
Abstract
Tumour necrosis factor (TNF) is a classical, pleiotropic pro-inflammatory cytokine. It is also the first ‘adipokine’ described to be produced from adipose tissue, regulated in obesity and proposed to contribute to obesity-associated metabolic disease. In this review, we provide an overview of TNF in the context of metabolic inflammation or metaflammation, its discovery as a metabolic messenger, its sites and mechanisms of action and some critical considerations for future research. Although we focus on TNF and the studies that elucidated its immunometabolic actions, we highlight a conceptual framework, generated by these studies, that is equally applicable to the complex network of pro-inflammatory signals, their biological activity and their integration with metabolic regulation, and to the field of immunometabolism more broadly.
Similar content being viewed by others


Main
Immune and metabolic responses are highly integrated and are fundamental for the proper function of multicellular organisms. Although such highly interconnected response systems are essential for survival, when improperly launched and poorly controlled, they can be uniformly damaging. This is certainly true for metabolic homeostasis and disease, and it is this interface that we will discuss here using TNF as a prototypical example.
Tumour necrosis factor alpha (TNF-α), also known as TNF superfamily member 2 (TNFSF2) or simply TNF, is a multifunctional cytokine that has well-established immunological roles in innate and adaptive immunity and in the normal physiological functions of immune cells, where its production and actions tend to be both temporally and spatially limited. By contrast, abnormally elevated and sustained production of TNF is associated with pathogenic inflammatory disease states including infection-linked sepsis and chronic autoimmune diseases. In recent decades, TNF has also come to be known as an adipokine, after the serendipitous observation of its elevated production in adipose tissue in obesity, which led to the appreciation of the inflammatory nature of obesity and associated metabolic pathologies. These and other critical observations stimulated the exponential growth in our understanding of adipose tissue biology and obesity and together fuelled a renaissance in the field of energy metabolism and the emergence of a new field of research: ‘immunometabolism’1. Immunometabolism focuses on elucidating both the effects of immune-derived signals on the metabolic reprograming of non-immune cells and the metabolic programs that underpin immune cell function. Similarly, new scientific terms and concepts (such as ‘adipokines’ and ‘metaflammation’) have emerged to define the unique combination of overlapping mediators and/or states that contribute to metabolic and immunometabolic homeostasis in health or during disease.
This article provides a brief introduction to the discovery of TNF as a classical, pleiotropic inflammatory mediator and the findings that elucidated its role in metabolism and metabolic disease. It is now clear that the metabolic functions of TNF and its impact on healthy metabolic homeostasis, as well as in metabolic disease (particularly in obesity-linked insulin resistance and glucose homeostasis), are extremely highly conserved: for example, all of its principal proximal and distal signalling elements have been clearly demonstrated in the fruit fly2,3,4, and blocking TNF activity can completely prevent the development of a Drosophila version of diabetes5. Nevertheless, we would also like to state at the outset that TNF and adipose tissue inflammation are presented here as a foundational framework with which to approach the metaflammatory nature of obesity and associated pathologies. We do not suggest that TNF has solitary causative or therapeutic potential, as the involvement of a plethora of additional inflammatory cytokines and mediators, as well as many other metabolic sites and immune effectors, has been shown and tested in metabolic diseases6,7,8,9. For a historic perspective on the broader immunological nature of metabolic diseases, readers are directed to a recent comprehensive review4.
History of TNF as a metabolic messenger
Discovery of TNF
TNF was amongst the first ‘cytokines’ to be identified. During the pre-molecular-biology era (before the 1980s), the discovery of clinically relevant factors relied on empirical and biochemical approaches to systematically isolate and screen principal bioactive factors, and so it was for the discovery of TNF (Fig. 1). Three independent clinical observations prompted the search for the key bioactive factor, and each paved the way to converge on the discovery of TNF. These observations also laid the groundwork for characterizing some of its key immunometabolic actions. The first observation was that burn injury could induce tumour regression; this can be traced back to the fourteenth century, when burn-induced tumour regression was practiced10, and to the use of Coley’s toxins (a mixture of heat-killed bacteria) for similar purposes in the nineteenth century. We now know that burns and infections cause inflammation and TNF production, as well as insulin resistance. Second, it was noted that a serum factor was capable of mediating the anorexia, tissue catabolism and fatal weight loss that characterize cachexia (often associated with cancer and some infectious diseases)11. Third, septic shock (following severe infections) was seen to involve the production of cytotoxic-lymphocyte-derived factors that stimulated pyrogenic activity (fevers) and hypertriglyceridaemia and suppressed lipid clearance. It is now known that these lymphocyte-derived factors included TNF. Consequently, TNF was historically recognized by three different names tumour necrosis factor alpha (TNF-α), cachectin and lymphotoxin—each name based on the cellular source and biological actions of the elusive factor being investigated.

Before its cloning (in 1984) and purification (in 1985), TNF was known by three names: TNF-α, cachectin and lymphotoxin. During this time, its production by activated macrophages and lymphocytes and effect on lipid metabolism were already established and under investigation in studies linking infection to altered carbohydrate and lipid metabolism. In parallel, the pathophysiology linking obesity to insulin resistance and metabolic diseases was being defined. After 1984, with the availability of purified proteins, TNF-binding receptors were identified and cloned and gene deletion models developed, along with the first anti-TNF monoclonal antibodies (mAbs). In 1993*, the elevated production of TNF in obese adipose tissue and its potential role in obesity-linked insulin resistance and diabetes were first reported, prompting a plethora of physiological, clinical and mechanistic studies aimed at dissecting the metabolic biology of TNF and its links to the immune response. This led to the implication of transcriptional regulation of key metabolic genes and crosstalk with insulin signalling involving IRS1 serine kinases, such as JNKs and IKKs, as key molecular players. In the 2000s, with the demonstration that adipose tissue macrophages were linked to metabolic dysfunction in obesity, additional pro-inflammatory immune cells and cytokines were implicated in adipose tissue inflammation. Although limited anti-TNF clinical studies in obesity-linked T2D produced both negative and positive results, other trials in patients with related inflammatory conditions confirmed a reduced diabetes risk. These observations are now consistent with those of numerous clinical studies using diverse anti-inflammatory strategies and showing association between beneficial metabolic outcomes, but further clinical studies are clearly warranted. Asterisk indicates several studies now recognized as milestone studies in Nature Milestones: Diabetes (https://www.nature.com/articles/d42859-021-00015-0, 2021). ATMs, adipose tissue macrophages; clin, clinical; CVD, cardiovascular disease; C/EBPα, CCAAT/enhancer-binding protein alpha (transcription factor); Glut4, insulin-sensitive glucose uptake transporter; IR, insulin resistance; IRS1, insulin receptor substrate 1; LPL, lipoprotein lipase; NASH, non-alcoholic steatohepatitis; preclin, preclinical; PPARγ, peroxisome proliferator-activated receptor gamma2 (lipogenic transcription factor); T2D, type 2 diabetes; TNF, tumour necrosis factor; TNFRs, TNF receptors; TNFR1, TNF receptor 1.
With advances in molecular biology, human ‘tumour necrosis factor’ and ‘lymphotoxin’ were cloned in 1984, and this was followed a year later by the purification and amino acid sequencing of murine ‘cachectin’12,13. These discoveries allowed confirmation that all three factors were the same protein and that it was distinct from, albeit structurally related to, another cytokine, lymphotoxin alpha14. The production of pure recombinant proteins then allowed the confirmation of previously attributed biological activities, exploration of mechanisms of action and generation of several classes of anti-TNF therapeutics. Further developments in genetic engineering also facilitated the development of genetically altered cellular and animal models of human disease, which have provided exceptionally exquisite platforms to manipulate and dissect the involvement of specific molecular players. These developments have proved invaluable for our current understanding of the molecular mechanisms that mediate the immunometabolic actions of TNF, as well as many other cytokines and immune mediators (in vitro and in vivo), their involvement in metabolic disease development and their production of novel targeted therapeutics for a variety of inflammatory conditions.
Discovery of TNF in obesity-linked insulin resistance and type 2 diabetes
The earliest clues that inflammation could be involved in obesity-linked metabolic dysfunction came in the 1960s from the observations that immune cells are present in adipose tissue in preclinical models of obesity and hyperglycaemia15,16 and in the 1980s from landmark studies demonstrating the inhibition of insulin action in cultured adipocytes by an activated macrophage-derived factor17,18 (Fig. 1). These early reports were largely overlooked until studies showed that TNF levels were elevated in adipose tissue in obese and insulin-resistant rodents and suggested that TNF had a role in mediating insulin resistance19, stimulating interest in this new biology. These observations formed what are now recognized as milestone studies in diabetes20 and were quickly clinically validated in human obesity and extended to establish the presence of similarly elevated levels in insulin-resistant muscle tissue21,22. In contrast to the localized elevation of TNF production, some early studies did not observe an increase in circulating TNF or its association with obesity or insulin resistance23,24. This resulted in a cautious, and arguably reluctant, acceptance of TNF’s role in human metabolic disease. However, numerous subsequent studies (summarized in a recent meta-analysis) have now established a clear positive association between TNF levels and type 2 diabetes (T2D) risk in humans25,26. Moreover, in multiple human gene-association studies, TNF polymorphisms have been linked to obesity, diabetes and other aspects of metabolic syndrome27. Collectively, these observations contextualized earlier clinical and preclinical studies in which in vivo administration of TNF was found to induce insulin resistance and impair glucose metabolism in healthy human subjects28,29,30 and in many preclinical animal models31,32,33.
