
Abstract
Improved nuclear spin-dependent parity violation measurements will enable experimental determination of poorly known electroweak coupling parameters. Here, we investigate the suitability of optically trapped linear polyatomic molecules as probes of nuclear spin-dependent parity violation. The presence of closely spaced, opposite-parity \(\ell\)-doublets is a general feature of such molecules, allowing parity-violation-sensitive pairs of levels to be brought to degeneracy in magnetic fields typically 100 times smaller than in diatomics. Assuming laser cooling and trapping of polyatomics at the current state-of-the-art for diatomics, we expect to measure nuclear spin-dependent parity-violating matrix elements iW with 70 times better sensitivity than the current best measurements. Our scheme should allow for 10% measurements of iW in nuclei as light as Be or as heavy as Yb, with averaging times on the order of 10 days and 1 s, respectively.
Similar content being viewed by others


Introduction
Measurements of nuclear spin-independent (NSI) and nuclear spin-dependent (NSD) parity violation (PV) are a means to probe standard model (SM) electroweak interactions on a tabletop scale1. NSI-PV has been measured in protons and a number of heavy atoms2,3,4,5,6,7,8,9 and found to be in good agreement with SM PV predictions due to the weak charge QW. However, the only non-zero measurement (14% relative uncertainty) of NSD-PV in an atomic system comes from Cs10, and this result implies constraints on SM meson-nucleon couplings which are in disagreement with other atomic PV measurements11,12. NSD-PV arises primarily from three interactions: vector electron-axial nucleon electroweak current coupling (VeAn), the nuclear anapole moment, and the combined effects of nuclear weak charge and normal hyperfine structure. The VeAn effect is described by two parameters C2u and C2d relating to spin-dependent Z0 boson exchange between an electron and an up or down quark, respectively. These parameters are among the most poorly measured in the SM, with relative uncertainties 300 and 70%, respectively13. PV measurements may also probe beyond standard model physics14,15. Searches for oscillating PV signals have been proposed as a means to detect axion-like particles, a leading dark matter candidate16.
A beam of cold diatomic molecules has been demonstrated17,18,19,20,21 to be a highly sensitive system for measuring NSD-PV effects. Mixing of opposite-parity quantum states from PV effects is amplified when the states have nearly the same energy6. The lowest two rotational states of diatomic molecules have opposite parity and may be brought to near degeneracy using a large magnetic field B. While this method is quite general, current systematic uncertainties are roughly 100 times too large to measure NSD-PV in the lightest nuclei where nuclear structure calculations are tractable22.
Recent advances in laser cooling have lead to optically trapped diatomic molecules with sub-Doppler temperature and single-molecule detection efficiency23, while similar strides with polyatomic molecules have followed closely behind24,25,26,27,28. Moreover, polyatomic molecules have been proposed as exquisite systems for precision measurements of fundamental symmetries29,30,31 and time variation of fundamental constants32,33.
Here, we show that linear asymmetric polyatomic molecules in an optical trap are well-suited for measurement of NSD-PV. Polyatomic molecules possess opposite-parity states 10–1000 times closer in energy than diatomics, requiring similarly smaller B-fields. Systematic uncertainties are reduced compared to beam experiments due to the lower magnetic field and a smaller interaction volume. Furthermore, these smaller fields may be produced without superconducting magnets, allowing trivial B-field reversal for detection and mitigation of systematic effects. We show that these molecules may be optically trapped using “magic” conditions where differential light shifts are small enough for a precise PV measurement. The obvious advantage of performing a precision measurement on trapped species compared to a beam is the increased interaction time τ. The sensitivity to any PV matrix element iW is \(\delta W/W = 1/\tau \sqrt {N_{\mathrm{m}}}\), where Nm is the total number of measurements (iW is purely imaginary due to conservation of time-reversal symmetry). Assuming optical trapping of polyatomic molecules at the current state-of-the-art for diatomics23,34,35, we expect at least a factor of 70 increase in PV sensitivity over the state-of-the-art BaF measurement20,36. Our method is applicable to all laser-coolable polyatomic molecules with 2Σ ground states, and possibly others.
