 0.712 for DiseaseCapsule, but only 0.512 for logistic regression (amounting to a difference of exactly 0.2, matching the range of the earlier results). Note that because test data were balanced (equal cases and controls), 0.5 means random performance. So, on these 644 genes, logistic regression matches random classifier rates, while DiseaseCapsule achieves excellent performance rates.
Breaking down results in terms of precision and recall confirms that DiseaseCapsule establishes decent performance rates (precision 0.715; recall 0.706), whereas logistic regression’s performance does not even match minimum standards (precision 0.522; recall 0.292(!)). For functional annotations of the 644 non-additive genes, see Supplementary Note 10.
Discussion
In this study, we have presented DiseaseCapsule, as a novel deep-learning-based approach to infer disease phenotype from individual genotype. As the major novelty, DiseaseCapsule captures non-linear, potentially arbitrarily complex functional relationships between genotype and phenotype across the whole genome. All prior approaches presented so far considered to evaluate variants in additive schemes (which reflects the common standard), presented approaches that consider non-additivity only within small local regions of the genome, or resorted to considering only a few genes, or some selected chromosomes when operating in more global ways.
DiseaseCapsule has come with two immediate theoretical advantages. First, because it operates across the whole genome, DiseaseCapsule does not need to focus exclusively on a few core disease-related genes, so it does not miss the weak effects of abundant peripheral genes. Second, DiseaseCapsule improves on capturing the hierarchical structures of the underlying genetic interactions thanks to the high degree of complexity that capsule networks can capture. This plays a particularly relevant role for ALS, because ALS is commonly hypothesized to have a complex genetic architecture.
In practical terms, DiseaseCapsule has outperformed all state-of-the-art approaches when predicting ALS from individual genotype: it has achieved 87% accuracy on hold-out test data. This translates into a relative increase of 28% over PRS, so it remains with 28% fewer misclassified people in comparison with the current clinical standard. This establishes substantial (arguably even drastic) advantages in comparison with what was possible earlier. Analysing results further has revealed that DiseaseCapsule achieves 89.4% recall, which reflects that DiseaseCapsule identifies 64% of the patients with ALS who remain undiscovered according to clinical standards (PRS 70.2%), so it may miss the advantages of early intervention according to current practice.
DiseaseCapsule has also redeemed its two major theoretical promises for application in clinical practice: sustainable use of training input, which reduces costs and efforts when raising clinical data, as well as advances in terms of interpretability of predictions. The latter point has become obvious through experiments based on inspecting the individual capsules of the network. The experiments have revealed 922 candidate genes for being associated with ALS, many of which had not been pointed out before following standard GWAS protocols; note that all of them appear to be plausible according to their functional annotations.
Realizing these advantages has required overcoming various non-negligible technical hurdles. First, integrating whole-genome data means dealing with feature spaces whose dimensions are in the millions, which corresponds to the amount of polymorphic sites in the human genome. Here we have developed a protocol that yields gene-specific principal components. These gene-specific principal components can then be combined in non-linear ways to reflect non-linear interactions across genes, where non-linearities can span the entire genome.
Secondly, capsule networks had never been applied to whole-genome genotype data before. We have enabled this by means of an architecture that uses fully connected, instead of convolutional layers as early layers in the capsule networks. This preserves to capture interactions between genes across the whole genome to a maximal degree, and appropriately accommodates the sequential nature of the input.
While determining the exact reasons for the superiority of DiseaseCapsule in predicting ALS from genotype still requires further investigation, some plausible hypotheses can be raised already.
First, as already alluded to above, capsule networks have the potential to learn the intrinsic hierarchical structures that underlie the data. The enhanced capability to analyse complex biological relationships that underlie diseases (see also ref. 34) explains the improved generalization over other models.
Second, DiseaseCapsule is able to pick up non-linear genetic interactions between variants (for example, epistasis) that have remained overlooked by the standard approaches.
To further investigate this hypothesis, we considered an objective function that addressed to find genes that supported classification in (the non-linear) DiseaseCapsule, but not in linear regression type schemes. Optimizing according to this objective function (as per a genetic algorithm) yielded a subset of 644 genes that significantly contributed to predicting ALS in DiseaseCapsule (accuracy 71.2%), while not working within the frame of logistic regression (accuracy 51.2%). Evidently, linear approaches remain blind to these 644 genes. Although these 644 genes still require further inspection, it is reasonable to assume that various genes among them have the potential to be of future use in exploring the heritability of ALS further.
Third, one can hypothesize that ALS follows an omnigenic model12 to a non-negligible degree. Results for PRS corroborate this idea. Gradually relaxing the threshold for inclusion of variants from 5 × 10−8 to 5 × 10−2 increases the accuracy by 20%. This means that numerous variants of small effect contribute to establishing ALS, in addition to the core disease-causing genes. DiseaseCapsule follows a whole-genome approach that does not put significance thresholds on individual variants (or genes) to appropriately take this into account.
