
Abstract
Dehydrohelicenes are some of the most attractive chiroptical materials with unique helical chirality. However, to our knowledge, there are no prior reports on their direct construction by asymmetric methods. In this work, sequential synthesis of aza-oxa-dehydro[7]helicenes via the electrochemical oxidative hetero-coupling of 3-hydoxycarbazoles and 2-naphthols followed by dehydrative cyclization and intramolecular C–C bond formation has been realized. In addition, an efficient enantioselective synthesis through chiral vanadium-catalyzed hetero-coupling and electrochemical oxidative transformations afforded heterodehydro[7]helicene without any racemization. The obtained dehydro[7]helicenes showed intense blue-colored circularly polarized luminescence (|glum| ≈ 2.5 × 10−3 at 433 nm). Thermodynamic and kinetic studies of the racemization barrier of heterodehydro[7]helicenes indicated significant chiral stability with ΔG‡> 140 kJ mol−1.
Similar content being viewed by others
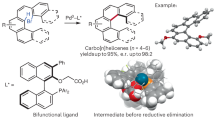
Introduction
Heterodehydrohelicenes, also known as quasi-heterocirculenes with helical chirality, can be classified as polycyclic heteroaromatics (PHAs)1 in which the two helical termini of a helicene are connected by a sigma bond2,3,4,5,6,7,8,9,10. Their unique helical chirality has led to extraordinary chiroptical responses11 which can be implemented in various material-based applications, such as organic light-emitting diodes (OLEDs) and field-effect transistors (FETs). Moreover, the superior chiroptical properties of heterodehydrohelicenes such as circular dichroism (CD) and circularly polarized luminescence (CPL) open the gate for various applications in optical information storage and transfer, in such cases the level of CPL can promote a further dimension to the information content transported through light12. In 1969, Zander and Franke first reported a aza-dehydro[6]helicene synthesized from the corresponding aza[6]helicene as a precursor through metal(Al)-mediated terminal ring closure2. Similar protocols have been subsequently applied to synthesize thiophene-based dehydro[5]3, [6] (using Al)4, and [7]helicenes (using Pd and Sn)5. Tanaka and Osuka have synthesized aza-dehydro[7]helicene derivatives with three pyrrole rings using bis(trifluoroacetoxy)iodobenzene (PIFA); these molecules exhibit interesting photophysical properties6. However, these heterodehydrohelicenes were difficult to isolate as optically pure forms due to their low racemization barriers2,3,4,5,6.
In 2017, Itami and Segawa first reported thia-dehydro[6]helicenes as saddle-helix molecules with tert-butyl substitutions at the terminal aromatic rings that prevent racemization; HPLC with a chiral stationary phase could be used for enantiomeric separation7. Tanaka, Osuka8, and Pittelkow9 have also reported heterodehydro[7]helicene modifications that lead to significantly high racemization barriers. In 2021, Maeda and Ema reported an aza-dehydro[7]helicene with intense CPL and Cotton effects10. Despite the immense potential exhibited by heterodehydrohelicenes, to our knowledge, there are no reports on their straightforward construction including asymmetric synthesis. This could be due to the limitations associated with the synthetic steps: low total yields, harsh reaction conditions (such as high temperature), easy racemization, and/or overuse of oxidants (narrow functional group tolerance). In 2016, we reported an efficient vanadium-catalyzed synthesis of oxa[9]helicenes13 via the oxidative coupling of arenol compounds14 followed by intramolecular dehydrative cyclization. Recently we also developed the catalytic enantioselective oxidative hetero-coupling of arenols using a chiral vanadium(V) complex15. Based on these radical-anion coupling mechanism, we envisioned applying an organic electrochemical method to develop more environmentally benign syntheses of heterodehydrohelicenes. Electrochemical syntheses have many advantages, since no oxidant is required and oxidative transformation can be conducted under mild reaction conditions16. Indeed, many metal- and oxidant-free electrochemical oxidative coupling reactions of arenols have been reported17. Also, some reports describing a cascade electrochemical approach for synthesizing PHAs.18 However, to our knowledge, there have been no reports on heterodehydrohelicene syntheses using arenol as a starting material under electrochemical conditions. Herein, we describe two efficient methods for synthesizing aza-oxa-dehydro[7]helicenes 3 possessing multiple heteroaromatic rings; the electrochemical sequential synthesis of 3 through the oxidative hetero-coupling of readily available arenols 1 and 2 followed by dehydrative cyclization and intramolecular C-C bond formation, and the enantioselective approach through chiral vanadium-catalyzed hetero-coupling and electrochemical oxidative transformations (Fig. 1).