Results
Relevant properties of polyatomic molecules
Consider the properties of linear asymmetric molecules with a 2Σ+ electronic ground state. If a bending vibrational mode (with vibrational constant ωb and quantum number vb) is excited and all other vibrational modes are in their ground state, the molecule’s rotational angular momentum N has a projection along the molecular axis \(\ell = \pm v_b, \pm (v_b - 2), \ldots , \pm 1\) or 0. Within this vibrational manifold, the effective Hamiltonian is
where Be is the rotational constant, γ is the spin-rotation (SR) constant, b and c are hyperfine (HF) constants, e is the electron charge, T2(∇E)⋅T2(Q) is a scalar product of rank-2 spherical tensors describing the electric field gradient ▽E at the nucleus with quadrupole moment Q, S is the electron spin, L is the electron orbital angular momentum, and I is the nuclear spin37,38. The upper (lower) sign corresponds to the parity P of the closely spaced “\(\ell\)-doublet” eigenstates \(\left| {N,v_b^\ell ,P = \pm } \right\rangle = \frac{1}{{\sqrt 2 }}\left( {\left| {N, + \ell } \right\rangle \pm ( - 1)^{N - \ell }\left| {N, - \ell } \right\rangle } \right)\). The \(\ell\)-doublet is the key property of polyatomic molecules absent from diatomics which we wish to exploit for a PV measurement. In linear modes, as well as diatomics, opposite-parity levels are spaced by roughly the rotational constant Be/2π ~1 GHz to 100 GHz. In an excited bending mode, opposite-parity states are spaced by only qb ≈ \(- 2B_{\mathrm{e}}^2/\omega _b\), which is on the order of \(q_b/2\pi \sim 10\,{\mathrm{MHz}}\) to 100 MHz. The relative spacing between levels may be tuned to degeneracy via the Zeeman interaction (last three terms of Eq. (1), with μB, μN the Bohr and nuclear magneton, respectively; and gS, gL, and gI the g-factors corresponding to S, L, and I, respectively). Examples are provided in Supplementary Note 1.
Nuclear spin-dependent parity violation
For a given electronic state of a molecule, the effective NSD-PV Hamiltonian is39
Here, Wp encodes the overlap of unpaired electrons with the nucleus and can be calculated with high accuracy by ab initio or semiempirical methods, \(\widehat {\mathbf{n}}\) is a unit vector along the molecular axis, and κ is the measurable parameter of interest. In a given nucleus, various NSD-PV effects contribute to κ = κ2 + κa + κQ. κ2 is proportional to the strength of the VeAn coupling, and is independent of nuclear mass A (for a typical nucleus, \(|\kappa _2| \simeq 0.05\)). κa is proportional to the nuclear anapole moment and is proportional to A2/3. κQ is due to the combined effects of nuclear weak charge and normal hyperfine structure, and is negligible compared to κ2 and κa40. Measurements in several nuclei are required to distinguish among the different NSD-PV effects.
Ultimately, the ability to precisely determine κ and its underlying contributions will be limited to the accuracy of theoretical values of Wp. Calculations of Wp have been performed on several diatomic molecules via Dirac-Hartree-Fock and relativistic density-functional41, quasirelativistic zero-order regular approximation29,42, and (with an estimated 1.5% accuracy) relativistic coupled-cluster43 methods. While such calculations are beyond the scope of this proposal, a semiempirical method may be used to calculate Wp for any species to ~10%, assuming the SR/HF constants in Eq. (1) are known17. The SR/HF constants relating to a typical metal atom M differ by a ~10% between laser-coolable monofluorides MF and MX molecules, where X is a suitable ligand with charge state −1 (e.g., OH, NC, or CCH). Thus, we expect similar 10% accuracy when estimating Wp for MX, using either MX SR/HF constants for a semiemprical calculation or a more detailed method described above for the corresponding MF. This approximation is in line with available theoretical values Wp(Ra) in RaF41,42 and RaOH29. In Table 1, we give a list of laser-coolable polyatomic molecules MX, with Wp from calculations on MX where available or MF otherwise (see also Supplementary Note 2).
In general, Eq. (2) should be summed over all nuclei i with spin Ii ≥ 1/2 in a molecule. The PV signal is easiest to interpret when the unpaired electron is centered on one atom in the molecule, i.e. \(W_{\mathrm{p}}^{(i)} \approx 0\) for all but one atom. A single-atom-centered unpaired electron is also a defining characteristic of laser-coolable molecules: this electron interacts negligibly with the nuclear vibration of the molecule, leading to electronic transitions which are highly diagonal in vibrational quantum number (and thus requires a small number of vibrational repump lasers for cooling)44. Laser cooling schemes have been proposed for molecules with an electron centered on atoms with a wide range of mass (as light as Be45 and B46, and as heavy as Yb47 and Tl48,49), and extension to polyatomic species, while technically more complicated, is straightforward27.