As already alluded to above, DiseaseCapsule requires less training data than other approaches to establish excellent performance. While the effects are obvious, the translation of the ‘viewpoint invariance property’ into the realm of genes and diseases still needs to be provided. It is reasonable to hypothesize that capsule networks capture the core effects regardless of their distribution across the ancestors of the individuals, and their possible interference. Instead, other approaches may become confused by interfering pathways, so they need to be presented with all possible combinations of interacting pathways before reasonable conclusions can be drawn.
Of course, several open questions have been remaining, some of which point to further promising avenues of research. Such immediate ideas are to also integrate epigenetic information, for example, or adapt models to haplotype data, so as to use phasing information whenever available.
In addition, it appears sensible to develop a formal protocol for machine-learning-based approaches by which to identify (combinations of) variants that are associated with the diseases/phenotypes one examines. While such formal association schemes do not yet exist, we have suggested clear steps towards that goal. For example, DiseaseCapsule has already been able to deliver 922 genes that may be associated with ALS, which deserve to be investigated further.
In addition, fortunately, the field of explainable deep neural networks is moving fast. So, it is reasonable to expect that one will understand the molecular mechanisms that drive diseases from examining the non-linear deep-learning-based approaches further in the nearer future. This then will aid practitioners in their assessment of strategies for preventing and treating the diseases.
Methods
Data: Project MinE
The data we used are from Project MinE (https://www.projectmine.com), a large-scale study that aims to reveal the epi-/genetic mechanisms that underlie ALS, in the frame of globally concerted collaboration45. The data we use in this project are from the Dutch cohort of the project. We have complied with all relevant ethical regulations, and informed consent was obtained from participants. The data contain 7,213 healthy (also known as control) individuals and 3,192 individuals affected with ALS. The cohort counts 5,208 females and 5,197 males. All participants of the study were genotyped using Illumina 2.5M single nucleotide polymorphism (SNP) array.
QC
First, we annotated SNPs according to dbSNP137 and mapped them to hg19 as the reference genome. We first performed QC so as to remove low-quality SNPs and individuals of low quality overall, by using PLINK 1.9 (refs. 56,57) (-geno 0.1 and -mind 0.1). We stratified individuals according to the genotyping platform, and subsequently performed a more stringent SNP QC (-geno 0.0, -maf 0.01, -hwe 1e-5 midp include-nonctrl). We kept only SNPs in autosomal regions, and filtered on the basis of differential missingness (-test-missing midp), excluding SNPs of a P value over 1 × 10−4. As a result, we obtained 4,370,685 SNPs, all of which are contained in the intersection the four batches (see below) the data consist of (Supplementary Fig. 2).
Batch structure and batch effects
The dataset consists of four batches, pertaining to technical identifiers C1, C3, C5 and C44. The number of samples and the ratio of cases versus controls can vary substantially across batches: C1 contains 225 cases and 380 controls, C3 is 130 cases and 49 controls, and C5 no cases but 5,155 controls, whereas C44 finally contains 2,387 cases and 1,629 controls (Supplementary Table 8). It is important to realize that C5 and C44, both of which are highly imbalanced—C5 contains no cases, while the majority of C44 are cases—cover approximately 92.5% of the samples, and thus dominate the dataset.
This points to the importance of removing batch effects. Otherwise, predicting cases and controls can be confounded with predicting C44 from C5, at still decent performance rates.
To remove artefacts by which to distinguish C44 from C5, we considered only the 5,155 and 1,629 healthy individuals from C5 and C44, respectively. Any SNP that supports discrimination between C5 and C44 healthy individuals can be identified as signalling batch-induced effects, and thus should be removed from further analysis.
To filter for batch-effect-transporting SNPs, we computed a 2 × 2 contingency table for each SNP, where rows represent alleles (major or minor allele) and columns represent batches (C5 or C44). Entries in this table reflect allele counts per batch. Subsequently, we performed a Pearson chi-squared test on each table, and thereby obtained a P value for each SNP. Small P values indicated that the particular SNP transports batch effects. To correct for multiple testing, we used the Benjamini–Hochberg procedure 58 and filtered all SNPs according to their adjusted P values, removing SNPs with an adjusted P value of less than 0.05. Filtering out 8,664 potentially batch-effect-signal-carrying SNPs this way, we remained with 4,362,021 SNPs from 22 autosomes.
Dimensionality reduction
GWAS for SNP selection
The dimension of the feature spaces amounts to 4,362,021, agreeing with the number of SNPs that passed quality and batch effect control. On the one hand, this means that the number of SNPs one can work with is sufficiently high to transport relevant meaning. On the other hand, it means that the number of features is too large for machine learning approaches to not overfit. Therefore, as usual, the dimensionality of the data needs to be reduced for machine learning approaches to generalize, while preserving the expressiveness of the original set of 4,362,021 features.
