
Abstract
Elucidation of both the three-dimensional structure and the dynamics of a protein is essential to understand its function. Technical breakthroughs in single-particle analysis based on cryo-electron microscopy (cryo-EM) have enabled the three-dimensional structures of numerous proteins to be solved at atomic or near-atomic resolution. However, the analysis of the dynamics of protein targets using cryo-EM is often challenging because of their large sizes and complex structural assemblies. Here, we describe DEFMap, a deep learning-based approach to directly extract the dynamics associated with the atomic fluctuations that are hidden in cryo-EM density maps. Using only cryo-EM density data, DEFMap provides dynamics that correlate well with data obtained from molecular dynamics simulations and experimental approaches. Furthermore, DEFMap successfully detects changes in dynamics that are associated with molecular recognition. This strategy combines deep learning, experimental data and molecular dynamics simulations, and may reveal a new multidisciplinary approach for protein science.
Similar content being viewed by others


Main
The physiological functions of proteins are associated with their three-dimensional (3D) structure and their dynamic behaviour in solution. High-resolution studies of the structural and dynamics properties of proteins are essential to elucidate the mechanisms underlying their biological functions, such as the regulation of cellular signalling mediated by protein–protein interactions and metabolic reactions catalysed by enzymes1,2. Various techniques have been developed to determine protein structures, such as X-ray crystallography, nuclear magnetic resonance (NMR) spectroscopy and cryo-electron microscopy (cryo-EM) single-particle analysis (SPA)3,4,5,6. Information about dynamics has been quantitatively obtained through several experimental and computational approaches, including NMR, hydrogen–deuterium exchange (HDX) mass spectrometry (MS)7, and molecular dynamics (MD) simulations8.
Recent breakthroughs in cryo-EM SPA3,9 have facilitated the determination of the structures of numerous biological molecules at atomic or near-atomic resolution10,11, including those of extremely large and complex macromolecules12,13,14,15 that have not been solved using conventional techniques. However, the investigation of the dynamics of such molecules is technically challenging owing to their large sizes and complex structural assemblies.
The 3D cryo-EM maps solved by SPA are reconstructed from numerous two-dimensional (2D) images of molecular particles identified in a micrograph3,4,5. Specimens used for cryo-EM SPA are prepared by rapidly freezing a solution in which proteins adopt variable conformations. Therefore, the dynamics properties of the proteins could be ‘hidden’ in the reconstructed cryo-EM maps. Local resolutions16,17 derived from the local map intensities in reconstructed 3D cryo-EM maps tend to correlate with the dynamics information associated with the atomic fluctuation; that is, lower local resolutions correspond to more flexible regions. However, local resolutions may be affected by artificial effects resulting from sample conditions. These artificial effects, including preferred orientations, compositional heterogeneity and local denaturation during sample preparation, could hamper the direct estimation of dynamics from the local resolutions.
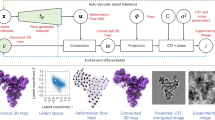
To address these challenges, we developed dynamics extraction from cryo-EM map (DEFMap), a deep learning-based approach that obtains the dynamics information of proteins from a cryo-EM map alone (Fig. 1a). We used all-atom MD simulations of cryo-EM SPA-targeted macromolecules and constructed a model based on a deep neural network to predict the dynamics information from cryo-EM density data. The performance of this model was validated with macromolecules that were not included in the training dataset by comparing the results with MD-derived and experimentally determined dynamics properties. Moreover, DEFMap identified the changes in dynamics associated with molecular recognition and their accompanying allosteric effects from a cryo-EM map alone without requiring additional experiments such as MD simulations. Our approach integrates multiple research areas such as deep learning, MD simulation and cryo-EM SPA, and facilitates the investigation of dynamics properties that are intractable using conventional techniques.

a, Complete workflow of DEFMap. Model training is carried out using macromolecules derived from EMDB and PDB (EMDB/PDB), which are conveniently handled in all-atom MD simulations. In the training-data construction stage, the dynamics properties are derived from the RMSF values (for heavy atoms) calculated from MD simulations (Methods, see the section on Molecular dynamics simulations). In the training stage, the 3D-CNN model in DEFMap learns the relationship between dynamics values and density data at the corresponding positions (Methods, see the sections on Data preparation for DEFMap and Construction of deep neural networks). In the prediction stage, for other cryo-EM maps that are not included in the training dataset, the trained model predicts dynamics values based on input density data (Methods, see the section on Postprocessing and visualization of output from the neural networks). In this study, 25 macromolecules were used to validate and train the DEFMap model, and 9 other macromolecules were used for dynamics predictions using the trained model and further structural analyses. b, A correlation plot between DynamicsDEFMap and DynamicsMD for EMD-3984 (Supplementary Table 2, entry 1). r denotes the correlation coefficient. DynamicsMD were calculated from the RMSF values derived from MD simulations. c, Comprehensive comparison of the correlation coefficients for DynamicsDEFMap with those for the local resolution estimates. The correlation coefficients are calculated against DynamicsMD, and then, a plot of correlation coefficients for DynamicsDEFMap against correlation coefficents for the standardized local resolutions is created. Each point denotes the individual map for the EMDB ID indicated by a label. The y = x line is represented by a black line; the points are located above the line, indicating the superior performance of DEFMap.