\(H_{\mathrm{p}}^{{\mathrm{eff}}}\) is a pseudoscalar interaction, which connects states with different parity P and the same lab frame angular momentum projection mF. We dub such states \(\left| {\tilde \eta ;m_F, + } \right\rangle ,\left| {\tilde \eta \prime ;m_F, - } \right\rangle\) a “PV pair”, with \(\tilde \eta\) denoting all other nominal quantum numbers when B = 0. So long as the \(\ell\)-doublet splitting is not smaller than all SR/HF splittings, PV pairs of a given rotational manifold N cross in an applied B-field when μBB ~ qb. This situation is common for light molecules due to their typically smaller HF interactions and larger qb arising from their larger rotational constant Be. Typical values of qb imply a modest field of B ~1 mT to 10 mT will bring a PV pair to degeneracy. For instances where the \(\ell\)-doublet splitting is smaller than all SR and HF intervals, states \(\left| {\tilde \eta ;m_F, \pm } \right\rangle\) remain split by ~qb for any applied B-field. In this case, the Zeeman interaction repels this \(\ell\)-doublet from a neighboring \(\left| {\tilde \eta \prime ;m_F, \pm } \right\rangle\) doublet, preventing the crossing of PV pairs \(\left| {\tilde \eta ;m_F, \pm } \right\rangle ,\left| {\tilde \eta \prime ;m_F, \mp } \right\rangle\). Examples are provided in Supplementary Note 1. Regardless of the hierarchy of \(\ell\)-doubling, SR, and HF interactions, PV crossings always occur between neighboring rotational levels at μBB ≈ Be, as in a diatomic molecule19, but require an experimentally more challenging B ~100 mT to 1000 mT19.
In some cases, it may be advantageous to measure iW in a more highly-excited rotational or vibrational state. For example, consider 171YbOH (I(171Yb) = 1/2). We may estimate the relevant parameters in Eq. (1) by reduced-mass-scaling (where appropriate) the constants Be, ωb=2, γ, b(H), and c(H) from the 174YbOH isotopologue (I(174Yb) = 0)50; by assuming constants b(Yb) and c(Yb) to be the same as in chemically similar 171YbF; and by taking \(q_b = - 2B_{\mathrm{e}}^2/\omega _2\). With these parameters, the \(\ell\)-doublet in \(\left| {v_b^\ell = 1^1,N = 1} \right\rangle\) is smaller than the SR and Yb HF interactions. For all possible PV pairs, the value of \(\left\langle {({\mathbf{S}} \times \widehat {\mathbf{I}}({\mathrm{Yb}})) \cdot \widehat {\mathbf{n}}} \right\rangle\) is only nonzero due to the small state mixing from the HF interaction with the H nucleus. However, in \(\left| {v_b^\ell = 1^1,N = 2,3} \right\rangle\), the \(\ell\)-doublet is larger than the Yb HF splitting and multiple PV pairs with \(\left\langle {({\mathbf{S}} \times \widehat {\mathbf{I}}({\mathrm{Yb}})) \cdot \widehat {\mathbf{n}}} \right\rangle\) ~0.1 exist.
Measurement procedure
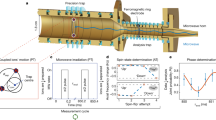
We now propose a procedure to trap polyatomic molecules and measure matrix elements iW of \(H_{\mathrm{p}}^{{\mathrm{eff}}}\). A cryogenic buffer gas beam source creates a slow, cold beam of the desired molecule species51,52. Molecules are sequentially laser-slowed53, trapped using a magneto-optical trap (MOT)54, and loaded into a red-detuned optical dipole trap (ODT) while performing Λ-enhanced cooling35. Then, one of the cooling laser frequencies is turned off in order to optically pump molecules into a single, optically dark SR/HF state. Stimulated Raman adiabatic passage completes state preparation by efficiently transferring to the \(\left| {v_b^\ell = 1^1,N,\eta ,m_F,P = ( - 1)^N} \right\rangle\) state55,56. Performing the measurement in an ODT allows for long interaction times while only requiring precise field control over a small volume.
The PV signal is measured by the Stark interference method, which has been examined in detail elsewhere6,7,19. We summarize the main points closely following the notation of Cahn et al.36. We apply a static magnetic field \({\mathbf{B}}\, = \,B\widehat {\mathbf{z}}\) to shift a particular PV pair to near degeneracy. We denote the time-dependent probability amplitudes of these states c±(t), and assume an initial state c−(0) = 1, c+ (0) = 0. An oscillating electric field \({\mathbf{E}} = E_0{\mathrm{cos}}(\omega _Et)\widehat {\mathbf{z}}\) is applied to drive the transition between the near-degenerate levels. The effective Hamiltonian \(H_ \pm ^{{\mathrm{eff}}}\) for the two-level system can be written36
Here Δ is the small detuning from degeneracy, d is the transition dipole moment, and α′ is the differential polarizability of the two states. In the limit where \(W \ll d{\kern 1pt} E_0,{\mathrm{\Delta }} \ll \omega _E\), and assuming for now that α′ = 0, the PV signal S = |c+(t)|2 is
From Eq. (4), we see that the combined PV and electric dipole transition probability (first term in square brackets) interferes with the standard electric dipole transition probability (second term in square brackets). The interference term changes sign under a reversal of either E, B, or Δ. The PV matrix element iW may be extracted through an asymmetry measurement19
where the ellipse denotes higher order terms in W/Δ. Detection using optical cycling should provide shot noise-limited readout57.