To this end, we performed a GWAS using PLINK v1.9 (ref. 56), and discarded SNPs that are very unlikely to carry disease-status-related signals. This reasoning is based on the fact that every SNP must carry an—albeit potentially rather weak—signal in its own right. Using only the training data (see below for descriptions of training, validation and test data), in agreement with the fact that regular GWAS makes use of disease status labels and thus classifies as part of the training process, we discarded all SNPs whose P values were greater than 0.05. Note that a threshold of 0.05 is considerably relaxed relative to the stringent threshold of 5 × 10−8 (ref. 59) that is used in regular GWAS. Unlike in regular GWAS, however, we would like to keep as many potentially associated SNPs. So, we do not discard all SNPs whose individual signals are too weak in their own right, knowing that weak individual signals can accumulate to strong signals where SNPs interact in possibly non-additive constellations in the deep learning models that we use. As a result, 505,333 SNPs were retained in this step (Supplementary Fig. 3); signals of all SNPs discarded were found to be too weak to potentially play a role.
We further annotated all 505,333 SNPs using ANNOVAR (24 October 2019; latest version)60, assigning them to genes (‘gene-based annotation’) based on the human reference genome (hg19). Genes were defined using the NCBI Reference Sequence (RefSeq) database. Each SNP could be assigned to at least one gene. When annotating SNPs with more than one gene, we kept track of the corresponding mapping relationships: if annotated as ‘intergenic’, SNPs were assigned to only the nearest gene. If annotated as non-intergenic with variant functions in different genes, we assigned the SNP to all related genes. As a result, SNPs were annotated with 18,279 genes overall, where the vast majority of genes have less than 200 SNPs annotated (Supplementary Fig. 4).
As usual, we also transformed genotype data according to minor allele information. Each SNP corresponds to a value i ∈ {0, 1, 2} in each individual, where i is the minor allele count at the particular polymorphic site in the individual.
PCA
PCA61 has been widely applied to exploit SNP data and demonstrated great effectiveness62. We recall that whole-genome PCA is not applicable for non-additive approaches to properly work, while gene-based PCA preserves to detect non-additive variant patterns across different genes. In agreement with that reasoning, we performed PCA for each collection of SNPs that became assigned to identical genes. In the following, we refer to that procedure as Gene-PCA.
Correspondingly, for each gene g, we constructed a matrix Ag ∈ {0, 1, 2}m×n where m = 10,405 is the total number of individual samples, and n ∈ 1, ..., 1,383 corresponds with the number of SNPs that became assigned to gene g; note that the maximum amount of SNPs assigned to a gene is 1,383, where however the number of genes with more than 200 SNPs assigned was small (see above). For an illustration, see Fig. 1.
PCA can remove noise and generate a robust compressed representation from input features. PCA is efficiently implemented on the basis of singular value decomposition: Ag is factorized into three matrices
Here Σ is a k × k (usually k ≪ n) rectangular diagonal matrix of singular values σk; importantly, k is the number of principal components one will use. U and V are matrices whose columns are orthogonal unit vectors called the left and the right singular vectors of A, respectively. To take into account that the input dimension n varies, we varied k, relative to n, accordingly: k = 8 for n > 20, k = 4 for 4 < n ≤ 20 and k = 1 for 1 ≤ n ≤ 4.
The reduction in dimension we achieve through this procedure is from 505,333 down to 75,584, where each of the 75,584 dimensions corresponds to a principal component computed for one of the 18,279 genes that had become annotated with potentially relevant SNPs, as discussed above.
The architecture and parameters of DiseaseCapsule
DiseaseCapsule is a neural network model that takes a real-valued vector of length 75,584 as input, and generates binary-valued output, 0 for control/healthy and 1 for ALS/disease. In general, DiseaseCapsule can be flexibly adapted to input vectors of varying sizes, as long as the length of the input vectors does not exceed a certain upper limit; the procedure based on GWAS filtering and Gene-PCA described above warrants this for whole-genome input.
The architecture of DiseaseCapsule follows the architecture of the capsule network that was described in the seminal paper, with modifications to accommodate that the input does not reflect rectangular, pixel-structured image data, and the output is binary. In detail, DiseaseCapsule consists of three layers: a fully connected layer (FC), a primary capsule layer (PrimaryCaps) and a phenotype capsule layer (PhenoCaps); for an illustration and details, see Fig. 2). The initial fully connected layer replaces the convolutional layers used in the seminal application, reflecting that convolution addresses to process rectangularly arranged image data. However, here we expect interactions between all genes across the whole genome, which the fully connected layer reflects.
The FC layer consists of 150 (regular) neurons followed by ReLU as activation function63. During training, a dropout rate of 0.5 was used for FC to prevent overfitting. Correspondingly, the output of FC is a 150 × 1 tensor, so virtually a 150-dimensional vector. This, in turn, is the input for the PrimaryCaps layer.