Results
Construction of DEFMap
To quantitatively extract the hidden dynamics information associated with the atomic fluctuations from only the density data in the cryo-EM map, DEFMap was constructed using a deep-learning method and MD simulations (Fig. 1a). The deep-learning method was designed to learn the relationship between local density data and the dynamics information. Although quantitative data on atomic fluctuations are required for training the neural network model, it is difficult to obtain adequate dynamics data from existing experimental methods. To overcome this limitation, we performed all-atom MD simulations using atomic structures that are derived from cryo-EM maps and are available in the Protein Data Bank (PDB)18; we calculated the root mean squared fluctuation (RMSF) representing atomic fluctuations as the dynamics information (Fig. 1a, see the training-data construction stage). With advances in MD techniques and computing performance, MD simulations have been widely used to elucidate the detailed dynamic behaviour of biological molecules8,19,20. Next, we trained the 3D convolutional neural network (3D-CNN)21,22,23 model to learn the relationship between cryo-EM density data and the logarithmic RMSF values in order to capture 3D patterns of the cryo-EM density data that reflect the protein dynamics (Fig. 1a, see the training stage). Three-dimensional CNNs have been used extensively to detect or classify patterns in 3D objects in various fields24,25,26 and have exhibited remarkable performance in their applications to 3D cryo-EM maps27,28,29,30. In this study, a sub-voxel (10 × 10 × 10 grid) that is extracted from the original voxels is provided as input data to the model, and the model is trained to predict the logarithmic RMSF value of the central voxel in each sub-voxel. Finally, using the trained 3D-CNN model, DEFMap can directly and quantitatively extract the hidden dynamics information in the form of the logarithmic RMSF values by using only the 3D cryo-EM density data of a new target protein (Fig. 1a, see the prediction stage). In the prediction stage, DEFMap does not require any MD calculations. For visualization in DEFMap, the residue-specific values averaged over each residue after normalization (termed DynamicsDEFMap) are mapped onto the corresponding atomic models.
Initially, to evaluate the performance of DEFMap, we retrieved 25 cryo-EM maps from the Electron Microscopy Data Bank (EMDB)31 (Supplementary Table 1) and used them to train the DEFMap 3D-CNN model (see Methods for details of the training dataset, MD simulation and training). Within the dataset, local resolutions calculated using the MonoRes implementation in Scipion16,32 tended to correlate with the normalized residue-specific logarithmic RMSF values obtained from MD (termed DynamicsMD) (Extended Data Fig. 1a and Supplementary Table 2; average correlation coefficient r = 0.510 ± 0.091). However, 10 out of 25 datasets were excluded from the evaluation because they exhibited inverse correlations. Using the same dataset, we performed 25-fold cross-validation33 in a unit of protein to accurately evaluate the performance of DEFMap within the dataset. We observed an improved correlation between the DynamicsDEFMap and DynamicsMD outputs (for the 15 datasets that exhibited a positive correlation in the local resolution estimates, r = 0.663 ± 0.135; for the 25 datasets, r = 0.665 ± 0.124; Fig. 1b,c and Supplementary Table 2). These results indicate that DEFMap can efficiently extract dynamics features from 3D cryo-EM density data. The direct benefit of using the DEFMap algorithm against using the input density data can be observed by comparing the correlation plot for DEFMap (Fig. 1b) with that for the corresponding map intensity (Extended Data Fig. 1b and Supplementary Table 2).
Correlation with dynamics from MD simulations and experiments
Subsequently, we trained the 3D-CNN model of DEFMap using the full training dataset (25 cryo-EM maps) and then tested DEFMap on three other cryo-EM maps (EMDB entry, PDB entry: EMD-4241, 6FE8 (ref. 34); EMD-7113, 6BLY (ref. 35); and EMD-20308, 6PCV (ref. 36)) to further evaluate its potential for dynamics analysis (Supplementary Table 3). The additional maps were selected for their distinct structural aspects in terms of secondary structure contents (α-helix, β-strand, others: 0.56, 0.06, 0.38 for EMD-4241, 6FE8; 0.05, 0.43, 0.52 for EMD-7113, 6BLY; and 0.30, 0.27, 0.43 for EMD-20308, 6PCV). In particular, the experimental dynamics of EMD-20308, 6PCV has been reported and is compared with DynamicsDEFMap later. The performance of DEFMap in extracting dynamics features for the structural fluctuations from cryo-EM maps is illustrated in Fig. 2. The dynamics values calculated by the trained DEFMap model correlated well with those derived from MD simulations at both the atomic level (r = 0.704, 0.726 and 0.673, respectively) and residue level (r = 0.727, 0.748 and 0.711, respectively) (Extended Data Fig. 2). The results showed that DEFMap could extract accurate dynamics information from the cryo-EM map data alone. Moreover, the mapping of DynamicsDEFMap onto the corresponding atomic models demonstrated that DEFMap captured conformational aspects such as rigidity in the protein interior and flexibility of the solvent-exposed secondary structure elements, with an accuracy similar to that of MD simulations (Fig. 2a–c, upper panels). The DynamicsDEFMap data matched the DynamicsMD data for the entire protein (Fig. 2a–c, lower panels). In some regions, DEFMap failed to extract accurate dynamics properties. We reasoned that low resolution of a local map hinders accurate extraction of dynamics data. In fact, overall map resolutions that were lower (particularly those >8 Å) resulted in inferior performance (Fig. 2d and Extended Data Fig. 3).

a–c, DynamicsMD and DynamicsDEFMap outputs for EMD-4241, 6FE8 (a), EMD-7113, 6BLY (b), and EMD-20308, 6PCV (c). The cryo-EM maps were preprocessed by a 5 Å low-pass filter. DynamicsDEFMap and DynamicsMD were mapped onto 3D atomic models using different colours as indicated in the colour bars (top panels). The bottom panels show the DynamicsMD (black) and DynamicsDEFMap (magenta) profiles as a function of the residue IDs, numbered in accordance with their order in the corresponding PDB file; r denotes the correlation coefficient between DynamicsMD and DynamicsDEFMap. d, Dependence of DEFMap performance on map resolution. The plot shows the r values obtained for different map resolutions (see Methods). The cryo-EM maps for EMD-4241 at 5 Å, 7 Å and 10 Å are shown at the bottom of the panel. e, A correlation plot of dynamics derived from DEFMap and from HDX-MS. The HDX exchange rates at 104 s for Rac exchanger 1 (EMD-20308, 6PCV) were normalized within the detected fragments and used as the experimental dynamics data. DynamicsDEFMap and DynamicsMD were converted to fragment-specific values by averaging them over each fragment in the atomic model. The DEFMap versus HDX-MS (r = 0.743), MD versus HDX-MS (r = 0.791) and DEFMap versus MD (r = 0.807) correlation plots are shown in the left, middle and right panels, respectively, along with their corresponding regression lines (orange). The visualization of the dynamics specific to the representative fragments on a 3D atomic model is shown in Extended Data Fig. 5.