Estimates of broadening and systematic uncertainties
Sensitivity to iW is expected to be limited by inhomogeneous broadening of Δ. For optically trapped molecules, the detuning uncertainty δΔ is dominated by the differential Stark shift due to the optical trap. Employing certain “magic” polarization conditions in an ODT58,59,60,61,62 will set α′ = 0, U′ = 0, and \(\delta {\mathrm{\Delta }} = \delta U\prime = \frac{{\partial U\prime }}{{\partial \theta }}\delta \theta\), where U is the trap depth, U′ is the differential trap depth, and θ is an angle related to the polarization. We investigated three such magic conditions for PV pairs in MgNC and YbOH and find \(\frac{{\partial U\prime }}{{\partial \theta }} = U/(\theta - \theta _{{\mathrm{magic}}})\) is typical. Figure 1 demonstrates magic conditions for one PV pair in MgNC. In producing magic conditions with a general elliptical polarization, we estimate \(\delta \theta /\theta \sim 10^{ - 3}\)61, while for linear polarized light, δθ/θ < 10−4 is possible using high-quality Glan-type polarizers. We, therefore, expect δU′/U ~ 10−4 using a magic angle trap58,59, where the ODT linear polarization is rotated by \(\theta _{{\mathrm{magic}}} = {\mathrm{cos}}^{ - 1}\left( {\frac{1}{{\sqrt 3 }}} \right) \approx 54.7^\circ\) with respect to the quantization axis. For U = 2π × 1 MHz, δΔ = 2π × 100 Hz.

Polarization sensitivity for magic optical trapping. The Differential ac Stark shift U′ is plotted as a function of polarization angle θ as defined in the figures for the nominal |N = 1, J = 1/2, F = 3, mF = 3, P = +〉, |N = 1, J = 3/2, F = 3, mF = 3, P = −〉 parity violation (PV) pair in 25MgNC. For each configuration U = 1.2 MHz for the PV pair when U′ = 0 at θmagic. The calculation is performed with B ≈ 8.072 mT such that the PV pair is degenerate in the absence of the trapping field. (Red) linear polarization in the x, z-plane, (blue) elliptical polarization in x, z-plane, (green) elliptical polarization in x, y-plane
An inhomogeneous B-field will also produce broadening; typically δΔ ≈ μBδB21. We require \(\delta B \ll 1/\delta U\mu _B \, \lesssim \, 1\, {\mathrm{nT}}\) for B-field inhomogeneity to not limit sensitivity. This implies δB/B = 10−7 for the largest fields we may require, B = 10 mT. For comparison, the recent BaF NSD-PV measurement demonstrated δB/B = 10−8, even with the much more experimentally challenging B ≈ 460 mT20,21. We expect that the ability to easily reverse a smaller field will further aid in detecting and eliminating B-field inhomogeneities. Moreover, the field must only be homogenous over a small, fixed interaction volume for an ODT. For measurements in a molecular beam, the interaction volume grows proportional to the interaction time; in a fountain or free fall configuration, the interaction volume grows proportional to the square of the interaction time. The smaller interaction volume in an ODT implies that systematic uncertainties involving field gradients should also be reduced. In order to prevent temporal fluctuations from limiting sensitivity, we will require fluctuations δB ≲ 1 nT for all timescales longer than 1/ωE, the shortest relevant timescale in the experiment.
From Eq. (3), we see that electric dipole and NSD-PV transition amplitudes are π/2 out of phase, and there is no interference in a static E-field. However, the presence of a non-reversing E-field Enr still poses an issue. In the molecule frame, a static Enr has significant frequency components at axial and radial ODT frequencies ωz, ωr, and multiples, sums, and differences thereof. Assuming uncorrelated trap oscillations, this effect will lead to an inhomogeneous broadening much smaller than that of the differential ac Stark shift. Accurate measurement of Enr is possible by Stark interference with a reversible pulsed dc field Er21, or by microwave depletion spectroscopy63. Finally, investigating multiple PV pairs in the same molecule provides strong systematic error rejection by varying the ratio \(\left\langle {({\mathbf{S}} \times \widehat {\mathbf{I}}) \cdot \widehat {\mathbf{n}}} \right\rangle /d\) by a calculable, possibly large, amount19 (see Supplementary Note 3). For example, in PV pairs with different signs of \(\left\langle {({\mathbf{S}} \times \widehat {\mathbf{I}}) \cdot \widehat {\mathbf{n}}} \right\rangle /d\), contributions to \({\cal{A}}\) from actual NSD-PV will switch sign, but contributions from Enr will not.