The PrimaryCaps is the first (‘low-level’) capsule layer. As such, it incorporates convolutional operations. It consists of 32 primary capsules. Each of these involves four different convolutional filters, implementing a 5 × 1 kernel, operating at a stride of 2, with no zero padding. This means that each convolutional filter computes a 73 × 1 tensor (that is a 73-dimensional vector) from the 150-dimensional input (FC output) vector. Using four convolutional filters per capsule results in 73 vectors of length 4 per capsule. This yields 32 × 73 = 2,336 vectors of length 4 as the output of PrimaryCaps. We refer to each of these vectors as ui, i = 1, ..., 2,336. Further, as is common, one refers to each array of 73 such four-dimensional vectors as one primary capsule. If needed, we index primary capsules using k ∈ {1, ..., 32}. Importantly, all 73 ui making part of one primary capsule k share their parameters when transiting to PhenoCaps, the last layer.
Finally, the output of PhenoCaps is used to derive predictions from. PhenoCaps consists of two 16-dimensional vectors vj, j = 1, 2, one referring to ‘ALS’ and one to ‘Healthy’. Each phenotype capsule receives input from all 32 primary capsules (that is, virtually from all 2,336 ui) making part of PrimaryCaps.
To transform PrimaryCaps output into PhenoCaps input, so-called pose matrices Wij are applied to the wi, which yields
In our case, pose matrices are 4 × 16-dimensional, so the uj∣i are 16-dimensional. This corresponds with the dimensionality of PhenoCaps. Pose matrices are learnt during training. As mentioned above, pose matrices are shared for all (73) i that refer to an identical primary capsule k. This means that there are 32 pose matrices to be learnt for each j = 1, 2.
Subsequently, we performed dynamic routing, as a key feature of capsule networks, intended to improve on the pooling operations. Dynamic routing is an iterative procedure that converges quickly. Here we used three iterations. For the corresponding details, see Extended Data Fig. 1. According to the routing procedure, the input sj to one of the PhenoCaps capsules evaluates as
where cij are the coupling coefficients, as determined through the dynamic routing procedure. In a rough description, first bij are computed by the iterative update \({b}_{ij}\leftarrow {b}_{ij}+{\hat{{{{\bf{u}}}}}}_{j| i}\cdot {{{{\bf{v}}}}}_{j}\) (and initialized as zero), which rewards if uj∣i and vj point in the same direction, which means that the output of primary capsule i agrees with phenotype capsule j. To turn bij into cij and ensure that ∑icij = 1, one eventually performs
to obtain the coupling coefficients.
Reflecting another key principle of capsule networks, one uses the squashing operation
as a non-linear activation function that can process vectors. This ensures that the length of the input vectors vj is between 0 and 1. Importantly, the length of capsule output vectors ui and vj indicates the probability that the capsule ‘is activated’.
To derive predictions from the vj, categorical cross entropy was employed as the loss function used during training.
Model training and testing
We randomly split the dataset into a set of samples used for training and validation set, which comprised 90% of individuals, and a test set, comprising the remaining 10% of individuals. Importantly, the test set is balanced, that is, the ratio of cases and controls is 1 (here: 520 cases and 520 controls), to ensure that models can be evaluated without misleading biases. For details, see Supplementary Fig. 5.
The entire dataset was used for Gene-PCA. As dimensionality reduction works in an unsupervised way, and thus does not require labels, this agrees with a generally applicable protocol. Subsequently, using the training-validation part of the data, fivefold cross-validation was performed to optimize the architecture and determine all other hyperparameters of the capsule network (DiseaseCapsule). To ensure a balanced evaluation during validation, validation splits were first randomly selected under the constraint of preserving a ratio of 1:1 between cases and controls. Subsequently, the cases in the remaining training data were upsampled to the same ratio, so as to avoid poor performance in prediction in particular for the minority class (Supplementary Fig. 5). This reflects a standard procedure in supervised machine learning. Upon having obtained all hyperparameters through cross-validation, the entire training-validation split, upsampled to ensure unbiased training, was used for training. This determines all parameters of the DiseaseCapsule network.
Specifically, we used the Adam algorithm64 to optimize all parameters in the frame of the usual backpropagation algorithm. We used an initial learning rate of 0.0001, and decayed it by γ = 0.8 in each epoch using an exponential scheduler. Optimization ran for 30 epochs, operating at a batch size of 128.
As for the simple three-layer perceptron (MLP) and the basic CNN (consisting of four convolution layers and two dense layers) that we used for benchmarking. For details, see Supplementary Fig. 6. Models were trained for 30 epochs with a batch size of 128 using the Adam optimizer, matching the procedure used for DiseaseCapsule.
Model interpretation
Relating coupling coefficients with phenotype recognition
When running DiseaseCapsule on the 1,040 test samples, coupling coefficients cij, i = 1, ..., 2,336, j = 1, 2 are determined individually for each of the test samples. This is due to the fact that coupling coefficients are determined during the forward step, and thus do not correspond to parameters to be learned during training. According to the general principles of capsule networks, one can interpret coupling coefficients cij as the degree of activation by which ui contributes to phenotype capsule j; large cij means that ui makes a crucial contribution to activating j.