The availability of genetically engineered mice lacking either TNF (in 1996) or its receptors (in 1993–1994) allowed the definitive demonstration that the action of TNF (via TNF receptor 1, TNFR1) was involved in obesity-linked insulin resistance in several preclinical models of obesity and T2D34,35,36,37. These studies were further complemented by numerous demonstrations that neutralization of TNF (with anti-TNF antibodies or peptides) in obese rodents could improve insulin sensitivity and glucose homeostasis19,38,39. The evidence for the role of TNF in disrupting insulin action and glucose metabolism is unequivocal and produced in numerous independent model systems and in humans. By contrast, the therapeutic effect of blocking TNF on human metabolism remains a topic of debate, being mostly based on early studies indicating that acute reduction of systemic TNF may not be sufficient to induce metabolic benefits. However, these studies were limited in scope, and subsequent cumulative evidence supports a beneficial impact of anti-TNF therapies on glucose metabolism (see https://www.metaflammation.org/). Although the proof-of-principle studies in humans do exist, it is not possible to reach definitive conclusions without performing large randomized clinical trials and taking into account the extensive experience now available with patient stratification, clinical use of monoclonal antibodies (mAbs)40 and alternative therapeutic entities41.
Emerging evidence of TNF in other metabolic and pro-inflammatory diseases
Evidence is also emerging for pathogenic roles of TNF in other metabolic diseases, such as non-alcoholic fatty liver disease (NAFLD): a spectrum of conditions linked to obesity that includes hepatosteatosis, non-alcoholic steatohepatitis (NASH) and cirrhosis. It is the latter two stages of NAFLD that can lead to end-stage liver disease and hepatocellular carcinoma and are associated with increased TNF expression and activity42,43. Mechanistically, hepatic TNF action has been implicated in promoting NASH progression in rodent models44. Polymorphisms in TNF genotypes also confer susceptibility to colorectal liver metastases in patients with NAFLD45. Additional observations suggest that obesity may promote liver inflammation and the development of hepatocellular carcinoma in a TNF-dependent manner46,47. Moreover, rodent models of NAFLD lacking TNF action exhibit improved insulin sensitivity and less pronounced liver steatosis and fibrosis48. However, hepatocyte-specific TNFR1 ablation improves insulin resistance but not NASH49, except in the context of dysbiosis44. A new anti-human-TNFR1 antibody has also been demonstrated to reduce liver steatosis, hepatocellular injury and fibrosis in a preclinical model of NAFLD50. Clinically, in patients with rheumatoid arthritis and NAFLD, treatment with TNF inhibitors also has beneficial effects on hepatic tissue51. However, in patients with multiple immune-related diseases, short-term anti-TNF treatment to prevent the development of cirrhosis or NASH may not be beneficial52. In this area, there is also a need for additional human studies and randomized clinical trials to address the indications and target patient populations that may benefit the most from such anti-inflammatory treatments.
Atherosclerosis is a pro-inflammatory and pro-atherogenic vascular disease that develops in the context of the metabolic syndrome with atherogenic dyslipidaemia (low high-density lipoprotein and high triglyceride levels), hyperglycaemia and elevated blood pressure. Preclinical studies in mouse models of atherosclerosis have provided compelling evidence for the pathogenic involvement of TNF. For example, pro-atherogenic mice lacking TNF exhibited reduced atherogenesis, endothelial adhesiveness and inflammatory markers when compared with controls53. Clinically, the use of anti-TNF therapy improves lipid profiles and reduces the incidence of heart failure and cardiovascular disease events40,54,55,56. Since TNF action is relevant to many aspects of cardiovascular function and disease, it is critical to conduct additional specialized randomized clinical studies to maximize the benefits and eliminate potential adverse effects40,57. Indeed, a very large randomized controlled trial targeting interleukin-1β (IL-1β) has led to tremendous renewed interest in the therapeutic utility of inflammatory interventions in atherosclerosis58.
Neuroinflammation associated with metabolic syndrome increases the likelihood and severity of several neurological disorders. Indeed, links between obesity, diabetes and Alzheimer’s disease are now well established59,60. Interestingly, neuroinflammation-related impaired insulin action in the brain involves the same molecular mechanisms as were reported earlier in peripheral tissues61,62 and is sometimes even referred to as a ‘type 3’ form of diabetes63. Beyond metabolic diseases, dysregulated TNF production is associated with additional disease states not currently considered classic metabolic diseases, including asthma and several different forms of cancer64,65, which are also strongly associated with obesity. In fact, these are exciting areas for further investigation and the application of immunometabolic approaches.
Finally, it is worth highlighting that dysregulated TNF expression, in which either too little or too much TNF is produced, has also been linked to differential susceptibility to several major infectious diseases, including tuberculosis and cerebral malaria66,67. Moreover, during the COVID-19 pandemic, obesity has emerged as one of the most important risk factors for predicting disease severity and death. Increased COVID-related mortality is also linked to older age and/or pre-existing health conditions, including obesity, T2D, cardiovascular diseases and chronic obstructive pulmonary disease, all conditions that involve metaflammation. Furthermore, TNF (and IL-6) levels also appear to be the most prominent cytokine levels that independently and significantly predict COVID-19 disease severity and death68. Anti-TNF therapy is already being trialled in patients with COVID-1969,70,71. Together, these findings suggest that one plausible model is that the abnormalities in the inflammatory state associated with obesity and the dysregulated production of inflammatory mediators, including TNF and others, have critical roles in multiple co-morbidities of obesity, beyond insulin resistance and diabetes.
Sites of TNF action in the metabolic syndrome
The collection of risk factors that define the metabolic syndrome includes central (visceral) obesity, dysregulated carbohydrate metabolism (insulin resistance, hyperglycaemia), dysregulated lipid metabolism (dyslipidaemia, hypertriglyceridaemia, reduced high-density lipoprotein cholesterol) and hypertension. Collectively, these factors are associated with an increased risk of developing non-communicable metabolic diseases such as T2D, NAFLD, cardiovascular disease, chronic kidney disease, airway disease and some cancers. All these diseases have additional features in common: they are all chronic conditions (often occurring over a period of years before clinical diagnosis); they all involve substantial tissue remodelling; they all co-exist with a state of low-grade chronic inflammation (or metaflammation); and this inflammation is also initially localized to specific tissues, often at the site of tissue remodelling.
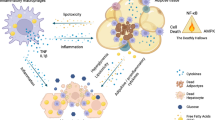
It is therefore not surprising that common pro-inflammatory molecular players including TNF and associated signalling networks have been implicated in promoting the pathogenesis of multiple metabolic diseases (Fig. 2). Since the initial observation of elevated adipose-derived TNF in obesity19, it has become clear that this archetypal pro-inflammatory cytokine can be causally linked to individual risk factors that define the metabolic syndrome and may serve to redirect fuel availability by reprograming energy metabolism. TNF promotes carbohydrate dysregulation (hyperglycaemia) in part by inhibiting insulin action, reducing glucose clearance (primarily by muscle and adipose tissue) and increasing hepatic glucose production. Promotion of hyperlipidaemia by TNF is mediated in part by stimulation of hepatic lipid synthesis31 and adipose lipolysis, along with suppression of triacylglycerol clearance and inhibition of insulin-stimulated de novo lipogenesis (in adipose tissue)72,73.