Other relaxation mechanisms are expected to lead to negligible broadening compared to differential ac Stark shifts. For example, in a beam experiment, δΔ is typically limited by interaction time τ, with τ ~ 100 μs20. In an ODT, τ may easily exceed 1 s. Trapped molecule lifetimes τtrap = 0.5 s to 25 s have been reported in a variety of traps23,55,64,65. With near-ideal vacuum conditions, we expect trap lifetimes τtrap ~ 10 s, limited by vibrational decay. The loss rate due to off-resonant scattering from the trapping laser can typically be made Rsc ≲ 1 s−1 by using standard mid-infrared wavelength fiber lasers. Typical inelastic collision cross sections are expected to be σin ≲ 10−9 cm3/s. Comparing with the trapping conditions of Cheuk et al.35, with N = 1300 molecules at density n = 6 × 108 cm−3, we estimate an inelastic molecule-molecule collision rate of Rin ≈ 0.5 s−1. Therefore, collisions will become important when trapped molecule number N ≳ 105, or with additional cooling.
Estimated sensitivity to parity violating effects
We now estimate the sensitivity of our method to NSD-PV matrix elements, \(\delta W = 1/\tau \sqrt {RNT}\), where R is the repetition rate, N the number of trapped molecules per measurement, and T the total measurement time. We assume molecules are trapped in a U = 2π × 1 MHz deep magic angle trap, with δθ/θ = 10−4. The combined effects of all relaxation times considered should allow for interaction times of τ ≲ 1 s, but optimum sensitivity is achieved by setting the interaction time equal to the coherence time of the system: \(\tau = \frac{1}{{\delta {\mathrm{\Delta }}}} \approx \frac{1}{U}\frac{\theta }{{\delta \theta }} \approx 1.6\,{\mathrm{ms}}\). Allowing tMOT = 50 ms to load the MOT and ttrans = 40 ms for state transfer, repetition rates R = 10 s−1 should be possible. We expect that with molecules produced from an isotopically enriched source, N ≈ 1000 for all species; this would be equivalent to the best to-date sample of directly cooled molecules in an ODT35. Under these conditions, our expected experimental sensitivity is δW ≈ 2π × 1 Hz/\(\sqrt {{\mathrm{Hz}}}\). This represents a factor of 70 improvement over the best to-date NSD-PV measurement in BaF20,21,36. More ambitiously, one could plausibly expect N = 105 to 106 could be achieved with improved loading and cooling efficiency31,66.
Discussion
With the proposed sensitivity, it should be possible to separate contributions to κ from nuclear anapole (κa ∝ A2/3) and VeAn (κ2 A-independent) effects by measuring NSD-PV in a variety of nuclei. With the only non-zero NSD-PV measurement to-date in the heavy 133Cs10, a precise measurement of PV in a light system would be especially illuminating. In light nuclei, κ ≈ κ2. As stated, κ2 depends upon C2u,d which are among the most poorly know SM parameters and are suppressed at tree level. Thus, a precise measurement of NSD-PV in light systems could potentially be sensitive to beyond SM physics above the 1 TeV scale14. In molecules such as BeNC and MgNC, the nuclear and molecular calculations are highly tractable. Furthermore, Wp(N) ~ Wp(C) ~ 2π × 10 mHz in these systems; a single species could provide a 10% measurement of κ for three nuclei (13C, 14N, and either 9Be, 10Be, or 25Mg) with T ≲ 100 hours per nucleus. The nuclear structure of 14N is of special interest and well studied due to the anomalously long 14C → 14N half-life and their role in radiocarbon dating22.
Because iW is enhanced by ≈ Z2A2/3 in heavy species41,42, RaOH appears especially promising29. However, the high mass is a hinderance to effective laser-slowing by the standard methods for molecules52,53. Moreover, the longest-lived Ra isotope possessing nuclear spin (225Ra) has a half-life of only 15 days. New techniques beyond those proposed here may be required to produce trapped RaOH in sufficient quantity for precision measurement. Nevertheless, MOTs of atomic 225Ra with typical N = 1000 have been produced for atomic electric dipole measurements67; with our expected sensitivity, only N = 300 total molecules would need to be detected for δW/W = 0.1.
We have shown that optically trapped polyatomic molecules offer a dramatically enhanced sensitivity to parity-violating effects and additional checks of systematic errors. Restriction to laser-coolable species still allows for measurement of NSD-PV in nuclei with a wide range of masses, necessary for the determination of key SM parameters and tests of beyond SM physics. The improved sensitivity should enable measurements of NSD-PV even in light nuclei where calculations are highly accurate.