Two methods for synthesizing dehydro[7]helicenes 3: (A) electrochemical sequential synthesis through the oxidative hetero-coupling of 1 and 2 followed by dehydrative cyclization and intramolecular C-C bond formation; (B) stepwise enantioselective synthesis through chiral vanadium-catalyzed hetero-coupling and electrochemical oxidative transformations.
Results and discussion
Screening of reaction conditions and evaluation of substrate scope
After examining various conditions and performing sedulous optimization (see Tables S1–S4 in Supplementary Method 3), a sequential protocol generating aza-oxa-dehydro[7]helicenes 3aa in 84% yield (81% isolated yield, current efficiency = 30%) was developed; hydroxybenzo[c]carbazole 1a and 7-methoxy-2-naphthol 2a were utilized as model substrates based on CV studies (see Fig. S18 in Supplementary Note 6), with fluorine-doped tin oxide (FTO)19 electrodes and Bu4NPF6 as the electrolyte (0.1 M) at 25 °C, in the presence of BF3·OEt2 (as an additive) in CH2Cl2 and no homocoupling products were observed (Table 1, entry 1)20. The structure of 3aa was confirmed by X-ray crystallography (see Supplementary Note 7). Other solvents [THF, MeCN, and MeOH (5 mL)] formed diol 4aa as the major side product in 9 to 22% yields (entries 2–4). The FTO electrodes showed high conversion of substrates (entry 1), while changing any one of the electrodes resulted in reduced conversion yields (entries 5–6). Doubling the current density reduced the yield of 3aa to 65% (entry 7); its reduction to 0.8 mA/cm2 did not significantly affect the yield but prolonged the reaction time (entry 8). High concentrations of the electrolyte, and other electrolytes such as LiClO4 (0.1 M) formed diol 4aa as the major side product (entries 9–10). Decreasing the amount of BF3·OEt2 or replacing it with other Brønsted acids (as acidic additives) produced low yields of aza-oxa-dehydro[7]helicene 3aa (4aa was isolated as the major side product in 5 to 20% yields) (entries 11–13). BF3·OEt2 transformed diol 4aa to helicene intermediate 5aa, which underwent facile anodic oxidation to form 3aa (see Scheme S1 in Supplementary Note 3). No reaction occurred in the absence of electricity (entry 14). Finally, to generalize this protocol, 3aa was synthesized using ElectraSyn® 2.0 (designed by IKA)21; it exhibited comparable results (entry 15).
Subsequently, the substrate scope of various hydroxycarbazoles 1 and 2-naphthols 2 were investigated under the optimal reaction conditions (Fig. 2). N-Aryl- and N-alkyl-substituted derivatives 1a–1f underwent facile conversion to aza-oxa-dehydro[7]helicenes 3aa–3fa in 78–86% yields. Different combinations of 2 with 7-benzyloxy or allyloxy groups 2b–2c were used to synthesize aza-oxa-dehydro[7]helicenes 3ab–3fc in 71–84% yields. A series of compounds 1 substituted by Me, Ph, and OMe groups at various positions on the aromatic ring formed products 3ga–3la in 76–84% yields. π-Expanded substrate 1i also reacted with 2a to form 3ia in 80% yield. Hydroxycarbazole 1m with an electron-withdrawing cyano group afforded 3ma in 67% yield.

Electrolysis conditions: FTO anode, FTO cathode, constant current = 3 mA (J = 1.2 mA/cm2), 1 and 2 (0.1 mmol), Bu4NPF6 (0.1 M), BF3·OEt2 (0.2 M), CH2Cl2 (5 mL), 25 °C, 10 h.
Two-pot synthesis and transformation of product
To establish the applicability of this method for concise synthesis, a two-pot protocol using commercially available substrates p-benzoquinone (6) and N-phenyl-2-naphthylamine (7) was tested; it produced aza-oxa-dehydro[7]helicene 3ba in 55% overall yield (Fig. 3A).

A Synthesis of dehydrohelicene 3ba from commercially available substrates. B AlCl3- medicated deprotection of Bn- group followed by Pd-catalyzed N-arylation. C Basic hydrolysis of cyano group to afford the corresponding carboxylic acid. D Demethylation of dehydrohelicene 3pa to give the corresponding circulene 8.
Transformations of aza-oxa-dehydro[7]helicenes 3 were also investigated (see Supplementary Method 6). The benzyl group on the nitrogen of 3fa was removed using AlCl3 to give 3na, which underwent Pd-catalyzed N-arylation to form 3ba (Fig. 3B). The carboxylic acid derivative 3oa was formed after the hydrolysis of 3ma under basic conditions (Fig. 3C). Furthermore, a treatment of 3pa with BBr3 at 25 °C afforded the corresponding aza-dioxa[8]circulene 8 in 92% yield (Fig. 3D).