The DEFMap performance for the maps that were preprocessed using several low-pass filters suggests that DEFMap is potentially useful in the case of intermediate-resolution maps (5–7 Å) for which de novo modelling of reliable atomic structures is substantially difficult. Further validation using an experimental map with an overall resolution of 6.20 Å and local resolutions ranging from 2 Å to 8 Å (EMDB entry, PDB entry: EMD-4772, 6R9T (ref. 37)), (Extended Data Fig. 4a and Supplementary Table 3) demonstrated that DynamicsDEFMap data exhibited a good correlation with DynamicsMD data (r = 0.646, Extended Data Fig. 4b). The performance of a model trained with datasets that were preprocessed to a resolution of 7 Å using low-pass filters was superior to that of the models trained with datasets that were preprocessed to resolutions of 5 Å and 6 Å using low-pass filtering (Extended Data Fig. 4c,d, respectively); this indicated that a model trained by datasets that were appropriately preprocessed in accordance with the target-map resolutions should be selected for the prediction.
To assess the potential of the current method, it is important to confirm the consistency of the DEFMap predictions with experimentally derived dynamics properties. Under appropriate conditions, the dynamics of large proteins (such as those targeted by cryo-EM) can also be experimentally determined using HDX-MS7; using this approach, the dynamics information at the peptide fragment level can be obtained by monitoring the effects of deuterium incorporation into protein amide groups (Supplementary Fig. 1). We compared the DynamicsDEFMap of EMD-20308 with the publicly available HDX-MS data36. The average DynamicsDEFMap data for each fragment detected in the HDX-MS experiments correlated well with the corresponding HDX rates (DynamicsHDX-MS, r = 0.743) and DynamicsMD data (r = 0.807), confirming that DEFMap captured the local dynamics accurately. Thus, DEFMap can provide insights equivalent to those obtained from experimental approaches (Fig. 2e and Extended Data Fig. 5).
DEFMap-based analysis of biologically relevant dynamics
We next assessed the potential of DEFMap to identify molecular binding sites and to investigate the allosteric effects of ligand binding. Ligand binding is a fundamental biological event and is often accompanied by the suppression of dynamics at the recognition interface. Therefore, we monitored the dynamics changes associated with ligand-induced perturbations in the cryo-EM density maps. In particular, we used DEFMap to detect ligand-induced dynamics changes for three pairs of apo and holo proteins that were not included in the training dataset (apo, holo: EMD-20080, EMD-20081 (ref. 38); EMD-9616, EMD-9622 (ref. 39); EMD-3957, EMD-3956 (ref. 40); Supplementary Table 4). We found good agreement between DynamicsDEFMap and DynamicsMD profiles for the aforementioned pairs (Extended Data Fig. 6). Moreover, the DEFMap-derived dynamics of the residues located near the binding partners were significantly suppressed upon ligand binding (Fig. 3a,b and Supplementary Table 5), demonstrating that DEFMap could detect conformational stabilization at the binding interface. Among the residues located at the interfaces (Supplementary Table 5), significant dynamics suppression was observed in the regions that have been extensively discussed in previous studies38,39,40. This finding suggests that DEFMap can identify the key interactions involved in complex formation by using the density data (Fig. 3b). We observed additional dynamics suppression for regions that were distant from the ligand-binding site in the EMD-20080, EMD-20081 pair (Arabidopsis defective in meristem silencing 3 (DMS3)–RNA-directed DNA methylation 1 (RDM1) complex with defective RNA-directed DNA methylation 1 (DRD1) peptide)38 (Fig. 3c). In the apo state, DMS3 dimers establish contact with an RDM1 dimer through their coiled-coil arms (Fig. 3c, middle panel). In the holo conformation, the binding partner, DRD1 peptide, is recognized by the RDM1 dimer, the coiled-coil arms of a DMS3 dimer (Fig. 3c, dimer 1) and a hinge domain of an opposite DMS3 dimer (Fig. 3c, dimer 2). DEFMap-based dynamics analysis showed that the binding of the DRD1 peptide induced suppression of dynamics at the RDM1–DMS3 interface and at the hinge domains in the DMS3 dimer 1 (Fig. 3c), indicating that ligand binding allosterically stabilizes these regions. Interestingly, the static models constructed from the cryo-EM maps displayed no substantial differences between the apo and holo proteins in these regions (Extended Data Fig. 7). This finding further highlights the potential of DEFMap to provide future directions for studies on the underlying mechanism by extracting dynamics information that cannot be inferred from static tertiary structures.

a, Ligand-induced dynamics suppression at the binding interfaces. The dynamics at the binding interfaces in apo (black) and holo (red) proteins were calculated by averaging DynamicsMD and DynamicsDEFMap for residues located within 5 Å of the binding partners. A two-sided t-test was performed to compare the difference between the dynamics in apo and holo proteins. The null hypothesis was that they have identical average dynamics values, and the significance threshold was 0.01. The error bars indicate standard deviations (*P < 0.01). b, Spatial distributions of ligand-induced dynamics changes. The dynamics changes were calculated by subtracting DynamicsDEFMap of the apo state from those of the holo state and mapping onto the holo atomic model using different colours, as indicated in the colour bar (lower values denote regions with suppressed dynamics). Binding partners and disordered regions in apo proteins are show in green and dark grey, respectively. In the upper panels, Cα atoms of residues located within 5 Å of the binding partners are represented as spheres, and the cryo-EM maps are shown in grey. The lower panels provide enlarged views of the regions indicated by dashed rectangles in the upper panels. The relevant residues discussed extensively in the literature are represented as sticks, with the subunit names given in superscript text. c, DEFMap detection of ligand-induced allosteric change. Enlarged views of the regions discussed in the main text are shown in the left- and right-hand side panels. The ligand-induced dynamics changes are indicated using different colours as shown in the colour bar.