As is common, we also determine coupling coefficients ckj that virtually measure the degree by which primary capsule k ‘activates’ phenotype capsule j. To obtain ckj from the cij, one sums all coupling coefficients cij across all ui that make part of k:
where j is either ‘Healthy’ or ‘ALS’, referring to one of the two Phenotype capsules. Recalling that ckj differ for each individual sample, we eventually average the ckj across the individuals to obtain a summarizing ckj one can work with.
Selecting and annotating genes decisive for classification
Evaluating combinations of primary and phenotype capsules according to ckj from equation (6) determines primary capsule 5 as the dominant driver to indicate that the phenotype capsule ‘ALS’ gets activated (Fig. 3a). In other words, genes that significantly contribute to activation of primary capsule 5 are potentially responsible for the development of ALS; in that, these genes are likely to be the predominant factor (‘Determining genes decisive for classification’ in Results). Thanks to capsule networks reflecting non-additive relationships, such genes can interact in arbitrary ways.
Computation of ckj involves running DiseaseCapsule on all 18,279 genes selected. Here we would like to determine the genes that play an important role in activating primary capsule 5 in their own right. To do so, we consider each gene g, g ∈ (1, 18,279) as exclusive input to the trained model. To implement this, we mask all other genes, that is, we set the values of all principal components that do not refer to the particularly picked gene g to zero. We do this for each individual sample. Subsequently, we run DiseaseCapsule on all the resulting ‘one-gene-only’ individual samples and note down the resulting coupling coefficients \({c}_{kj}^{g}\), for k = 5, j = ‘ALS’, for each of the individuals. Computation of \({c}_{kj}^{g}\) proceeds analogously to equation (6), when replacing the full individual sample with the ‘one-gene-only’ individual sample. Eventually, also here all individual \({c}_{kj}^{g}\) are averaged across the individual samples to obtain a summarizing \({c}_{kj}^{g}\) one can work with. We then kept all genes g whose \({c}_{kj}^{g}\) was above the 95-percentile (Fig. 4a), amounting to 922 genes, as core genes decisive for classification.
To annotate the biological functions of these 922 genes, we employed g:Profiler65 to perform common Gene Ontology and pathway enrichment analyses.
Selecting non-additive genes
Let S ⊂ G be a subset of genes selected from the set G of 18,279 genes overall. Let further ACCDC(S) be the training accuracy achieved by Gene-PCA + DiseaseCapsule (DC) and ACCLR(S) be the training accuracy of Gene-PCA + logistic regression (LR), as the best-performing linear approach, when running on only genes S. Running DC and LR on S is done by setting values of principal components referring to genes not from S to zero, in full analogy as for single genes g, as described above.
To determine a good subset of genes that predominantly interact in non-linear constellations, we make use of a genetic algorithm that seeks to determine
that is, the set of genes S that delivers the greatest gains in terms of classification performance in the non-linear DC over the linear LR. To implement the genetic algorithm, we consider subsets of genes as 18,279-dimensional binary-valued vectors (x1, x2, ..., x18,279) ∈ {0, 1}18,279 where xg = 1 if g ∈ S, that is, gene g belongs to S, and xg = 0 otherwise. Representing sets of genes S this way, one can implement the common evolutionary operations of genetic algorithms, like ‘selection’, ‘crossover’, ‘mutation’ or ‘fitness evaluation’. For solving the optimization problem equation (7), we employ ‘segregative genetic algorithms’, as available through the high-performance genetic algorithm toolbox Geatpy v2.6.0 (ref. 66).
Subsets S were initialized randomly, and the genetic algorithm was run at a population size of 30 and a maximum of generations of 200 as a stopping criterion. The subset of genes S that were determined to maximize equation (7) can be considered optimal in terms of interacting in exclusively non-additive ways to establish the ‘ALS’ signal in the frame of the DiseaseCapsule network.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data used in this study are publicly available. The ALS data used in this study are from the Dutch cohort of Project MinE8,11,45, and have been deposited at dbGaP Study Accession: phs003146.v1.p1. Amyotrophic Lateral Sclerosis online Database (ALSoD) is available at https://alsod.ac.uk/. The data of PD were downloaded from dbGaP Study Accession: phs000918.v1.p1 (refs. 46,47,48,49). Source data for Fig. 4 and Supplementary Fig. 1 are provided with this paper.
Code availability
The source code of DiseaseCapsule is publicly available on GitHub (https://github.com/HaploKit/DiseaseCapsule). Another related code for reproducing results and generating figures in this study is publicly available at Zenodo (https://doi.org/10.5281/zenodo.7118988)67.