Circular dichroism (CD) and circularly polarized luminescence (CPL) analysis of aza-oxa- dehydro[7]helicenes
HPLC using a chiral stationary phase (CHIRALPAK IC, hexane/i-PrOH=30:1), was used for the optical resolution of 3aa, 3ca and 3fa to analyze the racemization barriers of the obtained aza- oxa-dehydro[7]helicenes, (see Supplementary Note 2). The optical rotation values of optically pure 3aa first peak (M)-3aa: [α]D24 = –1464 (c = 0.0175 g/mL), second peak (P)-3aa: [α]D24 = +1462 (c = 0.0173 g/mL). The absolute configuration of (−)-3aa was determined as M through X-ray crystallographic analysis after recrystallization of the first peak of enantiomer 3aa. Eyring plots indicated the significant chiral stability of aza-oxa-dehydro[7]helicenes 3 (racemization barrier >140 kJ mol−1) (see Fig. S2–S10 in Supplementary Note 2); the t1/2 of compound 3aa was estimated to be greater than 9.5 × 103 years at 25 °C. Aza-oxa-dehydro[7]helicene 3aa showed higher chiral stability than that of corresponding aza-oxa[7]helicene 5aa (110 kJ mol−1). Subsequently, the chiroptical properties of the optically pure aza-oxa-dehydro[7]helicenes were investigated. All the helical dyes 3 showed absorption in the wavelength range of 340–404 nm and fluorescence maximum at 450 nm (see Figs. S11-S13 in Supplementary Note 4); CD and CPL11 signals were observed in these regions (see Figs. S14-S17 in Supplementary Note 5). Among synthesized aza-oxa-dehydro[7]helicenes, 3aa showed moderate quantum yield Φ = 0.25, and significant CPL activity with glum = 2.5 × 10−3 at 433 nm (Fig. 4).

Chiroptical properties of 3aa and 3ab (CD and CPL) were studied in CHCl3 (2 × 10−5 M). 3aa showed high CPL activity with glum = 2.5 × 10−3 at 433 nm and 3ab showed glum = 2.4 × 10−3 at 418 nm.
An efficient enantioselective synthesis of heterodehydro[7]helicenes via chiral vanadium complex catalyzed hetero-coupling and electrochemical oxidative transformations
Although many reports are found on the enantioselective synthesis of helicenes22,23,24,25,26, to our knowledge, there are no such reports for heterodehydrohelicenes. An efficient chemo- and enantioselective protocol for the oxidative hetero-coupling of hydroxycarbazoles and naphthols using a chiral vanadium(v) complex, producing axially chiral biaryl derivatives, has been previously reported15; based on this study, a stepwise enantioselective synthesis of diol 4 was examined, which was followed by their electrochemical transformation to the corresponding heterodehydrohelicenes (see Supplementary Method 7). Diol (R)-4ba was synthesized with a good optical purity using mononuclear vanadium complex (Ra,S)-9. Under electrooxidation conditions, (R)-4ba underwent a sequential dehydrative furan ring formation followed by the coupling of the two helical termini to afford the corresponding aza-oxa-dehydro[7]helicene, (M)-3ba (see Supplementary Note 1), in 87% yield without any loss in optical purity (Fig. 5A). Notably, the protocol was scaled up, as depicted in Fig. 5B, to form 0.62 g of (M)-3ba (current efficiency = 48%).

A Stepwise enantioselective synthesis of dehydrohelicene (M)-3ba using hybrid vanadium and electrochemistry. B Scaling up the enantioselective synthesis to semi-gram scale.
In conclusion, an efficient synthetic method utilizing an electrochemical reaction under mild conditions was developed for aza-oxa-dehydro[7]helicenes 3. The racemization barriers of the heterodehydrohelicenes synthesized in this study were significantly higher than those of the corresponding helicenes with sustained racemization half-lives. Furthermore, this paper reports the enantioselective synthesis of heterodehydrohelicenes for the first time, with a scaled-up reaction (to the gram-scale). Applications of the chiroptical properties are currently under investigation.
Methods
General methods
For synthetic details and analytical data of all reaction starting materials 1, see Supplementary Methods 1 and 2. For synthetic and analytical details of all dehydro[7]helicenes 3, see Supplementary method 4. For the general procedure of two-pot synthesis and derivatization of dehydro[7]helicene, see Supplementary Methods 5 and 6. Enantioselective synthesis and scaling-up details are highlighted in Supplementary Method 7. For NMR spectra seeSupplementary Data 1.