Practical application of DEFMap to large and complex molecular systems
Finally, we assessed the potential of DEFMap to rapidly provide novel biological insights from a cryo-EM map alone by targeting the novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and Zika virus. We applied DEFMap to the recently solved cryo-EM maps of the surface spike glycoprotein41 (Extended Data Fig. 8a, S protein) in SARS-CoV-2, the causative agent of the COVID-19 pandemic. Studies to develop molecular-targeted therapies for this disease are urgently required. We used DEFMap to visualize differences in dynamic properties between two distinct conformational states in the S protein, the host cell receptor-accessible and receptor-inaccessible forms (EMD-21457 and EMD-21452, respectively) (Fig. 4a and Extended Data Fig. 8a). The visualized dynamics indicated that the flexibilities of the heptad repeat 1 (HR1) motifs and their interacting β-rich regions are elevated upon detachment of the receptor-binding domain (Fig. 4b and Extended Data Fig. 8b). Their dynamics changes might be associated with the function42 (see Discussion). Wrapp et al. reported another cryo-EM map of the receptor-accessible form (EMD-21375 (ref. 43)), which adopts a structure similar to that of EMD-21457. The DynamicsDEFMap of EMD-21375 correlated well with that of EMD-21457 (r = 0.641), thus validating the DEFMap performance (Extended Data Fig. 9). Furthermore, we assessed the applicability of DEFMap to extremely large molecular systems such as a virus particle. We visualized the dynamics on the viral surface and in cross-sections of the Zika virus44 (ZIKV, EMD-8139) (Fig. 4b), which is associated with Guillain–Barré syndrome in adults and with microcephaly in fetuses. The protruding regions on the solvent-exposed surface were relatively more flexible than the buried regions. The dynamics on the viral surface differed among individual protomers in an asymmetric unit (Fig. 4c), suggesting that DEFMap can capture the dynamics of the individual protomers in the distinct environment. Similarly, we performed the dynamics analyses for several viral particles45,46,47, and the results indicated variable dynamics distributions on the viral surfaces (Extended Data Fig. 10). The dynamics in the aggregated state are inaccessible through HDX-MS because individual protomers are not separately recognizable in the detection stage. Furthermore, MD-based analyses are still relatively challenging and expensive for these large and complex systems, and are not feasible if the atomic structure is not available (for example, EMD-9053, a human enterovirus47; Extended Data Fig. 10, right panels).

a, Dynamics changes accompanied by a transition from the down conformation to the up conformation of the spike protein in SARS-CoV-2. The dynamics are mapped onto the atomic model in the up conformation using different colours as indicated in the colour bar. b, Mapping of DEFMap-derived dynamics onto the cryo-EM map of ZIKV. The solvent-exposed surface and the thin slice in the central section are shown in the left and right panels, respectively. The colour range is defined by minimum and maximum values in the inference. c, Enlarged view of an asymmetric unit of the ZIKV envelope proteins on the viral surface. The atomic model (PDB entry 5IZ7) is represented as ribbons. An asymmetric unit and each protomer component are indicated by regions within black and grey boundaries, respectively. The protruding loops exhibiting high flexibility (residues 151–161, 331–338 and 366–373) are shown as dashed white rectangles.
Discussion
This work provides a proof of concept that a deep-learning technique can efficiently extract features associated with in-solution behaviour from cryo-EM density data, which are modulated not only by intrinsic plasticity, but also by several artefacts attributed to sample conditions. DEFMap has the potential to facilitate the establishment of additional experimental or therapeutic strategies. For example, the allosteric effect observed in the RDM1–DMS3 complex can be further validated using HDX-MS experiments by comparing the HDX rates of the focused regions in the presence and absence of the DRD1 peptide. To give another example, the dynamics perturbation in HR1 motifs and their proximal regions of S protein in SARS-CoV-2 could be helpful in the establishment of a therapeutic strategy. A previous study on the S protein of SARS-CoV reported that the HR1 motifs undergo conformational changes to enable viral and host cell membrane fusion42. Considering that this phenomenon is triggered through host cell receptor binding (Extended Data Fig. 8b), the dynamics elevation is presumably associated with the conformational relaxation, which is favourable for subsequent conformational changes. The development of targeted drugs or antibodies to suppress these dynamics changes may be an effective strategy to counteract the viral infection.
A series of analyses have provided several clues to further improve the performance of DEFMap. The performance of the current model depended on the map resolutions (Fig. 2d and Extended Data Fig. 3), and this may reflect loss of detailed structural information in low-resolution maps. The dependence on resolution suggests that the performance of DEFMap will improve in line with the continuous progress in obtaining maps with higher resolution through the development of advanced equipment, such as cold field emission guns48,49, for cryo-EM data acquisition. From the viewpoint of processing density data, training the model with maps sharpened on the basis of their local resolution50 could improve the DEFMap performance owing to homogenization of the dataset properties. Also, additional model training using other macromolecules with various structural features and resolutions would increase the accuracy and the robustness of the model.
In conclusion, this study shows that DEFMap, a deep neural network-based approach, can successfully extract hidden dynamics information from static 3D cryo-EM density maps alone. The DEFMap approach is not restricted by molecular size and the complexity of the systems because the model infers the dynamics from local density data. We expect that the use of DEFMap as a complement to conventional cryo-EM SPA may accelerate the elucidation of mechanisms underlying biological events, and provide novel insights into protein behaviours in complicated systems and clues to establish therapeutic strategies. Furthermore, DEFMap may help researchers to readily (from preprocessing to visualization in less than one hour) access the dynamics properties of biological molecules, as this open-source tool does not require additional expensive and/or time-consuming experiments or in-depth expertise. DEFMap can save considerable computation time in comparison with MD; for example, for the three test proteins (EMD-4241, 6FE8; EMD-7113, 6BLY; and EMD-20308, 6PCV with molecular weights of 220.37, 228.62 and 231.48 kDa, respectively), the MD simulations using a single GPU (NVIDIA GeForce GTX 1080) took 10–20 h for the product runs, whereas the prediction times of DEFMap were only several minutes. We believe that in the future, DEFMap may accelerate data-driven structural investigations aiming to understand protein function and to develop strategies for molecular-targeted therapy against various diseases. This study bridges experimental data, deep-learning approaches and MD simulations, and enables accurate extraction of dynamics information from the data. The strategy provides a multidisciplinary avenue for study by integrating experimental science, simulation science and data science.