Both TNF and its receptors are conserved between mice and human. Similarly, many of the observations depicted have been reported in vitro (in human and rodent cellular models) and in vivo (both in preclinical rodent models of metabolic disease and through association studies in human disease). Notably, in most instances, such as metaflammation, TNF is likely to act locally within the same tissue in which its expression is elevated. Besides its direct action on context-dependent cellular metabolism, TNF also regulates the production of additional metabolic messengers: cytokines (↓ anti-inflammatory, ↑ pro-inflammatory), chemokines (↑ immune cell recruitment), hepatokines (liver: for example, ↓ fetuin A), adipokines (adipose: for example, adiponectin, leptin) and hormones (for example, ↑ GLP-1, ↑ GDF15). Collectively, these messengers represent a complex extracellular network of signals involved in regulating whole-body energy homeostasis and disease development. At the tissue level, the biological actions include promoting immune cell recruitment (most tissues), cell activation (Kupffer cells, adipose tissue macrophages, hepatic and pancreatic stellate cells), fibrosis (liver, adipose tissue, kidney and atherosclerotic vasculature), cell proliferation (liver, vascular smooth muscle cells), apoptosis (most tissues) and differentiation (for example, osteoclastogenesis in bone), as well as in inhibiting cell differentiation (for example, adipogenesis in adipose tissue, myogenesis in muscle, osteoblastogenesis in bone). Ultimately, these actions are key in supporting the local tissue-remodelling events that are characteristic of chronic metabolic diseases linked to metaflammation. In non-adipose tissues, TNF can induce ectopic fat deposition by increasing lipogenesis (for example, in liver) and/or decreasing fatty acid oxidation (FAO) in muscle, liver and heart. In obesity-related metabolic diseases, TNF may also promote ectopic fat deposition in key metabolic tissues (liver, pancreas and muscle) indirectly through its anti-adipogenic actions to further limit adipose tissue expansion. Not shown above: additional tissues reported to have TNF-induced metabolic activity include brown adipose tissue (energy expenditure), gut (dysbiosis and absorption), bone (RA) and skin. AA, amino acid; ECs, endothelial cells; FFA, free fatty acid; HGP, hepatic glucose production; NAFLD, non-alcoholic fatty liver; NEFA, non-esterified fatty acid; NOX, NADPH-dependent oxidases; OxPhos, oxidative phosphorylation; PPP, pentose phosphate pathway; PT, proximal tubules; RCT, reverse cholesterol transport; ROS, reactive oxygen species; TNF, tumour necrosis factor.
TNF also acts indirectly to regulate whole-body energy metabolism. Its pro-inflammatory actions alter the production of other adipokines, metabokines, lipokines and cytokines. TNF also promotes lipolysis and increases free fatty acid levels, which can influence adverse metabolic outcomes, including insulin resistance. On the other hand, TNF can suppress adiponectin, an adipokine with anti-inflammatory and insulin-sensitizing effects. Although TNF has well-established roles as a potent catabolic and anti-adipogenic cytokine, how can this action be reconciled with the fact that it is elevated in obesity—an anabolic state? One critical determinant of this outcome is spatial and temporal context and the levels of exposure. Another framework that may offer a solution is the concept of limited adipose tissue expansion. We know that adipose tissue does not have unlimited capacity to expand and infinitely store surplus fuel safely (as neutral lipids). Consequently, in the face of chronic nutritional surplus, its maximum storage capacity will eventually be exceeded, resulting in impaired clearance of toxic lipids, which inevitably accumulate in tissues not designed to store them safely. Hence, metabolic stress and tissue dysfunction ensue. Given its role as an anti-adipogenic cytokine whose expression is elevated in obese adipose tissue, TNF could be considered an excellent molecular mediator of the mechanisms involved in limiting adipose tissue expansion in obesity-linked metabolic disease73,74,75. Removal of this barrier might provide further lipid storage capacity in adipose tissue. Of course, other possibilities exist, such as a potential mechanism that exists to adapt to nutrient deprivation.
Cholesterol metabolism is also regulated by TNF. In the liver, TNF promotes cholesterol and apolipoprotein biosynthesis while decreasing cholesterol catabolism and excretion as bile acids. In immune cells (such as macrophages and lymphocytes), TNF also inhibits reverse cholesterol transport76. Importantly, the recognized differences in cholesterol–lipoprotein metabolism between rodents and humans have substantially limited the translation of many mechanistic insights to clinical therapeutics. From a mechanistic perspective, hypertension is arguably the least well understood component of the metabolic syndrome. However, as one of several inflammatory mediators, TNF promotes hypertension in multiple ways, for example by inducing vascular insulin resistance and reducing vasodilation, but also by increasing intravascular fluid and vasoconstriction and promoting sympathetic overactivity77. Again, these actions are not exclusive to TNF but rather can be generated through the action of a multitude of inflammatory mediators.
Globally, it is likely that the catabolic actions of TNF (and other pro-inflammatory cytokines) serve a primary metabolic role to mobilize stored fuel (initially to ensure glucose and free fatty acid availability in the circulation and then through proteolysis). This process may be required to support energy-demanding immune functions associated with inflammation, such as microbicidal activity (via production of reactive oxygen species and nitrogen radicals), production of pro-inflammatory cytokines and chemokines, and immune cell migration and proliferation. Hence, repartitioning of fuel and biological resources may be the physiological process that supports host defence. However, in the absence of inflammatory resolution and under prolonged stress (such as chronic nutritional overload), these actions become pathological. As most pathological conditions are related to an ancestral role in physiology, this dual activity spectrum of inflammatory mediators is critical in remodelling and interpreting their context-dependent roles in chronic disease settings. Indeed, most adaptive measures are essential for survival, but only when they can be limited temporally and spatially—otherwise, they become uniformly destructive and pathological.
Molecular mechanisms of TNF action
TNF production and TNF receptors
TNF is produced by a range of immune and non-immune cells and is the first cytokine to appear within minutes of any injury or stress by pro-inflammatory stimuli78. However, its production can be regulated at multiple levels: transcriptional79, post-transcriptional80,81, translational82 and post-translational83. It exists as two signalling-competent forms. The first is a 26-kDa transmembrane trimer (mbTNF in Fig. 3), which can signal through both classical paracrine and retrograde signalling84,85. The second is a 17-kDa soluble trimer (sTNF), which is produced by the action of cell surface proteases such as TNF-α-converting enzyme (TACE/ADAM1786). Interestingly, Tace+/– heterozygote mice have been shown to be protected from obesity-induced insulin resistance87.

TNF signalling complexes can elicit distinct actions in a cell type- and context-dependent manner. TNF can also switch from inducing gene activation to inducing cell death, depending on the bioavailability of specific proteins. Both sTNF and mbTNF can bind TNF receptors 1 and 2, and each receptor can elicit both common and distinct signals. Activation of TNFR1 by sTNF has been best characterized in relation to metaflammation. This involves the recruitment of TNF receptor signalling complex I (TNF-RSCI), which is dependent on ubiquitinated RIPK1. Signals mediated by TNF-RSCI lead to activation of MAP kinase cascades (including IRS-1 serine kinases: IKKβ, JNK, Erk and p38) and downstream transcription factors whose activity promote cell survival. TNFR1 can also elicit both apoptotic and necroptotic cell death signals via cytosolic TNF–RSC IIa, IIb and IIc (not shown) when non-ubiquitinated RIPK1 and RIPK3 are abundant or are promoted by deubiquitinases such as CYLD and A20 or by reduction of ubiquitin ligases such as LUBAC and cIAP1/2. In contrast, activation of TNFR2 results in assembly of the TNFR2 signalling complex, which is less well defined but can lead to alternative NFκB activation as well as activation of common MAPK cascades. TNF-induced metabolic reprogramming includes reduced insulin signalling (mediated in part by TNF-activated IRS-1 serine kinases, depicted in orange boxes). TNF action can also indirectly regulate autocrine or intercellular metabolic reprogramming (and systemic metabolism) via altered production and secretion of extracellular messengers. Some of these, in turn, have been shown to directly regulate insulin signalling. Putative therapeutic targets shown to improve insulin sensitivity: blue thunderbolt, processes inhibited by anti-TNF therapeutics; *, inhibited by salicylates; **, levels reduced by the glucosylceramide synthase inhibitor AMP-DNM. Scissors symbol indicates protease activity. Dotted lines indicate multi-step processes. TNF-RSC1, TNF(R1) receptor signalling complex1; pTFs, p38-phosphorylated transcription factors (for example, ATF1/2, p53, CHOP, C/EBPβ and MEF); FAN, factor activating neutral sphingomyleinase; TRADD, TNFRSF1A-associated via death domain; TRAF1/2, TNF receptor-associated factors 1/2; cIAP, baculoviral IAP repeat-containing 2; RIPK1, receptor (TNFRSF)-interacting serine-threonine kinase 1; TAK1, TGF-β activated kinase 1 (MAP3K7); TAB, TAK1-binding protein; APP3; aminopeptidase 3 IKK, inhibitor of κB kinase; NIK, NFκB-inducing kinase; LUBAC, linear ubiquitin chain assembly complex; ECM, extracellular matrix; icTNF, intracellular TNF; PRR, pattern recognition receptors; TCF7L2, transcription factor 7-like 2.
Signal transduction by both mbTNF and sTNF occurs following binding to one of two homotrimeric cell-surface receptors, TNFR1 (TNFRSF1, CD120a or p55/p60 (mouse/human)) and TNFR2 (TNFRSF1B, CD120b or p75/p80 (mouse/human)). In healthy tissues, TNFR1 is constitutively and ubiquitously expressed, whereas TNFR2 expression is restricted to lymphocytes and endothelial cells but can be induced in response to TNFR1 activation and signalling88. TNFR2 is also reported to mediate ‘ligand passing’ and may thereby regulate the association between TNF and TNFR189. Both TNF receptors can also be cleaved at the cell surface by TACE to generate soluble receptors, and adipose TNFR2 expression as well as serum TNFR2 levels are elevated in human obesity24,90,91,92. These secreted TNFRs may modify TNF bioactivity in the circulation and facilitate TNF excretion93, among other functions94.