Here, we have only considered linear asymmetric molecules. Symmetric top molecules possess k-doublets of opposite parity (similar to \(\ell\)-doublets) even in their vibrational ground state. For most 2A1 states (analogue of 2Σ), the k-doublet splitting (~10 kHz) is smaller than SR/HF and thus not suitable for Zeeman tuning PV pairs to degeneracy for a NSD-PV measurement. However, the close spacing and miniscule differential Zeeman and ac Stark shifts of k-doublets may make symmetric top molecules in a magnetic or optical trap ideal for measuring NSI-PV.
Data availability
The data that support the findings of this study are available from the corresponding author on request.
References
Khriplovich, I. B. & Lamoreaux, S. K. CP Violation without Strangeness: Electric Dipole moments of Particles, Atomics, and Molecules (Springer-Verlag 1997).
The Jefferson Lab Qweak Collaboration. Precision measurement of the weak charge of the proton. Nature 557, 207–211 (2018).
Macpherson, M. J. D., Zetie, K. P., Warrington, R. B., Stacey, D. N. & Hoare, J. P. Precise measurement of parity nonconserving optical rotation at 876 nm in atomic bismuth. Phys. Rev. Lett. 67, 2784–2787 (1991).
Meekhof, D. M., Vetter, P., Majumder, P. K., Lamoreaux, S. K. & Fortson, E. N. High-precision measurement of parity nonconserving optical rotation in atomic lead. Phys. Rev. Lett. 71, 3442–3445 (1993).
Vetter, P. A., Meekhof, D. M., Majumder, P. K., Lamoreaux, S. K. & Fortson, E. N. Precise test of electroweak theory from a new measurement of parity nonconservation in atomic thallium. Phys. Rev. Lett. 74, 2658–2661 (1995).
Nguyen, A. T., Budker, D., DeMille, D. & Zolotorev, M. Search for parity nonconservation in atomic dysprosium. Phys. Rev. A 56, 3453–3463 (1997).
Tsigutkin, K. et al. Observation of a large atomic parity violation effect in ytterbium. Phys. Rev. Lett. 103, 071601 (2009).
Antypas, D. et al. Isotopic variation of parity violation in atomic ytterbium. Nat. Phys. 15, 120–123 (2019).
Khriplovich, I. B. Parity Nonconservation in Atomic Phenomena. (Gordon and Breach Science Publishers, USA, 1991).
Wood, C. S. et al. Measurement of parity nonconservation and an anapole moment in cesium. Science 275, 1759 (1997).
Haxton, W. C. & Wieman, C. E. Atomic parity nonconservation and nuclear anapole moments. Annu. Rev. Nucl. Part. Sci. 51, 261–293 (2001).
Johnson, W. R., Safronova, M. S. & Safronova, U. I. Combined effect of coherent Z exchange and the hyperfine interaction in the atomic parity-nonconserving interaction. Phys. Rev. A 67, 062106 (2003).
The Jefferson Lab PVDIS Collaboration, Wang et al. Measurement of parity violation in electron quark scattering. Nature 506, 67 EP – (2014).
Langacker, P., Luo, M. & Mann, A. K. High-precision electroweak experiments: A global search for new physics beyond the standard model. Rev. Mod. Phys. 64, 87–192 (1992).
Dzuba, V. A., Flambaum, V. V. & Stadnik, Y. V. Probing low-mass vector bosons with parity nonconservation and nuclear anapole moment measurements in atoms and molecules. Phys. Rev. Lett. 119, 223201 (2017).
Stadnik, Y. V. & Flambaum, V. V. Axion-induced effects in atoms, molecules, and nuclei: parity nonconservation, anapole moments, electric dipole moments, and spin-gravity and spin-axion momentum couplings. Phys. Rev. D. 89, 043522 (2014).
Kozlov, M. G. Semiempirical calculations of P and P, T odd effects in diatomic molecules-radicals. Sov. Phys. JETP 62, 1114–1118 (1985). [Zh. Eksp. Teor. Fiz.89,1933(1985)].
Flambaum, V. & Khriplovich, I. On the enhancement of parity nonconserving effects in diatomic molecules. Phys. Lett. A 110, 121 (1985).
DeMille, D., Cahn, S. B., Murphree, D., Rahmlow, D. A. & Kozlov, M. G. Using molecules to measure nuclear spin-dependent parity violation. Phys. Rev. Lett. 100, 023003 (2008).
Altunta, E., Ammon, J., Cahn, S. B. & DeMille, D. Demonstration of a sensitive method to measure nuclear-spin-dependent parity violation. Phys. Rev. Lett. 120, 142501 (2018).
Altunta, E., Ammon, J., Cahn, S. B. & DeMille, D. Measuring nuclear-spin-dependent parity violation with molecules: Experimental methods and analysis of systematic errors. Phys. Rev. A 97, 042101 (2018).