Methods
Molecular dynamics simulations
All-atom MD simulations for macromolecules in the current datasets (34 molecules) were performed to obtain RMSF values that were used to train a deep-learning model in DEFMap or evaluate their performance. The selected macromolecules included proteins or complexes with DNA or RNA, which are conveniently handled in the simulations. The initial coordinates used in the simulations were obtained from PDB (https://www.rcsb.org) and were processed using the Structure Preparation module of the Molecular Operating Environment (MOE) software (Chemical Computing Group), version 2016.08 (https://www.chemcomp.com/). In brief, loops were modelled for disordered regions containing less than seven residues, and other non-natural amino termini and carboxy termini were capped with acetyl and formyl groups, respectively. Hydrogen atoms were added and topology files were generated using the pdb2gmx module in the GROMACS package 2016.551. All MD simulations were carried out with periodic boundary conditions (PBC), using GROMACS51 on an NVIDIA GeForce GTX 1080 GPU. The periodic cell was in the shape of an octahedron. The Amber ff99SB-ILDN force field was used for proteins, nucleotides and ions52, and the TIP3P potential was used for water53. Water molecules were placed around the substrate model within a distance of 10 Å, and counter ions (NaCl) were added to neutralize the system. Electrostatic interactions were calculated using the particle mesh Ewald (PME) method54 with a cut-off radius of 10 Å, and a nonbonded cut-off of 10 Å was applied for van der Waals interactions. The P-LINCS algorithm was used to constrain all bond lengths to their equilibrium values55. After energy minimization of the fully solvated models, the resulting systems were equilibrated for 100 ps under constant number of molecules, volume and temperature (NVT) conditions, followed by a 100 ps run under constant number of molecules, pressure and temperature (NPT) conditions, with the heavy atoms of the macromolecules held in fixed positions. The temperature was maintained at 298 K by velocity rescaling with a stochastic term56, and Parrinello–Rahman pressure coupling57 was used to maintain the pressure at 1 bar, with the temperature and pressure time constants set to 0.1 ps and 2 ps, respectively. Subsequently, production runs of 20 ns were carried out under NPT conditions without positional restraints. After PBC corrections, the generated trajectories were aligned using overall Cα atoms, and the RMSF values (Å) for heavy atoms were calculated using the rmsf module of GROMACS. Logarithmic RMSFs were then used to represent dynamics properties for the purpose of efficient training in DEFMap.
Data preparation for DEFMap
Twenty-five cryo-EM density maps were selected and downloaded from EMDB and PDB (Supplementary Table 1, average overall resolution = 3.62 ± 0.46) to train the 3D-CNN model. Their resolutions were relatively high compared to the average overall resolution of cryo-EM maps deposited in 2019 (5.6 Å). To evaluate the potential of DEFMap for dynamics analysis, ten additional cryo-EM density maps were selected and downloaded from EMDB and PDB (Supplementary Tables 3,4). To prepare sub-voxelized density data as input data for DEFMap, we carried out the data preparation as follows. Initially, the maps were rescaled to 1.5 Å per pixel and low-pass filtered with a cut-off of 5.0 Å using EMAN2.3 (ref. 58). Subsequently, the intensities were standardized within each map, and any negative values were eliminated. Each grid point in the maps was associated with the MD-derived logarithmic RMSF of the nearest atom in the voxelized coordinates. The resulting maps were sub-voxelized to generate the input density data, with 103 voxels distributed over 153 Å3. Here, the shape of the input data is 10 × 10 × 10 grid with a single channel. The training data were augmented by x-, y- and z-axis rotations of 90°, 180° and 270°, which resulted in a tenfold increase in the data. The preprocessed maps with an overall resolution of 5 Å were primarily used in this study. To investigate the dependence of DEFMap performance on map resolution (Fig. 2d), the cryo-EM maps used for training and the other three datasets were low-pass-filtered to resolutions of 6 Å, 7 Å, 8 Å, 9 Å and 10 Å, and the resulting maps were used as training datasets and prediction targets (for example, DynamicsDEFMap for the 10.0 Å test map was extracted using the model trained by the 10.0 Å maps). The atomic models were voxelized through high-throughput molecular dynamics (HTMD)59. All preprocessing procedures were performed using Python.
Construction of deep neural networks
The architecture of the neural network used in DEFMap included 3D convolutional blocks and dense blocks. The 3D convolutional blocks consisted of three 3D convolutional layers with Leaky ReLU activation, max pooling and dropout. The kernel size for convolution, the maximum pooling size and the dropout ratio were set to 4 × 4 × 4, 2 × 2 and 0.2, respectively. Different filter sizes (64, 128 and 256) were applied to the three 3D convolutional layers. The dense blocks consisted of two dense layers of 1,024 units with Leaky ReLU activation and a dense layer of a unit with identity activation. The mean squared error was used as the loss function. In total, the model has 5,774,785 trainable parameters. An overview of the neural networks is provided in Supplementary Fig. 2. The epochs, learning rate and batch size hyperparameters were set to 50, 0.00005 and 128, respectively, and early stopping with a patience interval of three epochs was used to prevent overfitting. To evaluate the performance of DEFMap, 25-fold cross-validation was performed using the 25 cryo-EM maps in a unit of protein. Specifically, we selected a protein as a test set and assigned the remaining 24 sets to training and validation (75% for training and 25% for validation), and we repeated this procedure 25 times. All of the learning curves for the 25-fold cross-validation are provided in Supplementary Fig. 3a. Subsequently, we trained the neural network model using all of the sub-voxels of the 25 cryo-EM maps (60% for training, 40% for validation) for further evaluation. The learning curve of the training set is provided in Supplementary Fig. 3b. All of the models were trained using an NVIDIA Tesla V100 GPU with 16 GB of memory. The Keras60 library 2.2.4 with TensorFlow 1.13.1 as backend was used for the calculations.