In adipocytes, TNFR1 is required for TNF-induced processes such as insulin resistance36,95,96,97 and lipolysis98, and for anti-adipogenesis75,84. These metabolic actions can be reversed in vivo by targeting TNFR1 with small RNAs99. Consequently, much focus has been given to dissecting the molecular signals downstream of TNFR173. However, TNFR2 is also implicated in obesity-related metabolic actions100. Some studies have even suggested a requirement for yet another, unidentified TNF receptor101. Both TNFR1 and TNFR2 can trigger distinct and common signalling pathways (Fig. 3), and their biological activity is likely to be interconnected102. In vivo, the roles of each TNF receptor are undoubtedly cell and tissue specific and highly context dependent.
Intracellular TNF signalling complexes and crosstalk with other signalling networks
Ligand binding and activation of TNFR1 promotes the assembly of an intracellular receptor-proximal TNFR1 signalling complex (TNF–RSCI). The subsequent propagation and spatio-temporal regulation of downstream signals is governed by the availability and recruitment of specific ubiquitination machinery (Fig. 3). The activation of serine kinase cascades associated with cell survival and inflammation (that is, IκB kinases (IKKs), p38 and Janus kinase (JNK) signalling) requires the presence of ubiquitinated receptor-interacting protein kinase 1 (RIPK1). However, a shift in the balance away from pro-inflammatory and toward cell death signals occurs when RIPK1 is inactivated103 or deubiquitinase activity (for example, by CYLD lysine 63 deubiquitinase, CYLD) is increased, or in the absence of the linear ubiquitin chain assembly complex (LUBAC)102. These TNF-induced cell death pathways are promoted via distinct secondary signalling complexes (TNF–RSCIIa, –RSCIIb and –RSCIIc). Consistent with this scheme, the therapeutic reduction of RIPK1 in obesogenic mice reduces adiposity and results in a favourable metabolic profile, and human variants of the RIPK1 gene promoter have also been linked to increased obesity and related metabolic traits103.
Similarly, signals mediated by TNF–RSC I are crucial mediators of metabolic reprogramming, in part through crosstalk with other intracellular signalling networks including insulin–IGF1 receptor signalling and Wnt–β-catenin signalling (Fig. 3). Further complexity arises from the fact that many isoforms of kinases can be involved (for example, at least four isoforms of p38 and IKK), which can differ in their temporal or cell-specific expression and functional profiles. An illustrative example is p38, for which conflicting studies existed regarding its metabolic role. However, recent systematic evaluation of the activation and function of each isoform has clearly demonstrated a detrimental role of abnormal p38 activity in metabolic homeostasis104,105. Although JNK is the most robust kinase to be implicated in disrupting insulin action and glucose metabolism in numerous studies106,107,108, this kinase, too, exists in at least three isoforms and multiple sub-isoforms109,110. Nonetheless, activation of JNK consistently results in impaired glucose metabolism, whereas blocking its pathological activity is beneficial in the context of metabolic disease. One mechanism by which JNK has been shown to be critical for TNF-induced insulin resistance is via serine phosphorylation of a key scaffolding protein involved in insulin receptor signalling, namely insulin receptor substrate 1 (IRS-1)111,112,113,114. Given that IRS-1 possesses over 50 putative phosphoserine/phosphothreonine sites114, it is unsurprising that many other serine kinases have also been implicated in mediating insulin resistance by targeting IRS1. Examples include IKKβ115,116,117, protein kinase C proteins (PKCs)118,119,120, extracellular signal–related kinases 1 and 2 (ERK1/2)121,122, mitogen-activated kinase kinase kinase kinase 4 (MAP4K4)123,124 and IKKε125,126. It is likely that other mechanisms are in place in different tissues, and further insights are emerging rapidly109,110.
One of the most intractable questions about adipose tissue inflammation has been, what are the triggers for TNF production and metaflammation during obesity and how does metaflammation connect to metabolic deterioration? Recently, we demonstrated that adipocyte calcium homeostasis plays a critical role in these pathologies. TNF can impair insulin receptor signalling in adipocytes by stimulating endoplasmic reticulum Ca2+ release, increasing cytosolic free Ca2+ levels and activating Ca2+/calmodulin-dependent protein kinase II (CamKII; Fig. 3). These events are mediated in part by a JNK-dependent increase in inositol triphosphate receptor (IP3R) Ca2+ channels127. Moreover, adipocyte-specific loss of functional IP3R1 and 2 protected mice fed a high-fat diet against adipose tissue inflammation and insulin resistance, despite significant diet-induced weight gain. These observations further elucidate the involvement of CamKII in the downregulation of insulin-stimulated glucose uptake128 and the link between gene variants of IP3R and ‘metabolically healthy’ obesity129,130.
Another mechanism through which TNF can promote insulin resistance is by regulating intracellular lipid metabolism, in particular by promoting the accumulation and/or release of pathogenic pro-inflammatory lipids (ceramides, the ganglioside GM3, diacylglycerol)131,132,133. Notably, synthesis of the Ca2+-releasing messenger inositol triphosphate (IP3) also produces diacylglycerol. This membrane-anchored lipid is required for the activation of both classic and novel PKCs, and its elevated levels have also been linked to both obesity and lipid-induced insulin resistance133.
In other mechanistic studies addressing TNF-induced anti-adipogenesis, we have shown that TNF signalling can also crosstalk with, and effectively ‘hijack’, the Wnt–β-catenin signalling network. This crosstalk promotes TNF-induced β-catenin–transcription factor 7 like 2 (TCF7L2) transcriptional activity and may uncouple the link between nutritional availability and titrated adipocyte hyperplasia in vivo75,134,135,136. As such, it represents a molecular basis for the ‘limited adipose tissue expandability’ hypothesis and is supported by clinical evidence137,138,139,140,141. Collectively these observations demonstrate that TNF signalling does crosstalk with multiple intracellular signalling networks, and this concept of interconnectivity is likely to be fundamental to our understanding of its pleiotropic actions.
Transcriptional targets of TNF signalling
A key mechanism by which chronic TNF signalling elicits metabolic reprogramming of cells is by regulating gene expression. Multiple transcription factors have been reported to be activated by TNF signalling, including classically activated nuclear factor kappa B (NFκB; RelA:p50), non-classically activated NFκB (RelB:p52), activator protein 1 (AP-1; cFos:cJun), activating transcription factor 2 (ATF2), myocyte enhancer factor 2 (MEF2), cAMP response element–binding protein (CREB), CCAAT/enhancer-binding protein (C/EBPα) and TCF7L273,104. However, thanks to targeted gene ablation studies combined with transcriptomic profiling, receptor-specific transcriptional targets of TNF signalling have been determined73, confirming a primary role of NFκB target genes in the metabolic reprogramming of TNFR1-stimulated adipocytes and myotubes142,143. Notably, TNF can also synergize with interferon gamma (IFNγ) to sustain NFκB activation144, particularly during classical activation of pro-inflammatory macrophages. TNF-induced transcription is also required for neutrophil priming of NADPH oxidase145, as well as priming of NLR-family pyrin domain-containing-3 (NLRP3) inflammasome components146. Conversely, transcriptional repression has also been linked to metabolic actions of TNF, for example through inhibition of lipogenic peroxisome proliferator-activated receptor gamma2–retinoid X receptor alpha (PPARγ–RXRα) heterodimers. Interesting, thiazolidinediones, a potent class of insulin-sensitizing drugs, are synthetic ligands for PPARγ and can reverse TNF-induced insulin resistance147. Similarly, almost all approved anti-diabetics suppress inflammation.
Globally, TNF-induced transcriptional changes can be directly linked to numerous endpoint actions, including metabolic reprograming of carbohydrates, lipids and amino acids; feedback regulation and/or priming of intracellular signalling networks; production of additional metabolic messengers (cytokines, chemokines, metabokines, lipokines, adipokines); and cytoskeletal or extracellular matrix changes. It is this cellular reprogramming that determines cell-specific fate (that is, survival, activation, proliferation, differentiation, or death) and ultimately is key to tissue remodelling associated with chronic inflammation (Fig. 3).