References
Miller, R. G. et al. Practice parameter update: the care of the patient with amyotrophic lateral sclerosis: drug, nutritional, and respiratory therapies (an evidence-based review): report of the quality standards subcommittee of the American Academy of Neurology. Neurology 73, 1218–1226 (2009).
Brown, R. H. & Al-Chalabi, A. Amyotrophic lateral sclerosis. N. Engl. J. Med. 377, 162–172 (2017).
Kiernan, M. C. et al. Amyotrophic lateral sclerosis. Lancet 377, 942–955 (2011).
Lautrup, S., Sinclair, D. A., Mattson, M. P. & Fang, E. F. Nad+ in brain aging and neurodegenerative disorders. Cell Metab. 30, 630–655 (2019).
de la Rubia, J. E. et al. Efficacy and tolerability of eh301 for amyotrophic lateral sclerosis: a randomized, double-blind, placebo-controlled human pilot study. Amyotroph. Lateral Scler. Frontotemporal Degen. 20, 115–122 (2019).
Al-Chalabi, A. et al. An estimate of amyotrophic lateral sclerosis heritability using twin data. J. Neurol. Neurosurg. Psychiatry 81, 1324–1326 (2010).
Parone, P. A. et al. Enhancing mitochondrial calcium buffering capacity reduces aggregation of misfolded sod1 and motor neuron cell death without extending survival in mouse models of inherited amyotrophic lateral sclerosis. J. Neurosci. 33, 4657–4671 (2013).
Van Rheenen, W. et al. Common and rare variant association analyses in amyotrophic lateral sclerosis identify 15 risk loci with distinct genetic architectures and neuron-specific biology. Nat. Genet. 53, 1636–1648 (2021).
Nguyen, H. P., Van Broeckhoven, C. & van der Zee, J. Als genes in the genomic era and their implications for ftd. Trends Genet. 34, 404–423 (2018).
Ryan, M., Heverin, M., McLaughlin, R. L. & Hardiman, O. Lifetime risk and heritability of amyotrophic lateral sclerosis. JAMA Neurol. 76, 1367–1374 (2019).
Van Rheenen, W. et al. Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat. Genet. 48, 1043–1048 (2016).
Boyle, E. A., Li, Y. I. & Pritchard, J. K. An expanded view of complex traits: from polygenic to omnigenic. Cell 169, 1177–1186 (2017).
Génin, E. Missing heritability of complex diseases: case solved? Hum. Genet. 139, 103–113 (2020).
Shi, H., Kichaev, G. & Pasaniuc, B. Contrasting the genetic architecture of 30 complex traits from summary association data. Am. J. Hum. Genet. 99, 139–153 (2016).
Tam, V. et al. Benefits and limitations of genome-wide association studies. Nat. Rev. Genet. 20, 467–484 (2019).
Moore, J. H. The ubiquitous nature of epistasis in determining susceptibility to common human diseases. Hum. Hered. 56, 73–82 (2003).
Jiao, S. et al. Genome-wide search for gene–gene interactions in colorectal cancer. PLoS ONE 7, e52535 (2012).
Hung, H. et al. Detection of gene–gene interactions using multistage sparse and low-rank regression. Biometrics 72, 85–94 (2016).
Ferrario, P. G. & König, I. R. Transferring entropy to the realm of gxg interactions. Brief. Bioinformatics 19, 136–147 (2018).
Hornik, K., Stinchcombe, M. & White, H. Multilayer feedforward networks are universal approximators. Neural Netw. 2, 359–366 (1989).
Montufar, G. F., Pascanu, R., Cho, K. & Bengio, Y. On the number of linear regions of deep neural networks. Adv. Neural Inf. Process. Syst. 27, 2924–2932 (2014).
Krizhevsky, A., Sutskever, I. & Hinton, G. E. Imagenet classification with deep convolutional neural networks. Adv. Neural Inf. Process. Syst. 25, 1097–1105 (2012).
LeCun, Y., Bengio, Y. & Hinton, G. Deep learning. Nature 521, 436–444 (2015).
Alzubaidi, L. et al. Review of deep learning: concepts, CNN architectures, challenges, applications, future directions. J. Big Data 8, 1–74 (2021).
Simonyan, K. & Zisserman, A. Very deep convolutional networks for large-scale image recognition. In 3rd International Conference on Learning Representations 1–14 (Computational and Biological Learning Society, 2015); https://arxiv.org/pdf/1409.1556.pdf
He, K., Zhang, X., Ren, S. & Sun, J. Deep residual learning for image recognition. In Proc. IEEE Conference on Computer Vision and Pattern Recognition 770–778 (IEEE, 2016); https://doi.ieeecomputersociety.org/10.1109/CVPR.2016.90
Huang, G., Liu, Z., Van Der Maaten, L. & Weinberger, K. Q. Densely connected convolutional networks. In Proc. IEEE Conference on Computer Vision and Pattern Recognition 4700–4708 (IEEE, 2017); https://ieeexplore.ieee.org/document/8099726
Chakraborty, S. et al. Interpretability of deep learning models: a survey of results. In 2017 IEEE Smartworld, Ubiquitous Intelligence & Computing, Advanced & Trusted Computed, Scalable Computing & Communications, Cloud & Big Data Computing, Internet of People and Smart City Innovation (smartworld/SCALCOM/UIC/ATC/CBDcom/IOP/SCI) 1–6 (IEEE, 2017); https://ieeexplore.ieee.org/document/8397411
Hestness, J. et al. Deep learning scaling is predictable, empirically. CoRR abs/1712.00409 (2017).