Postprocessing and visualization of output from the neural networks
Postprocessing of the atomic dynamics values calculated by DEFMap was carried out for further validation and analysis. First, the output values were normalized and then averaged over each residue. Next, the residue-specific values (DynamicsDEFMap) were assigned to the atomic models as temperature factors with HTMD59 and were visualized using the PyMOL61 and UCSF Chimera62 programs. To map the dynamics onto the viral particle cryo-EM maps, icosahedral symmetry was applied to the native output values (without normalization) using E2PROC3D.py from the EMAN2.3 package. All postprocessing procedures were performed using Python.
Data availability
All of the datasets used in this study are publicly available in the Protein Data Bank (PDB) at https://www.rcsb.org/ and the Electron Microscopy Data Bank (EMDB) at https://www.ebi.ac.uk/pdbe/emdb/. Detailed descriptions of the datasets are provided in Supplementary Tables 1,2,3,4. The models and the preprocessed input data used in 25-fold cross-validation are available in the Zenodo public repository at https://doi.org/10.5281/zenodo.4317158.
Code availability
The Python code used to implement DEFMap is available on GitHub at https://github.com/clinfo/DEFMap (https://doi.org/10.5281/zenodo.4317158). DEFMap is available under the MIT License.
References
Boehr, D. D., Nussinov, R. & Wright, P. E. The role of dynamic conformational ensembles in biomolecular recognition. Nat. Chem. Biol. 5, 789–796 (2009).
Kohen, A. Role of dynamics in enzyme catalysis: substantial versus semantic controversies. Acc. Chem. Res. 48, 466–473 (2015).
Cheng, Y. Single-particle cryo-EM—how did it get here and where will it go. Science 361, 876–880 (2018).
Cheng, Y., Grigorieff, N., Penczek, P. A. & Walz, T. A primer to single-particle cryo-electron microscopy. Cell 161, 438–449 (2015).
Murata, K. & Wolf, M. Cryo-electron microscopy for structural analysis of dynamic biological macromolecules. Biochim. Biophys. Acta Gen. Subj. 1862, 324–334 (2018).
Nitta, R., Imasaki, T. & Nitta, E. Recent progress in structural biology: lessons from our research history. Microscopy 67, 187–195 (2018).
Masson, G. R. et al. Recommendations for performing, interpreting and reporting hydrogen deuterium exchange mass spectrometry (HDX-MS) experiments. Nat. Methods 16, 595–602 (2019).
Hollingsworth, S. A. & Dror, R. O. Molecular dynamics simulation for all. Neuron 99, 1129–1143 (2018).
Kühlbrandt, W. Cryo-EM enters a new era. eLife 3, e03678 (2014).
Cheng, Y. Single-particle cryo-EM at crystallographic resolution. Cell 161, 450–457 (2015).
Merk, A. et al. Breaking cryo-EM resolution barriers to facilitate drug discovery. Cell 165, 1698–1707 (2016).
Gremer, L. et al. Fibril structure of amyloid-β(1–42) by cryo-electron microscopy. Science 358, 116–119 (2017).
Kato, T., Makino, F., Miyata, T., Horváth, P. & Namba, K. Structure of the native supercoiled flagellar hook as a universal joint. Nat. Commun. 10, 1–8 (2019).
Kujirai, T. et al. Structural basis of the nucleosome transition during RNA polymerase II passage. Science 362, 595–598 (2018).
Li, X. et al. A unified mechanism for intron and exon definition and back-splicing. Nature 573, 375–380 (2019).
Vilas, J. L. et al. MonoRes: automatic and accurate estimation of local resolution for electron microscopy maps. Structure 26, 337–344.e4 (2018).
Kucukelbir, A., Sigworth, F. J. & Tagare, H. D. Quantifying the local resolution of cryo-EM density maps. Nat. Methods 11, 63–65 (2014).
Berman, H. M. et al. The protein data bank. Nucleic Acids Res. 28, 235–242 (2000).
Hollingsworth, S. A. et al. Cryptic pocket formation underlies allosteric modulator selectivity at muscarinic GPCRs. Nat. Commun. 10, 3289 (2019).
Plattner, N., Doerr, S., De Fabritiis, G. & Noé, F. Complete protein–protein association kinetics in atomic detail revealed by molecular dynamics simulations and Markov modelling. Nat. Chem. 9, 1005–1011 (2017).
Ji, S., Xu, W., Yang, M. & Yu, K. 3D convolutional neural networks for human action recognition. IEEE Trans. Pattern Anal. Mach. Intell. 35, 221–231 (2013).
Maturana, D. & Scherer, S. VoxNet: a 3D convolutional neural network for real-time object recognition. In Proc. IEEE/RSJ International Conference on Intelligent Robots and Systems (IROS) 922–928 (IEEE, 2015).
Zhirong, W. et al. 3D ShapeNets: a deep representation for volumetric shapes. In Proc. IEEE Conference on Computer Vision and Pattern Recognition (CVPR) 1912–1920 (IEEE, 2015).
Chen, H., Dou, Q., Yu, L., Qin, J. & Heng, P.-A. VoxResNet: deep voxelwise residual networks for brain segmentation from 3D MR images. NeuroImage 170, 446–455 (2018).
Kamnitsas, K. et al. Efficient multi-scale 3D CNN with fully connected CRF for accurate brain lesion segmentation. Med. Image Anal. 36, 61–78 (2017).
Qi, D. et al. Automatic detection of cerebral microbleeds from MR Images via 3D convolutional neural networks. IEEE Trans. Med. Imaging 35, 1182–1195 (2016).
Avramov, T. K. et al. Deep learning for validating and estimating resolution of cryo-electron microscopy density maps. Molecules 24, 1181 (2019).
Maddhuri Venkata Subramaniya, S. R., Terashi, G. & Kihara, D. Protein secondary structure detection in intermediate-resolution cryo-EM maps using deep learning. Nat. Methods 16, 911–917 (2019).