TNF signalling and/or inflammation as a therapeutic target
Informed by naturally occurring modulators of TNF activity, studies along several lines of investigation continue to explore the therapeutic potential of targeting TNF production and signalling to mitigate its pathogenic effects. These include inhibitors of TNF synthesis (erythropoietin148, synthetic compounds149,150,151 and TACE inhibitors152), neutralizing TNF peptides and antibodies (sTNFR1, sTNFR2 and anti-TNF mAbs), high-affinity antagonistic peptides for TNFRs153 and naturally occurring microRNAs as putative nutraceuticals99. Among these, the most successful has undoubtedly been the development and use of anti-TNF mAbs to treat autoimmune chronic inflammatory diseases (such as Crohn’s disease, ulcerative colitis, rheumatoid arthritis, juvenile idiopathic arthritis, psoriatic arthritis, ankylosing spondylitis and plaque psoriasis, among others)154. Despite these promising clinical developments and the availability of biosimilars, there has been limited progress in applying anti-TNF therapy to treat metabolic diseases, although numerous interventional studies have validated its beneficial impact on glucose metabolism (https://www.metaflammation.org). These may be related to the lack of placebo-controlled randomized clinical trials supporting such a role or to potential additional considerations that are required for approval by the US Food and Drug Administration and UK National Institute for Health and Care Excellence, such as cost, long-term use and route of administration.
Although TNF is the focus of this review and was the first adipokine demonstrated to play a role in obesity-linked insulin resistance, it would be naive to believe that it acts alone in vivo or indeed is the sole factor involved in metabolic disease development. Indeed, several facts point to the opposite conclusion: (i) TNF-induced signal transduction is intimately integrated with extracellular cues and signalling networks, for example synergizing with IFN signalling and undergoing crosstalk with growth factor signalling including insulin–IGF and Wnt–-catenin signalling; (ii) its actions result in the production of additional cytokines and chemokines; and (iii) intervention with many other anti-inflammatory mediators generates metabolic benefits. Hence a reductionist approach focusing solely on TNF as the single mediator is unlikely to provide the enigmatic single magic bullet for future therapeutics, but rather may guide researchers toward the most effective strategies based on this conceptual framework.
Critical considerations for future research
The substantial body of knowledge that has led to our current understanding of TNF as a metabolic messenger has also revealed some important lessons that may prove useful for future immunometabolic research and the application of this knowledge for diagnostic and/or therapeutic use. First, the action of TNF (particularly during inflammation), and of inflammatory mediators in general, is highly context dependent in regard to time, space, dose, combinatorial action, and so on. The action of TNF or any other multi-functional cytokine is governed by many factors, not least the site, amounts and duration of production and energy status. Second, in the context of metaflammation (and physiologically during host defence), TNF and many other mediators possess properties of a classical cytokine, with activity as a paracrine and/or autocrine signal and the ability to generate local action with minute amounts of production. This observation contrasts with the higher levels and hormone-like actions seen in the context of cytokine storm, sepsis and cachexia—highlighting the diversity and scale of systemic exposure and functional outcomes. Third, the ratio of TNF levels to soluble TNF receptor levels is critical for determining net local or systemic bioactivity; this also applies to other cytokines and their endogenous antagonists (such as IL-1 and IL-1Ra). Last, resolution of inflammation is also a critical determinant of many of these contextual factors. Future immunometabolic targeting strategies should consider local sites of production and duration of action, as well as redundancies, and perhaps most importantly, patient stratification. Moreover, in designing intervention strategies, particularly for chronic conditions, it may be useful to consider targeting early stages of disease, when impact on tissue remodelling is more likely to be reversible. Finally, and perhaps most pertinently, modulation of metabolic inflammation, including anti-TNF therapies, should be considered as part of a combinatorial approach.
Our knowledge of TNF as a metabolic messenger has implications that go beyond understanding its solitary role in metabolic disease pathology. Indeed, common emerging features now firmly establish a fundamental aspect of immunometabolism—the roles of immune-derived signals in metabolic reprogramming and during tissue remodelling and function. Such rapidly growing mechanistic insights will undoubtedly continue to stimulate exciting research avenues and inform new, innovative strategies for developing targeted therapeutics against chronic metabolic diseases, a cluster of conditions that constitutes the greatest threat to global health.
References
Mathis, D. & Shoelson, S. E. Immunometabolism: an emerging frontier. Nat. Rev. Immunol. 11, 81–83 (2011).
Padmanabha, D. & Baker, K. D. Drosophila gains traction as a repurposed tool to investigate metabolism. Trends Endocrinol. Metab. 25, 518–527 (2014).
Mattila, J. & Hietakangas, V. Regulation of carbohydrate energy metabolism in Drosophila melanogaster. Genetics 207, 1231–1253 (2017).
Hotamisligil, G. S. Foundations of immunometabolism and implications for metabolic health and disease. Immunity 47, 406–420 (2017).
Agrawal, N. et al. The Drosophila TNF Eiger is an adipokine that acts on insulin-producing cells to mediate nutrient response. Cell Metab. 23, 675–684 (2016).
Fève, B. & Bastard, J. P. The role of interleukins in insulin resistance and type 2 diabetes mellitus. Nat. Rev. Endocrinol. 5, 305–311 (2009).
Tack, C. J., Stienstra, R., Joosten, L. A. & Netea, M. G. Inflammation links excess fat to insulin resistance: the role of the interleukin-1 family. Immunol. Rev. 249, 239–252 (2012).
Donath, M. Y. Targeting inflammation in the treatment of type 2 diabetes: time to start. Nat. Rev. Drug Discov. 13, 465–476 (2014).
Hotamisligil, G. S. Inflammation, metaflammation and immunometabolic disorders. Nature 542, 177–185 (2017).
Libert, C. Cytokine anniversary: TNF trailblazers five centuries apart. Nature 523, 158 (2015).
Tracey, K. J. & Cerami, A. Metabolic responses to cachectin/TNF. A brief review. Ann. N.Y. Acad. Sci. 587, 325–331 (1990).
Pennica, D. et al. Human tumour necrosis factor: precursor structure, expression and homology to lymphotoxin. Nature 312, 724–729 (1984).
Beutler, B. et al. Identity of tumour necrosis factor and the macrophage-secreted factor cachectin. Nature 316, 552–554 (1985).
Ruddle, N. H. Lymphotoxin and TNF: how it all began—a tribute to the travelers. Cytokine Growth Factor Rev. 25, 83–89 (2014).
Hellman, B. Studies in obese-hyperglycemic mice. Ann. N. Y. Acad. Sci. 131, 541–558 (1965).
Hausberger, F. X. Pathological changes in adipose tissue of obese mice. Anat. Rec. 154, 651–660 (1966).
Pekala, P., Kawakami, M., Vine, W., Lane, M. D. & Cerami, A. Studies of insulin resistance in adipocytes induced by macrophage mediator. J. Exp. Med. 157, 1360–1365 (1983).
Mahoney, J. R. Jr et al. Lipopolysaccharide-treated RAW 264.7 cells produce a mediator that inhibits lipoprotein lipase in 3T3-L1 cells. J. Immunol. 134, 1673–1675 (1985).
Hotamisligil, G. S., Shargill, N. S. & Spiegelman, B. M. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science 259, 87–91 (1993).
Bordon, Y. TNF short-circuits the insulin receptor. Nature Milestones: Diabetes, 12. https://www.nature.com/articles/d42859-021-00015-0 (2021).
Kern, P. A. et al. The expression of tumor necrosis factor in human adipose tissue. Regulation by obesity, weight loss, and relationship to lipoprotein lipase. J. Clin. Invest. 95, 2111–2119 (1995).
Saghizadeh, M., Ong, J. M., Garvey, W. T., Henry, R. R. & Kern, P. A. The expression of TNF alpha by human muscle. Relationship to insulin resistance. J. Clin. Invest. 97, 1111–1116 (1996).
Hube, F. & Hauner, H. The role of TNF-alpha in human adipose tissue: prevention of weight gain at the expense of insulin resistance? Horm. Metab. Res. 31, 626–631 (1999).
Mohamed-Ali, V. et al. Production of soluble tumor necrosis factor receptors by human subcutaneous adipose tissue in vivo. Am. J. Physiol. 277, E971–E975 (1999).
Lasselin, J. et al. Adipose inflammation in obesity: relationship with circulating levels of inflammatory markers and association with surgery-induced weight loss. J. Clin. Endocrinol. Metab. 99, E53–E61 (2014).
Liu, C. et al. Adiponectin, TNF-α and inflammatory cytokines and risk of type 2 diabetes: a systematic review and meta-analysis. Cytokine 86, 100–109 (2016).
Sookoian, S. C., González, C. & Pirola, C. J. Meta-analysis on the G-308A tumor necrosis factor alpha gene variant and phenotypes associated with the metabolic syndrome. Obes. Res. 13, 2122–2131 (2005).
Van der Poll, T. et al. Tumor necrosis factor mimics the metabolic response to acute infection in healthy humans. Am. J. Physiol. 261, E457–E465 (1991).
Plomgaard, P. et al. Tumor necrosis factor-alpha induces skeletal muscle insulin resistance in healthy human subjects via inhibition of Akt substrate 160 phosphorylation. Diabetes 54, 2939–2945 (2005).
Plomgaard, P., Fischer, C. P., Ibfelt, T., Pedersen, B. K. & van Hall, G. Tumor necrosis factor-alpha modulates human in vivo lipolysis. J. Clin. Endocrinol. Metab. 93, 543–549 (2008).