Maris, P. et al. Origin of the anomalous long lifetime of 14C. Phys. Rev. Lett. 106, 202502 (2011).
Anderegg, L. et al. Laser cooling of optically trapped molecules. Nat. Phys. 14, 890–893 (2018).
Prehn, A., Ibrügger, M., Glöckner, R., Rempe, G. & Zeppenfeld, M. Optoelectrical cooling of polar molecules to submillikelvin temperatures. Phys. Rev. Lett. 116, 063005 (2016).
Isaev, T. A. & Berger, R. Polyatomic candidates for cooling of molecules with lasers from simple theoretical concepts. Phys. Rev. Lett. 116, 063006 (2016).
Kozyryev, I., Baum, L., Matsuda, K., Hemmerling, B. & Doyle, J. M. Radiation pressure force from optical cycling on a polyatomic molecule. J. Phys. B: At., Mol. Opt. Phys. 49, 134002 (2016).
Kozyryev, I., Baum, L., Matsuda, K. & Doyle, J. M. Proposal for laser cooling of complex polyatomic molecules. ChemPhysChem 17, 3641–3648 (2016).
Kozyryev, I. et al. Sisyphus laser cooling of a polyatomic molecule. Phys. Rev. Lett. 118, 173201 (2017).
Isaev, T. A., Zaitsevskii, A. V. & Eliav, E. Laser-coolable polyatomic molecules with heavy nuclei. J. Phys. B: At., Mol. Opt. Phys. 50, 225101 (2017).
Kozyryev, I., Lasner, Z. & Doyle, J. M. Enhanced sensitivity to ultralight bosonic dark matter in the spectra of the linear radical SrOH. Preprint at https://arxiv.org/abs/1805.08185 (2018).
Kozyryev, I. & Hutzler, N. R. Precision measurement of time-reversal symmetry violation with laser-cooled polyatomic molecules. Phys. Rev. Lett. 119, 133002 (2017).
Kozlov, M. G. Linear polyatomic molecules with ∏ ground state: Sensitivity to variation of the fundamental constants. Phys. Rev. A 87, 032104 (2013).
Prehn, A., Ibrugger, M., Rempe, G. & Zeppenfeld, M. High-resolution spectroscopy on cold electrically trapped formaldehyde. Preprint at https://arxiv.org/abs/1807.06618 (2018).
Park, J. W., Yan, Z. Z., Loh, H., Will, S. A. & Zwierlein, M. W. Second-scale nuclear spin coherence time of ultracold 23Na40K molecules. Science 357, 372–375 (2017).
Cheuk, L. W. et al. Λ-enhanced imaging of molecules in an optical trap. Phys. Rev. Lett. 121, 083201 (2018).
Cahn, S. B. et al. Zeeman-tuned rotational level-crossing spectroscopy in a diatomic free radical. Phys. Rev. Lett. 112, 163002 (2014).
Hirota, E. High-Resolution Spectroscopy of Transient Molecules. (Springer-Verlag, Berlin, 1985).
Brown, J. M. & Carrington, A. Rotational Spectroscopy of Diatomic Molecules (Cambridge Univ. Press, 2003).
Flambaum, V. & Khriplovich, I. P-odd nuclear forces - a source of parity violation in atoms. Sov. Phys. JETP 52, 835 (1980).
Sheng, D., Orozco, L. A. & Gomez, E. Preliminary studies for anapole moment measurements in rubidium and francium. J. Phys. B: At., Mol. Opt. Phys. 43, 074004 (2010).
Borschevsky, A., Iliaš, M., Dzuba, V. A., Flambaum, V. V. & Schwerdtfeger, P. Relativistic study of nuclear-anapole-moment effects in diatomic molecules. Phys. Rev. A 88, 022125 (2013).
Isaev, T. A. & Berger, R. Electron correlation and nuclear charge dependence of parity-violating properties in open-shell diatomic molecules. Phys. Rev. A 86, 062515 (2012).
Hao, Y. et al. Nuclear anapole moment interaction in baf from relativistic coupled-cluster theory. Phys. Rev. A 98, 032510 (2018).
Di Rosa, M. Laser-cooling molecules. Eur. Phys. J. D. - At., Mol., Opt. Plasma Phys. 31, 395–402 (2004).
Lane, I. C. Ultracold fluorine production via Doppler cooled BeF. Phys. Chem. Chem. Phys. 14, 15078–15087 (2012).
Hendricks, R., Holland, D., Truppe, S., Sauer, B. E. & Tarbutt, M. Vibrational branching ratios and hyperfine structure of BH and its suitability for laser cooling. Front. Phys. 2, 51 (2014).
Lim, J. et al. Laser cooled YbF molecules for measuring the electron’s electric dipole moment. Phys. Rev. Lett. 120, 123201 (2018).