Mostosi, P., Schindelin, H., Kollmannsberger, P. & Thorn, A. Automated interpretation of cryo-EM density maps with convolutional neural networks. bioRxiv, 644476 (2019).
Xu, K., Wang, Z., Shi, J., Li, H. & Zhang, Q. C. A2-Net: molecular structure estimation from cryo-EM density volumes. In Proc. AAAI Conference on Artificial Intelligence 33, 1230–1237 (AAAI, 2019).
Lawson, C. L. et al. EMDataBank.org: unified data resource for CryoEM. Nucleic Acids Res. 39, D456–D464 (2010).
de la Rosa-Trevín, J. M. et al. Scipion: a software framework toward integration, reproducibility and validation in 3D electron microscopy. J. Struct. Biol. 195, 93–99 (2016).
Goodfellow, I., Bengio, Y. & Courville, A. Deep Learning (MIT Press, 2016).
Zhang, W., Lukoyanova, N., Miah, S., Lucas, J. & Vaughan, C. K. Insights into centromere DNA bending revealed by the Cryo-EM structure of the core centromere binding factor 3 with Ndc10. Cell Rep. 24, 744–754 (2018).
Sun, Y. et al. Molecular basis for the recognition of the human AAUAAA polyadenylation signal. Proc. Natl Acad. Sci. USA 115, E1419–E1428 (2018).
Cash, J. N. et al. Cryo–electron microscopy structure and analysis of the P-Rex1–Gβγ signaling scaffold. Sci. Adv. 5, eaax8855 (2019).
Dedden, D. et al. The architecture of Talin1 reveals an autoinhibition mechanism. Cell 179, 120–131.e13 (2019).
Wongpalee, S. P. et al. CryoEM structures of Arabidopsis DDR complexes involved in RNA-directed DNA methylation. Nat. Commun. 10, 3916 (2019).
Lan, P. et al. Structural insight into precursor tRNA processing by yeast ribonuclease P. Science 362, eaat6678 (2018).
Abascal-Palacios, G., Ramsay, E. P., Beuron, F., Morris, E. & Vannini, A. Structural basis of RNA polymerase III transcription initiation. Nature 553, 301–306 (2018).
Walls, A. C. et al. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 181, 281–292.e6 (2020).
Walls, A. C. et al. Tectonic conformational changes of a coronavirus spike glycoprotein promote membrane fusion. Proc. Natl Acad. Sci. USA 114, 11157–11162 (2017).
Wrapp, D. et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 367, 1260–1263 (2020).
Kostyuchenko, V. A. et al. Structure of the thermally stable Zika virus. Nature 533, 425–428 (2016).
Wu, W. et al. Expression of quasi-equivalence and capsid dimorphism in the Hepadnaviridae. PLoS Comput. Biol. 16, e1007782 (2020).
Cao, L. et al. Structural basis for neutralization of hepatitis A virus informs a rational design of highly potent inhibitors. PLoS Biol. 17, e3000229 (2019).
Liu, Y. et al. Molecular basis for the acid-initiated uncoating of human enterovirus D68. Proc. Natl Acad. Sci. USA 115, E12209–E12217 (2018).
Hamaguchi, T. et al. A new cryo-EM system for single particle analysis. J. Struct. Biol. 207, 40–48 (2019).
Kato, T. et al. CryoTEM with a cold field emission gun that moves structural biology into a new stage. Microsc. Microanal. 25, 998–999 (2019).
Ramírez-Aportela, E. et al. Automatic local resolution-based sharpening of cryo-EM maps. Bioinformatics 36, 765–772 (2019).
Abraham, M. J. et al. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1-2, 19–25 (2015).
Lindorff-Larsen, K. et al. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 78, 1950–1958 (2010).
Jorgensen, W. L. & Thomas, L. L. Perspective on free-energy perturbation calculations for chemical equilibria. J. Chem. Theory Comput. 4, 869–876 (2008).
Darden, T., York, D. & Pedersen, L. Particle mesh Ewald: an N⋅Log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 (1993).
Hess, B. P-LINCS: a parallel linear constraint solver for molecular simulation. J. Chem. Theory Comput. 4, 116–122 (2008).
Bussi, G., Donadio, D. & Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 014101 (2007).
Parrinello, M. & Rahman, A. Polymorphic transitions in single crystals: a new molecular dynamics method. J. Appl. Phys. 52, 7182–7190 (1981).
Tang, G. et al. EMAN2: an extensible image processing suite for electron microscopy. J. Struct. Biol. 157, 38–46 (2007).
Doerr, S., Harvey, M. J., Noé, F. & De Fabritiis, G. HTMD: high-throughput molecular dynamics for molecular discovery. J. Chem. Theory Comput. 12, 1845–1852 (2016).
Chollet, F. Keras. https://keras.io (2015).
Schrödinger LLC. The PyMOL Molecular Graphics System (version: 2.2.0) (2018).
Pettersen, E. F. et al. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Acknowledgements
This work was supported by MEXT as ‘Priority Issue on the Post K computer (Building Innovative Drug Discovery Infrastructure Through Functional Control of Biomolecular Systems)’ and as ‘Program for Promoting Researches on the Supercomputer Fugaku (Application of Molecular Dynamics Simulation to Precision Medicine Using Big Data Integration System for Drug Discovery)’. S.M. was supported by JSPS KAKENHI Grant Number JP17K15106.
Author information
Authors and Affiliations
Contributions
S.M., S.I., K.T. and Y.O. designed the study. S.M. and S.I. performed the preprocessing of cryo-EM maps, designed the neural networks and carried out model training and DEFMap analyses. S.M. and M.A. performed the MD calculations. S.M. and T.K. prepared the original cryo-EM map data. K.T. and Y.O. conceived the project. S.M. and S.I. wrote the manuscript. All of the authors discussed the research, edited the manuscript and approved its final version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Machine Intelligence thanks Joseph Davis, Carlos O. S. Sorzano and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Correlation plot of the cryo-EM map local resolution (a) and the given density data (b) with DynamicsMD for EMD-3948 (entry 1 in Supplementary Table 2).