Feingold, K. R. et al. Effect of tumor necrosis factor (TNF) on lipid metabolism in the diabetic rat. Evidence that inhibition of adipose tissue lipoprotein lipase activity is not required for TNF-induced hyperlipidemia. J. Clin. Invest. 83, 1116–1121 (1989).
Ling, P. R., Bistrian, B. R., Mendez, B. & Istfan, N. W. Effects of systemic infusions of endotoxin, tumor necrosis factor, and interleukin-1 on glucose metabolism in the rat: relationship to endogenous glucose production and peripheral tissue glucose uptake. Metabolism 43, 279–284 (1994).
Miles, P. D. et al. TNF-alpha-induced insulin resistance in vivo and its prevention by troglitazone. Diabetes 46, 1678–1683 (1997).
Uysal, K. T., Wiesbrock, S. M., Marino, M. W. & Hotamisligil, G. S. Protection from obesity-induced insulin resistance in mice lacking TNF-α function. Nature 389, 610–614 (1997).
Ventre, J. et al. Targeted disruption of the tumor necrosis factor-alpha gene: metabolic consequences in obese and nonobese mice. Diabetes 46, 1526–1531 (1997).
Uysal, K. T., Wiesbrock, S. M. & Hotamisligil, G. S. Functional analysis of tumor necrosis factor (TNF) receptors in TNF-alpha-mediated insulin resistance in genetic obesity. Endocrinology 139, 4832–4838 (1998).
da Costa, R. M. et al. TNF-α induces vascular insulin resistance via positive modulation of PTEN and decreased Akt/eNOS/NO signaling in high fat diet-fed mice. Cardiovasc. Diabetol. 15, 119 (2016).
Borst, S. E. & Bagby, G. J. Neutralization of tumor necrosis factor reverses age-induced impairment of insulin responsiveness in skeletal muscle of Sprague-Dawley rats. Metabolism 51, 1061–1064 (2002).
Liang, H. et al. Blockade of tumor necrosis factor (TNF) receptor type 1-mediated TNF-alpha signaling protected Wistar rats from diet-induced obesity and insulin resistance. Endocrinology 149, 2943–2951 (2008).
McKellar, G. E., McCarey, D. W., Sattar, N. & McInnes, I. B. Role for TNF in atherosclerosis? Lessons from autoimmune disease. Nat. Rev. Cardiol. 6, 410–417 (2009).
Salomon, B. L. Insights into the biology and therapeutic implications of TNF and regulatory T cells. Nat. Rev. Rheumatol. 17, 487–504 (2021).
Crespo, J. et al. Gene expression of tumor necrosis factor alpha and TNF-receptors, p55 and p75, in nonalcoholic steatohepatitis patients. Hepatology 34, 1158–1163 (2001).
Kugelmas, M., Hill, D. B., Vivian, B., Marsano, L. & McClain, C. J. Cytokines and NASH: a pilot study of the effects of lifestyle modification and vitamin E. Hepatology 38, 413–419 (2003).
Henao-Mejia, J. et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 482, 179–185 (2012).
Divella, R. et al. Synergism of adipocytokine profile and ADIPOQ/TNF-α polymorphisms in NAFLD-associated MetS predict colorectal liver metastases outgrowth. Cancer Genomics Proteomics 16, 519–530 (2019).
Park, E. J. et al. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 140, 197–208 (2010).
Nakagawa, H. et al. ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer Cell 26, 331–343 (2014).
Tomita, K. et al. Tumour necrosis factor alpha signalling through activation of Kupffer cells plays an essential role in liver fibrosis of non-alcoholic steatohepatitis in mice. Gut 55, 415–424 (2006).
Bluemel, S., Wang, Y., Lee, S. & Schnabl, B. Tumor necrosis factor alpha receptor 1 deficiency in hepatocytes does not protect from non-alcoholic steatohepatitis, but attenuates insulin resistance in mice. World J. Gastroenterol. 26, 4933–4944 (2020).
Wandrer, F. et al. TNF-Receptor-1 inhibition reduces liver steatosis, hepatocellular injury and fibrosis in NAFLD mice. Cell Death Dis. 11, 212 (2020).
Verhoeven, F. et al. Safety of TNF inhibitors in rheumatic disease in case of NAFLD and cirrhosis. Semin. Arthritis Rheum. 50, 544–548 (2020).
Tang, K. T., Dufour, J. F., Chen, P. H., Hernaez, R. & Hutfless, S. Antitumour necrosis factor-α agents and development of new-onset cirrhosis or non-alcoholic fatty liver disease: a retrospective cohort. BMJ Open Gastroenterol. 7, e000349 (2020).
Ohta, H. et al. Disruption of tumor necrosis factor-alpha gene diminishes the development of atherosclerosis in ApoE-deficient mice. Atherosclerosis 180, 11–17 (2005).
Avgerinou, G. et al. Anti-tumor necrosis factor α treatment with adalimumab improves significantly endothelial function and decreases inflammatory process in patients with chronic psoriasis. Int. J. Cardiol. 151, 382–383 (2011).
Tam, L. S., Kitas, G. D. & González-Gay, M. A. Can suppression of inflammation by anti-TNF prevent progression of subclinical atherosclerosis in inflammatory arthritis? Rheumatology (Oxford) 53, 1108–1119 (2014).
Spinelli, F. R. et al. Decrease of asymmetric dimethyl arginine after anti-TNF therapy in patients with rheumatoid arthritis. Drug Dev. Res. 75, S67–S69 (2014).
Francisco, V. et al. Adipokines: linking metabolic syndrome, the immune system, and arthritic diseases. Biochem. Pharmacol. 165, 196–206 (2019).
Everett, B. M. et al. Inhibition of Interleukin-1β and reduction in atherothrombotic cardiovascular events in the CANTOS trial. J. Am. Coll. Cardiol. 76, 1660–1670 (2020).
Nguyen, J. C., Killcross, A. S. & Jenkins, T. A. Obesity and cognitive decline: role of inflammation and vascular changes. Front. Neurosci. 8, 375 (2014).
Pugazhenthi, S., Qin, L. & Reddy, P. H. Common neurodegenerative pathways in obesity, diabetes, and Alzheimer’s disease. Biochim. Biophys. Acta, Mol. Basis Dis. 1037–1045, 2017 (1863).
Bomfim, T. R. et al. An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer’s disease–associated Aβ oligomers. J. Clin. Invest. 122, 1339–1353 (2012).
Ferreira, S. T., Clarke, J. R. & Bomfim, T. R. & De Felice, F. G. Inflammation, defective insulin signaling, and neuronal dysfunction in Alzheimer’s disease. Alzheimers Dement. 10, S76–S83 (2014).
de la Monte, S. M. & Wands, J. R. Alzheimer’s disease is type 3 diabetes-evidence reviewed. J. Diabetes Sci. Technol. 2, 1101–1113 (2008).
Aggarwal, B. B. et al. in Cytokines as Potential Therapeutic Targets for Inflammatory Skin Diseases (Ernst Schering Research Foundation Workshops, vol. 56) (eds. Numerof, R., Dinarello, C. A. & Asadullah, K.) 161–186 (Springer, 2006).
Sethi, G., Sung, B. & Aggarwal, B. B. TNF: a master switch for inflammation to cancer. Front. Biosci. 13, 5094–5107 (2008).
Lou, J., Lucas, R. & Grau, G. E. Pathogenesis of cerebral malaria: recent experimental data and possible applications for humans. Clin. Microbiol. Rev. 14, 810–820 (2001).
Jacobs, M. et al. Tumor necrosis factor is critical to control tuberculosis infection. Microbes Infect. 9, 623–628 (2007).
Del Valle, D. M. et al. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat. Med. 26, 1636–1643 (2020).
Robinson, P. C., Richards, D., Tanner, H. L. & Feldmann, M. Accumulating evidence suggests anti-TNF therapy needs to be given trial priority in COVID-19 treatment. Lancet Rheumatol 2, e653–e655 (2020).
Feldmann, M. et al. Trials of anti-tumour necrosis factor therapy for COVID-19 are urgently needed. Lancet 395, 1407–1409 (2020).
Mahase, E. Covid-19: anti-TNF drug adalimumab to be trialled for patients in the community. Br. Med. J. 371, m3847 (2020).
Sethi, J. K. & Hotamisligil, G. S. The role of TNF alpha in adipocyte metabolism. Semin. Cell Dev. Biol. 10, 19–29 (1999).
Cawthorn, W. P. & Sethi, J. K. TNF-alpha and adipocyte biology. FEBS Lett. 582, 117–131 (2008).
Sethi, J. K. & Vidal-Puig, A. J. Thematic review series: adipocyte biology. Adipose tissue function and plasticity orchestrate nutritional adaptation. J. Lipid Res. 48, 1253–1262 (2007).