Hunter, L. R., Peck, S. K., Greenspon, A. S., Alam, S. S. & DeMille, D. Prospects for laser cooling TlF. Phys. Rev. A 85, 012511 (2012).
Norrgard, E. B. et al. Hyperfine structure of the B 3∏1 state and predictions of optical cycling behavior in the X → B transition of TlF. Phys. Rev. A 95, 062506 (2017).
Nakhate, S., Steimle, T. C., Pilgram, N. H. & Hutzler, N. R. The pure rotational spectrum of YbOH. Chem. Phys. Lett. 715, 105–108 (2019).
Maxwell, S. E. et al. High-flux beam source for cold, slow atoms or molecules. Phys. Rev. Lett. 95, 173201 (2005).
Hutzler, N. R., Lu, H.-I. & Doyle, J. M. The buffer gas beam: an intense, cold, and slow source for atoms and molecules. Chem. Rev. 112, 4803–4827 (2012).
Barry, J. F., Shuman, E. S., Norrgard, E. B. & DeMille, D. Laser radiation pressure slowing of a molecular beam. Phys. Rev. Lett. 108, 103002 (2012).
Barry, J. F., McCarron, D. J., Norrgard, E. N., Steinecker, M. H. & DeMille, D. Magneto-optical trapping of a diatomic molecule. Nature 512, 286 (2014).
Chotia, A. et al. Long-lived dipolar molecules and Feshbach molecules in a 3d optical lattice. Phys. Rev. Lett. 108, 080405 (2012).
Panda, C. D. et al. Stimulated Raman adiabatic passage preparation of a coherent superposition of ThO H3Δ1 states for an improved electron electric-dipole-moment measurement. Phys. Rev. A 93, 052110 (2016).
Lasner, Z. & DeMille, D. Statistical sensitivity of phase measurements via laser-induced fluorescence with optical cycling detection. Phys. Rev. A 98, 053823 (2018).
Romalis, M. V. & Fortson, E. N. Zeeman frequency shifts in an optical dipole trap used to search for an electric-dipole moment. Phys. Rev. A 59, 4547–4558 (1999).
Kotochigova, S. & DeMille, D. Electric-field-dependent dynamic polarizability and state-insensitive conditions for optical trapping of diatomic polar molecules. Phys. Rev. A 82, 063421 (2010).
Neyenhuis, B. et al. Anisotropic polarizability of ultracold polar 40K87 molecules. Phys. Rev. Lett. 109, 230403 (2012).
Kim, H., Han, H. S. & Cho, D. Magic polarization for optical trapping of atoms without Stark-induced dephasing. Phys. Rev. Lett. 111, 243004 (2013).
Rosenband, T., Grimes, D. D. & Ni, K.-K. Elliptical polarization for molecular stark shift compensation in deep optical traps. Opt. Express 26, 19821–19825 (2018).
O’Leary, B. In search of the electrons electric dipole moment in thorium monoxide: an improved upper limit, systematic error models, and apparatus upgrades. Ph.D. thesis, Yale University (2016).
Norrgard, E. B., McCarron, D. J., Steinecker, M. H., Tarbutt, M. R. & DeMille, D. Submillikelvin dipolar molecules in a radio-frequency magneto-optical trap. Phys. Rev. Lett. 116, 063004 (2016).
Williams, H. J. et al. Magnetic trapping and coherent control of laser-cooled molecules. Phys. Rev. Lett. 120, 163201 (2018).
De Marco, L. et al. A degenerate Fermi gas of polar molecules. Science 363, 853–856 (2019).
Parker, R. H. et al. First measurement of the atomic electric dipole moment of 225Ra. Phys. Rev. Lett. 114, 233002 (2015).
Acknowledgements
The authors thank E. Altuntaş, D. DeMille, N. Hutzler, and I. Kozyryev for insightful conversations into polyatomic molecular structure and PV measurements. We thank E. Altuntaş and E. Shirley for their careful reading of the manuscript. E.B.N. and D.S.B. acknowledge support from the National Research Council Postdoctoral Research Associateship Program.
Author information
Authors and Affiliations
Contributions
E.B.N. conceived the research proposal. E.B.N., D.S.B., S.E., J.A.F., N.N.K. and J.S. contributed to analysis and interpretation of the data, and in preparing the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Norrgard, E.B., Barker, D.S., Eckel, S. et al. Nuclear-spin dependent parity violation in optically trapped polyatomic molecules. Commun Phys 2, 77 (2019). https://doi.org/10.1038/s42005-019-0181-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42005-019-0181-1
This article is cited by
-
An optical tweezer array of ultracold polyatomic molecules
Nature (2024)
-
Absolute frequency metrology of buffer-gas-cooled molecular spectra at 1 kHz accuracy level
Nature Communications (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