Ching, T. et al. Opportunities and obstacles for deep learning in biology and medicine. J. R. Soc. Interface 15, 20170387 (2018).
Wainberg, M., Merico, D., Delong, A. & Frey, B. J. Deep learning in biomedicine. Nat. Biotechnol. 36, 829–838 (2018).
Sabour, S., Frosst, N. & Hinton, G. E. Dynamic routing between capsules. Adv. Neural Inf. Process. Syst. 30, 3856–3866 (2017).
Sabour, S., Frosst, N. & Hinton, G. Matrix capsules with em routing. In 6th International Conference on Learning Representations, ICLR 2018 (OpenReview.net, 2018); https://openreview.net/pdf?id=HJWLfGWRb
Camacho, D. M., Collins, K. M., Powers, R. K., Costello, J. C. & Collins, J. J. Next-generation machine learning for biological networks. Cell 173, 1581–1592 (2018).
Wang, L. et al. An interpretable deep-learning architecture of capsule networks for identifying cell-type gene expression programs from single-cell rna-sequencing data. Nat. Mach. Intell. 2, 693–703 (2020).
Kourou, K., Exarchos, T. P., Exarchos, K. P., Karamouzis, M. V. & Fotiadis, D. I. Machine learning applications in cancer prognosis and prediction. Comput. Struct. Biotechnol. J. 13, 8–17 (2015).
Curbelo Montañez, C. A., Fergus, P., Chalmers, C. & Hind, J. Analysis of extremely obese individuals using deep learning stacked autoencoders and genome-wide genetic data. In Computational Intelligence Methods for Bioinformatics and Biostatistics: 15th International Meeting, CIBB 2018, Caparica, Portugal, September 6–8, 2018, Revised Selected Papers 15 (eds Raposo, M. et al.) 262–276 (Springer, 2020).
He, B. et al. Ai-enabled in silico immunohistochemical characterization for Alzheimer’s disease. Cell Rep. Methods 2, 100191 (2022).
Chen, D. et al. A stacking framework for multi-classification of alzheimer’s disease using neuroimaging and clinical features. J. Alzheimer’s Dis. 87, 1627–1636 (2022).
Xie, C. et al. Amelioration of Alzheimer’s disease pathology by mitophagy inducers identified via machine learning and a cross-species workflow. Nat. Biomed. Eng. 6, 76–93 (2022).
Li, X., Liu, L., Zhou, J. & Wang, C. Heterogeneity analysis and diagnosis of complex diseases based on deep learning method. Sci. Rep. 8, 1–8 (2018).
Greenside, P., Shimko, T., Fordyce, P. & Kundaje, A. Discovering epistatic feature interactions from neural network models of regulatory dna sequences. Bioinformatics 34, i629–i637 (2018).
Yin, B. et al. Using the structure of genome data in the design of deep neural networks for predicting amyotrophic lateral sclerosis from genotype. Bioinformatics 35, i538–i547 (2019).
Zhang, S. et al. Genome-wide identification of the genetic basis of amyotrophic lateral sclerosis. Neuron 110, 992–1008 (2022).
Consortium, P. M. A. S. et al. Project mine: study design and pilot analyses of a large-scale whole-genome sequencing study in amyotrophic lateral sclerosis. Eur. J. Hum. Genet. 26, 1537 (2018).
Auer, P. L. et al. Imputation of exome sequence variants into population-based samples and blood-cell-trait-associated loci in African Americans: NHLBI GO Exome Sequencing Project. Am. J. Hum. Genet. 91, 794–808 (2012).
International Parkinson’s Disease Genomics Consortium (IPDGC) & Wellcome Trust Case Control Consortium 2 (WTCCC2). A two-stage meta-analysis identifies several new loci for Parkinson’s disease. PLoS Genet. 7, e1002142 (2011).
Nalls, M. A. et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat. Genet. 46, 989–993 (2014).
Nalls, M. A. et al. Neurox, a fast and efficient genotyping platform for investigation of neurodegenerative diseases. Neurobiol. Aging 36, 1605.e7–1605.e12 (2015).
Leal, S. S. & Gomes, C. M. Calcium dysregulation links als defective proteins and motor neuron selective vulnerability. Front. Cell. Neurosci. 9, 225 (2015).
Van Spronsen, M. & Hoogenraad, C. C. Synapse pathology in psychiatric and neurologic disease. Curr. Neurol. Neurosci. Rep. 10, 207–214 (2010).