Cawthorn, W. P., Heyd, F., Hegyi, K. & Sethi, J. K. Tumour necrosis factor-alpha inhibits adipogenesis via a beta-catenin/TCF4(TCF7L2)-dependent pathway. Cell Death Differ. 14, 1361–1373 (2007).
Voloshyna, I. et al. Infliximab reverses suppression of cholesterol efflux proteins by TNF-α: a possible mechanism for modulation of atherogenesis. BioMed Res. Int. 2014, 312647 (2014).
Yanai, H. et al. The underlying mechanisms for development of hypertension in the metabolic syndrome. Nutr. J. 7, 10 (2008).
Fong, Y. et al. Antibodies to cachectin/tumor necrosis factor reduce interleukin 1 beta and interleukin 6 appearance during lethal bacteremia. J. Exp. Med. 170, 1627–1633 (1989).
Falvo, J. V., Tsytsykova, A. V. & Goldfeld, A. E. Transcriptional control of the TNF gene. Curr. Dir. Autoimmun. 11, 27–60 (2010).
Anderson, P. Post-transcriptional regulation of tumour necrosis factor alpha production. Ann. Rheum. Dis. 59, i3–i5 (2000).
Stamou, P. & Kontoyiannis, D. L. Posttranscriptional regulation of TNF mRNA: a paradigm of signal-dependent mRNA utilization and its relevance to pathology. Curr. Dir. Autoimmun. 11, 61–79 (2010).
Mazumder, B., Li, X. & Barik, S. Translation control: a multifaceted regulator of inflammatory response. J. Immunol. 184, 3311–3319 (2010).
Liu, J., Qian, C. & Cao, X. Post-translational modification control of innate immunity. Immunity 45, 15–30 (2016).
Xu, H., Sethi, J. K. & Hotamisligil, G. S. Transmembrane tumor necrosis factor (TNF)-alpha inhibits adipocyte differentiation by selectively activating TNF receptor 1. J. Biol. Chem. 274, 26287–26295 (1999).
Xu, H., Uysal, K. T., Becherer, J. D., Arner, P. & Hotamisligil, G. S. Altered tumor necrosis factor-alpha (TNF-alpha) processing in adipocytes and increased expression of transmembrane TNF-alpha in obesity. Diabetes 51, 1876–1883 (2002).
Black, R. A. et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature 385, 729–733 (1997).
Serino, M. et al. Mice heterozygous for tumor necrosis factor-alpha converting enzyme are protected from obesity-induced insulin resistance and diabetes. Diabetes 56, 2541–2546 (2007).
Medler, J. & Wajant, H. Tumor necrosis factor receptor-2 (TNFR2): an overview of an emerging drug target. Expert Opin. Ther. Targets 23, 295–307 (2019).
Tartaglia, L. A., Pennica, D. & Goeddel, D. V. Ligand passing: the 75-kDa tumor necrosis factor (TNF) receptor recruits TNF for signaling by the 55-kDa TNF receptor. J. Biol. Chem. 268, 18542–18548 (1993).
Hotamisligil, G. S., Arner, P., Atkinson, R. L. & Spiegelman, B. M. Differential regulation of the p80 tumor necrosis factor receptor in human obesity and insulin resistance. Diabetes 46, 451–455 (1997).
Rönnemaa, T., Pulkki, K. & Kaprio, J. Serum soluble tumor necrosis factor-alpha receptor 2 is elevated in obesity but is not related to insulin sensitivity: a study in identical twins discordant for obesity. J. Clin. Endocrinol. Metab. 85, 2728–2732 (2000).
Good, M. et al. TNF and TNF receptor expression and insulin sensitivity in human omental and subcutaneous adipose tissue—influence of BMI and adipose distribution. Diab. Vasc. Dis. Res. 3, 26–33 (2006).
Kohno, T. et al. A second tumor necrosis factor receptor gene product can shed a naturally occurring tumor necrosis factor inhibitor. Proc. Natl Acad. Sci. USA 87, 8331–8335 (1990).
Aderka, D., Engelmann, H., Maor, Y., Brakebusch, C. & Wallach, D. Stabilization of the bioactivity of tumor necrosis factor by its soluble receptors. J. Exp. Med. 175, 323–329 (1992).
Peraldi, P., Hotamisligil, G. S., Buurman, W. A., White, M. F. & Spiegelman, B. M. Tumor necrosis factor (TNF)-alpha inhibits insulin signaling through stimulation of the p55 TNF receptor and activation of sphingomyelinase. J. Biol. Chem. 271, 13018–13022 (1996).
Sethi, J. K. et al. Characterisation of receptor-specific TNFalpha functions in adipocyte cell lines lacking type 1 and 2 TNF receptors. FEBS Lett. 469, 77–82 (2000).
Martins, L. B. et al. Paradoxical role of tumor necrosis factor on metabolic dysfunction and adipose tissue expansion in mice. Nutrition 50, 1–7 (2018).
Sethi, J. K., Xu, H. Y. & Hotamisligil, G. S. Roles of tumor necrosis factor receptor 1 and receptor 2 in insulin receptor signaling. Diabetes 48, A219–A219 (1999).
Aquilano, K. et al. Adipocyte metabolism is improved by TNF receptor-targeting small RNAs identified from dried nuts. Commun. Biol. 2, 317 (2019).
Liu, L. S., Spelleken, M., Röhrig, K., Hauner, H. & Eckel, J. Tumor necrosis factor-alpha acutely inhibits insulin signaling in human adipocytes: implication of the p80 tumor necrosis factor receptor. Diabetes 47, 515–522 (1998).
Wu, S., Dong, K., Wang, J. & Bi, Y. Tumor necrosis factor alpha improves glucose homeostasis in diabetic mice independent with tumor necrosis factor receptor 1 and tumor necrosis factor receptor 2. Endocr. J. 65, 601–609 (2018).
Wajant, H. & Siegmund, D. TNFR1 and TNFR2 in the control of the life and death balance of macrophages. Front. Cell Dev. Biol. 7, 91 (2019).
Karunakaran, D. et al. RIPK1 gene variants associate with obesity in humans and can be therapeutically silenced to reduce obesity in mice. Nat. Metab. 2, 1113–1125 (2020).
Sabio, G. & Davis, R. J. TNF and MAP kinase signalling pathways. Semin. Immunol. 26, 237–245 (2014).
Leiva, M., Matesanz, N., Pulgarín-Alfaro, M., Nikolic, I. & Sabio, G. Uncovering the role of p38 family members in adipose tissue physiology. Front. Endocrinol. 11, 572089 (2020).
Hirosumi, J. et al. A central role for JNK in obesity and insulin resistance. Nature 420, 333–336 (2002).
Tuncman, G. et al. Functional in vivo interactions between JNK1 and JNK2 isoforms in obesity and insulin resistance. Proc. Natl Acad. Sci. U.S.A. 103, 10741–10746 (2006).
Waeber, G. et al. The gene MAPK8IP1, encoding islet-brain-1, is a candidate for type 2 diabetes. Nat. Genet. 24, 291–295 (2000).
Weston, C. R. & Davis, R. J. The JNK signal transduction pathway. Curr. Opin. Cell Biol. 19, 142–149 (2007).
Hotamisligil, G. S. & Davis, R. J. Cell signaling and stress responses. Cold Spring Harb. Perspect. Biol. 8, a006072 (2016).
Hotamisligil, G. S. et al. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha- and obesity-induced insulin resistance. Science 271, 665–670 (1996).
Kanety, H., Feinstein, R., Papa, M. Z., Hemi, R. & Karasik, A. Tumor necrosis factor alpha-induced phosphorylation of insulin receptor substrate-1 (IRS-1). Possible mechanism for suppression of insulin-stimulated tyrosine phosphorylation of IRS-1. J. Biol. Chem. 270, 23780–23784 (1995).
Aguirre, V. et al. Phosphorylation of Ser307 in insulin receptor substrate-1 blocks interactions with the insulin receptor and inhibits insulin action. J. Biol. Chem. 277, 1531–1537 (2002).
Copps, K. D. & White, M. F. Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia 55, 2565–2582 (2012).
Gao, Z. et al. Serine phosphorylation of insulin receptor substrate 1 by inhibitor kappa B kinase complex. J. Biol. Chem. 277, 48115–48121 (2002).
Austin, R. L., Rune, A., Bouzakri, K., Zierath, J. R. & Krook, A. siRNA-mediated reduction of inhibitor of nuclear factor-kappaB kinase prevents tumor necrosis factor-alpha-induced insulin resistance in human skeletal muscle. Diabetes 57, 2066–2073 (2008).
Jiang, S. & Messina, J. L. Role of inhibitory κB kinase and c-Jun NH2-terminal kinase in the development of hepatic insulin resistance in critical illness diabetes. Am. J. Physiol. Gastrointest. Liver Physiol. 301, G454–G463 (2011).
Li, Y. et al. Protein kinase C Theta inhibits insulin signaling by phosphorylating IRS1 at Ser(1101). J. Biol. Chem. 279, 45304–45307 (2004).