Normalized residue-averaged values are shown in the correlation plot. The regression lines are coloured orange; r denotes the correlation coefficient. The average correlation coefficient for the density data of 25 cryo-EM maps used in training is 0.459 ± 0.179 (Supplementary Table 2).
Extended Data Fig. 2 Correlation plots between MD-derived and DEFMap-determined dynamics for proteins not included in the training dataset.
a,b, Normalized atomic (a, blue) and residue-averaged (b, purple) values are shown in the correlation plots; the regression lines are coloured orange; r denotes the correlation coefficient.
Extended Data Fig. 3 The map resolution dependence of the dynamics extraction accuracy for test macromolecules (Supplementary Tables 3 and 4).
a,b, Correlation coefficient values between the MD- and DEFMap-derived atomic-specific (a) and residue-specific (b) dynamics at different resolutions. The cryo-EM maps of the training and the evaluation datasets were preprocessed to identical resolutions with low-pass filters.
Extended Data Fig. 4 DEFMap performance for a map with the intermediate resolution (EMDB/PDB entry, EMD-4772/6R9T).
a, Distribution of local resolution. The local resolution is calculated using the MonoRes implementation in Scipion and mapped onto the original map using different colours as indicated in the colour bars. b,c,d, The correlation plots between DynamicsDEFMap and DynamicsMD for the intermediate-resolution map. The target cryo-EM maps are processed by low-pass filters with cutoff values of 7 Å (b), 5 Å (c), and 6 Å (d). The predictions are carried out using the models trained by the datasets preprocessed to the identical resolutions. The regression lines are coloured orange; r denotes the correlation coefficient.
Extended Data Fig. 5 The spatial distribution of the fragment-specific dynamics in the atomic models (EMDB/PDB entry, EMD-20308/6PCV).
The representative fragments-specific dynamics for Rac exchanger 1 complexed with G protein beta gamma subunits are mapped onto the atomic model using different colours as indicated in the colour bar (HDX-MS, left; MD, middle; DEFMap, right). The fragments not detected in the HDX-MS experiments are coloured grey.
Extended Data Fig. 6 Residue-specific dynamics profiles for apo/holo proteins.
a,b, DynamicsMD (black) and DynamicsDEFMap (magenta) datasets for 6OIS/EMD-20080 and 6OIT/EMD-20081 (a), 6AGB/EMD-9616 and 6AH3/EMD-9622 (b), and 6EU2/EMD-3957 and 6EU1/EMD-3956 (c), respectively, plotted against the residue IDs, numbered according to their order in the coordinate files. r denotes the correlation coefficient between DynamicsMD and DynamicsDEFMap.
Extended Data Fig. 7 Atomic models of residues showing allosteric dynamics changes induced by ligand binding in the DMS3-RDM1 complex with DRD1 peptide.
a,b, The main chains of the hinge domain in DMS3 dimer 1 (a) and in the RDM1-DMS3 dimer 2 binding interface (b) are coloured according to the extent of ligand-induced changes in dynamics using the indicated colour bar. The side chains of residues showing the dynamics changes of less than -0.5 are represented as sticks (apo, magenta; holo, cyan).
Extended Data Fig. 8 Conformational changes of the spike protein of SARS-CoV-2.
a, Overall architecture of the spike protein of SARS-CoV-2. The spike protein mediates the entry of the virus into host cells. The protein is composed of two subunits: S1 subunit is involved in the recognition of the host cell receptor and S2 subunit is involved in the viral and membrane fusion process. The receptor binding domain in the S1 subunit exhibits a hinge-like motion and the ‘down’ and ‘up’ conformations are shown in grey and magenta, respectively. The conformational transition from the ‘down’ to ‘up’ states is indicated by a red arrow in the right-hand panel. The structural components associated with the structural transition from prefusion to post-fusion forms, the central helix, β-rich region, and the heptad-repeat 1 (HR1) motif, are indicated. b, Conformational rearrangement of the HR1 motif in the state transition from the prefusion to post-fusion forms. The post-fusion form of the spike protein in SARS-CoV-2 is shown in green by superimposing with the prefusion form. The conformational rearrangement of the HR1 motif is indicated by a yellow arrow. The prefusion form is coloured according to the indicated colour bar.
Extended Data Fig. 9 A correlation plot between DynamicsDEFMap calculated using EMD21375 and EMD-21375.
The regression lines are coloured orange; r denotes the correlation coefficient.
Extended Data Fig. 10 DEFMap-based dynamics visualization of virus particles.
The DEFMap-derived dynamics data are mapped onto the cryo-EM maps of the viral particles by applying the icosahedral symmetry. The colour range is defined by the minimum and maximum values in each inference. The scale bar represents 50 Å and is indicated by black lines. EMDB/PDB entries are indicated. The atomic model corresponding to EMD-9053 (right) is not available.
Supplementary information
Supplementary Information
Supplementary Figs. 1–3 and Tables 1–5.
Rights and permissions
About this article

Cite this article
Matsumoto, S., Ishida, S., Araki, M. et al. Extraction of protein dynamics information from cryo-EM maps using deep learning. Nat Mach Intell 3, 153–160 (2021). https://doi.org/10.1038/s42256-020-00290-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s42256-020-00290-y
This article is cited by
-
Low-data interpretable deep learning prediction of antibody viscosity using a biophysically meaningful representation
Scientific Reports (2023)
-
A robust normalized local filter to estimate compositional heterogeneity directly from cryo-EM maps
Nature Communications (2023)
-
The transformational role of GPU computing and deep learning in drug discovery
Nature Machine Intelligence (2022)
-
Visualization of judgment regions in convolutional neural networks for X-ray diffraction and scattering images of aliphatic polyesters
Polymer Journal (2021)
-
Experimental demonstration of the novel “van-Hove integral method (vHI)” for measuring diffusive dynamics by elastic neutron scattering
Scientific Reports (2021)