Lepeta, K. et al. Synaptopathies: synaptic dysfunction in neurological disorders—a review from students to students. J. Neurochem. 138, 785–805 (2016).
Ikemoto, A., Nakamura, S., Akiguchi, I. & Hirano, A. Differential expression between synaptic vesicle proteins and presynaptic plasma membrane proteins in the anterior horn of amyotrophic lateral sclerosis. Acta Neuropathol. 103, 179–187 (2002).
Burk, K. & Pasterkamp, R. J. Disrupted neuronal trafficking in amyotrophic lateral sclerosis. Acta Neuropathol. 137, 859–877 (2019).
Südhof, T. C. Neuroligins and neurexins link synaptic function to cognitive disease. Nature 455, 903–911 (2008).
Chang, C. C. et al. Second-generation plink: rising to the challenge of larger and richer datasets. Gigascience 4, s13742-015-0047-8 (2015).
Purcell, S. & Chang, C. Plink 1.9 beta. PLINK 1.9 http://www.cog-genomics.org/plink/1.9/ (2015).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B 57, 289–300 (1995).
Consortium, I. H. et al. A haplotype map of the human genome. Nature 437, 1299 (2005).
Wang, K., Li, M. & Hakonarson, H. Annovar: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164 (2010).
Pearson, K. LIII. on lines and planes of closest fit to systems of points in space. Lond. Edinb. Dublin Philos. Mag. J. Sci. 2, 559–572 (1901).
Price, A. L., Zaitlen, N. A., Reich, D. & Patterson, N. New approaches to population stratification in genome-wide association studies. Nat. Rev. Genet. 11, 459–463 (2010).
Nair, V. & Hinton, G. E. Rectified linear units improve restricted Boltzmann machines. In International Conference on Machine Learning (eds Fürnkranz, J. et al.) 807–814 (Omnipress, 2010).
Kingma, D. P. & Ba, J. Adam: a method for stochastic optimization. In 3rd International Conference on Learning Representations (Ithaca, NY: arXiv.org, 2015).
Raudvere, U. et al. g: Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 47, W191–W198 (2019).
Jazzbin et al. geatpy: the genetic and evolutionary algorithm toolbox with high performance in Python. Geatpy http://www.geatpy.com/ (2020).
Luo, X., Kang, X. & Schönhuth, A. Diseasecapsule: v1.0.0. Zenodo https://doi.org/10.5281/zenodo.7118988 (2022).
Acknowledgements
X.L. and X.K. were supported by the Chinese Scholarship Council. A.S. was supported by the Dutch ALS Foundation via grant AV20190010. A.S. was also supported by the Dutch Scientific Organization through Vidi grant 639.072.309 during the early stages of the project. A.S. further received funding from the European Union’s Horizon 2020 research and innovation programme under Marie Skłodowska-Curie grant agreements no. 956229 (ALPACA) and no. 872539 (PANGAIA). In addition, we thank B. Yin and S. Ray for their helpful discussions in the early stage of this project. We thank M. Balvert for helpful discussions in the early stages and insightful suggestions when revising the manuscript. We also thank K. van der Eijck and J. Veldink for providing data and helpful information about the mechanisms of ALS. Finally, we thank the International Parkinson’s Disease Genomics Consortium (IPDGC), NINDS, NIA and DoD USAMRAA proposal, 10064005/11348001, for providing the PD data.
Funding
Open access funding provided by Universität Bielefeld.
Author information
Authors and Affiliations
Contributions
X.L., X.K. and A.S. developed the method. X.L. and X.K. implemented the code and conducted the data analysis. X.L., X.K. and A.S. wrote the manuscript. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Peer review
Peer review information
Nature Machine Intelligence thanks Evandro F. Fang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 The schematic diagram of the dynamic routing algorithm.
The PrimaryCaps layer outputs 32 × 73 = 2336 vectors ui(i = 1, . . . , 2336), which are then transformed into \({\hat{{{{\bf{u}}}}}}_{j| i}\)(j = 1, 2) by premultiplying pose matrices Wij. Note that pose matrices are learned during training. The right half of this figure shows the iterative dynamic routing procedures. The coupling coefficients cij indicate the probability that primary capsule i agrees with phenotype capsule j. The input sj to one of the phenotype capsules is subsequently processed by using the squashing operation. The output vj is used to update the bij and cij until the model converges.
Supplementary information
Supplementary Information
Supplementary notes, discussion, Figs. 1–7 and Tables 1–9.
Supplementary Data 1
Source data for Supplementary Fig. 1.
Source data
Source Data Fig. 4
Statistical source data.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Luo, X., Kang, X. & Schönhuth, A. Predicting the prevalence of complex genetic diseases from individual genotype profiles using capsule networks. Nat Mach Intell 5, 114–125 (2023). https://doi.org/10.1038/s42256-022-00604-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s42256-022-00604-2