Kim, J. K. et al. PKC-theta knockout mice are protected from fat-induced insulin resistance. J. Clin. Invest. 114, 823–827 (2004).
Nawaratne, R. et al. Regulation of insulin receptor substrate 1 pleckstrin homology domain by protein kinase C: role of serine 24 phosphorylation. Mol. Endocrinol. 20, 1838–1852 (2006).
De Fea, K. & Roth, R. A. Modulation of insulin receptor substrate-1 tyrosine phosphorylation and function by mitogen-activated protein kinase. J. Biol. Chem. 272, 31400–31406 (1997).
Engelman, J. A., Berg, A. H., Lewis, R. Y., Lisanti, M. P. & Scherer, P. E. Tumor necrosis factor alpha-mediated insulin resistance, but not dedifferentiation, is abrogated by MEK1/2 inhibitors in 3T3-L1 adipocytes. Mol. Endocrinol. 14, 1557–1569 (2000).
Bouzakri, K. & Zierath, J. R. MAP4K4 gene silencing in human skeletal muscle prevents tumor necrosis factor-alpha-induced insulin resistance. J. Biol. Chem. 282, 7783–7789 (2007).
Danai, L. V. et al. Inducible deletion of protein kinase Map4k4 in obese mice improves insulin sensitivity in liver and adipose tissues. Mol. Cell. Biol. 35, 2356–2365 (2015).
Chiang, S. H. et al. The protein kinase IKKepsilon regulates energy balance in obese mice. Cell 138, 961–975 (2009).
Patel, M. N. et al. Hematopoietic IKBKE limits the chronicity of inflammasome priming and metaflammation. Proc. Natl Acad. Sci. U.S.A. 112, 506–511 (2015).
Guney, E. et al. Aberrant Ca2+ homeostasis in adipocytes links inflammation to metabolic dysregulation in obesity. Preprint in bioRxiv at https://doi.org/10.1101/2020.10.28.360008 (2020).
Illario, M. et al. Calcium-calmodulin-dependent kinase II (CaMKII) mediates insulin-stimulated proliferation and glucose uptake. Cell. Signal. 21, 786–792 (2009).
Shungin, D. et al. New genetic loci link adipose and insulin biology to body fat distribution. Nature 518, 187–196 (2015).
Mao, L., Fang, Y., Campbell, M. & Southerland, W. M. Population differentiation in allele frequencies of obesity-associated SNPs. BMC Genomics 18, 861 (2017).
Wiegmann, K., Schütze, S., Machleidt, T., Witte, D. & Krönke, M. Functional dichotomy of neutral and acidic sphingomyelinases in tumor necrosis factor signaling. Cell 78, 1005–1015 (1994).
Aerts, J. M. et al. Pharmacological inhibition of glucosylceramide synthase enhances insulin sensitivity. Diabetes 56, 1341–1349 (2007).
Petersen, M. C. & Shulman, G. I. Mechanisms of insulin action and insulin resistance. Physiol. Rev. 98, 2133–2223 (2018).
Sethi, J. K. & Vidal-Puig, A. Wnt signalling and the control of cellular metabolism. Biochem. J. 427, 1–17 (2010).
Lagathu, C. et al. Dact1, a nutritionally regulated preadipocyte gene, controls adipogenesis by coordinating the Wnt/beta-catenin signaling network. Diabetes 58, 609–619 (2009).
Lagathu, C. et al. Secreted frizzled-related protein 1 regulates adipose tissue expansion and is dysregulated in severe obesity. Int. J. Obes. 34, 1695–1705 (2010).
Isakson, P., Hammarstedt, A., Gustafson, B. & Smith, U. Impaired preadipocyte differentiation in human abdominal obesity: role of Wnt, tumor necrosis factor-alpha, and inflammation. Diabetes 58, 1550–1557 (2009).
Gustafson, B. et al. Inflammation and impaired adipogenesis in hypertrophic obesity in man. Am. J. Physiol. Endocrinol. Metab. 297, E999–E1003 (2009).
Roubert, A. et al. The influence of tumor necrosis factor-α on the tumorigenic Wnt-signaling pathway in human mammary tissue from obese women. Oncotarget 8, 36127–36136 (2017).
Guo, C. et al. Genetic ablation of tumor necrosis factor-alpha attenuates the promoted colonic Wnt signaling in high fat diet-induced obese mice. J. Nutr. Biochem. 77, 108302 (2020).
Verjee, L. S. et al. Unraveling the signaling pathways promoting fibrosis in Dupuytren’s disease reveals TNF as a therapeutic target. Proc. Natl Acad. Sci. U.S.A. 110, E928–E937 (2013).
Ruan, H., Hacohen, N., Golub, T. R., Van Parijs, L. & Lodish, H. F. Tumor necrosis factor-alpha suppresses adipocyte-specific genes and activates expression of preadipocyte genes in 3T3-L1 adipocytes: nuclear factor-kappaB activation by TNF-alpha is obligatory. Diabetes 51, 1319–1336 (2002).
Bhatnagar, S. et al. Tumor necrosis factor-α regulates distinct molecular pathways and gene networks in cultured skeletal muscle cells. PLoS One 5, e13262 (2010).
Cheshire, J. L. & Baldwin, A. S. Jr. Synergistic activation of NF-kappaB by tumor necrosis factor alpha and gamma interferon via enhanced I kappaB alpha degradation and de novo I kappaBbeta degradation. Mol. Cell. Biol. 17, 6746–6754 (1997).
Wright, H. L., Thomas, H. B., Moots, R. J. & Edwards, S. W. RNA-seq reveals activation of both common and cytokine-specific pathways following neutrophil priming. PLoS One 8, e58598 (2013).
McGeough, M. D. et al. TNF regulates transcription of NLRP3 inflammasome components and inflammatory molecules in cryopyrinopathies. J. Clin. Invest. 127, 4488–4497 (2017).
Peraldi, P., Xu, M. & Spiegelman, B. M. Thiazolidinediones block tumor necrosis factor-alpha-induced inhibition of insulin signaling. J. Clin. Invest. 100, 1863–1869 (1997).
Cerami, A. TNF and EPO: major players in the innate immune response: their discovery. Ann. Rheum. Dis. 71, i55–i59 (2012).
Moreira, A. L. et al. Thalidomide exerts its inhibitory action on tumor necrosis factor alpha by enhancing mRNA degradation. J. Exp. Med. 177, 1675–1680 (1993).
Kruys, V., Marinx, O., Shaw, G., Deschamps, J. & Huez, G. Translational blockade imposed by cytokine-derived UA-rich sequences. Science 245, 852–855 (1989).
Belarbi, K. et al. TNF-α protein synthesis inhibitor restores neuronal function and reverses cognitive deficits induced by chronic neuroinflammation. J. Neuroinflammation 9, 23 (2012).
Wong, E. et al. Harnessing the natural inhibitory domain to control TNFα converting enzyme (TACE) activity in vivo. Sci Rep. 6, 35598 (2016).
Cui, X. et al. Trivalent soluble TNF receptor, a potent TNF-α antagonist for the treatment collagen-induced arthritis. Sci Rep. 8, 7327 (2018).
Shepard, H. M., Phillips, G. L., D Thanos, C. & Feldmann, M. Developments in therapy with monoclonal antibodies and related proteins. Clin. Med. (Lond.) 17, 220–232 (2017).
Acknowledgements
J.K.S. is funded by the Welcome Trust (grant number 206453/Z/17/Z) and the UK National Institute for Health Research Southampton Biomedical Research Centre. G.S.H. is funded by grants from the US National Institutes of Health (grant numbers DK123458 and HL125753), the Juvenile Diabetes Research Foundation and Lab1636.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Metabolism thanks Mark Febbraio and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: George Caputa; Ashley Castellanos-Jankiewicz.
Rights and permissions
About this article

Cite this article
Sethi, J.K., Hotamisligil, G.S. Metabolic Messengers: tumour necrosis factor. Nat Metab 3, 1302–1312 (2021). https://doi.org/10.1038/s42255-021-00470-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s42255-021-00470-z
This article is cited by
-
Central inhibition of stearoyl-CoA desaturase has minimal effects on the peripheral metabolic symptoms of the 3xTg Alzheimer’s disease mouse model
Scientific Reports (2024)
-
Effects of isorhamnetin on liver injury in heat stroke-affected rats under dry-heat environments via oxidative stress and inflammatory response
Scientific Reports (2024)
-
Cellular heterogeneity in TNF/TNFR1 signalling: live cell imaging of cell fate decisions in single cells
Cell Death & Disease (2024)
-
Tissue Accumulation, Cytotoxicity, Oxidative Stress, and Immunotoxicity in African Catfish, Clarias gariepinus Exposed to Sublethal Concentrations of Hexavalent Chromium
Biological Trace Element Research (2024)
-
Adipocyte p53 coordinates the response to intermittent fasting by regulating adipose tissue immune cell landscape
Nature Communications (2024)