General procedure for the sequential preparation of heterodehydrohelicenes 3
A solution of benzo[c]carbazol-10-ol derivatives 1 (0.1 mmol), 2-naphthols 2 (0.1 mmol), tetrabutylammonium hexafluorophosphate(V) (193.7 mg, 0.5 mmol) and BF3・EtO2 (0.2 M) in CH2Cl2 (5.0 mL) was transferred into an undivided electrolysis cell. This cell is equipped with two FTO electrodes (1.0 × 2.5 cm2), which are connected to DC power supply (see Fig. S1 in Supplementary Method 2). At rt, a constant current electrolysis with a current density of 1.20 mA/cm2 was applied. After stirring for 10 h, the electrolysis was stopped and purification of the crude products by column chromatography (SiO2, EtOAc/hexane) provided the desired heterodehydro[7]helicene 3.
Data availability
Additional data supporting the findings described in this manuscript are available in Supplementary Information and Supplementary Data 1. All CIFs are available in Supplementary Data 2-6. The X-ray crystallographic coordinate for structures reported in this study have been deposited at the Cambridge Crystallographic Data Center (CCDC) under deposition numbers CCDC-2091483 (3aa), CCDC-2128351 (5aa), CCDC-158892 (3ma), CCDC-2183557 (8), and CCDC 2057234 (10aa). These data can be obtained free of charge from The Cambridge Crystallographic Data Center via www.ccdc.cam.ac.uk/data_request/cif. The authors declare that all other data supporting the findings of this study are available within the article and Supplementary Information files, and also are available from the corresponding author upon reasonable request.
References
Borissov, A. et al. Recent advances in heterocyclic nanographenes and other polycyclic heteroaromatic compounds. Chem. Rev. 122, 565–788 (2022).
Zander, M. & Franke, W. H. Über carbazolo-carbazole. Chem. Ber. 102, 2728–2738 (1969).
Wynberg, H., Groen, M. B. & Schadenberg, H. Synthesis and resolution of some heterohelicenes. J. Org. Chem. 36, 2797–2809 (1971).
Dopper, J. H., Oudman, D. & Wynberg, H. Dehydrogenation of heterohelicenes by a Scholl type reaction. dehydrohelicenes. J. Org. Chem. 40, 3398–3401 (1975).
Rajca, A. et al. Intramolecular cyclization of thiophene-based [7]helicenes to quasi- [8]circulenes. J. Org. Chem. 74, 9105–9111 (2009).
Chen, F., Tanaka, T., Mori, T. & Osuka, A. Synthesis, structures, and optical properties of azahelicene derivatives and unexpected formation of azahepta[8]circulenes. Chem. Eur. J. 24, 7489–7497 (2018).
Fujikawa, T., Segawa, Y. & Itami, K. Laterally π-extended dithia[6]helicenes with heptagons: saddle-helix hybrid molecules. J. Org. Chem. 82, 7745–7749 (2017).
Matsuo, Y., Chen, F., Kise, K., Tanaka, T. & Osuka, A. Facile synthesis of fluorescent hetero[8]circulene analogues with tunable solubilities and optical properties. Chem. Sci. 10, 11006–11012 (2019).
Lousen, B. et al. Compressing a non‐planar aromatic heterocyclic [7]helicene to a planar hetero[8]circulene. Chem. Eur. J. 26, 4935–4940 (2020).
Maeda, C., Nomoto, S., Akiyama, K., Tanaka, T. & Ema, T. Facile synthesis of azahelicenes and diaza[8]circulenes through the intramolecular Scholl reaction. Chem. Eur. J. 27, 15699–15705 (2021).
Mori, T. Chiroptical properties of symmetric double, triple, and multiple helicenes. Chem. Rev. 121, 2373–2412 (2021).
Peeters, E. et al. Circularly polarized electroluminescence from a polymer light-emitting diode. J. Am. Chem. Soc. 119, 9909–9910 (1997).
Sako, M. et al. Efficient enantioselective synthesis of oxahelicenes using redox/acid cooperative catalysts. J. Am. Chem. Soc. 138, 11481–11484 (2016).
Sako, M., Takizawa, S. & Sasai, H. Chiral vanadium complex-catalyzed oxidative coupling of arenols. Teterahedron 76, 131645 (2020).
Sako, M. et al. Chemo- and enantioselective hetero-coupling of hydroxycarbazoles catalyzed by a chiral vanadium(v) complex. Org. Chem. Front. 8, 4878–4885 (2021).
Cheng, X. et al. Recent applications of homogeneous catalysis in electrochemical organic synthesis. CCS Chem. 4, 1120–1152 (2022).
Röckl, J. L., Pollok, D., Franke, F. & Waldvogel, S. R. A decade of electrochemical dehydrogenative C,C-coupling of aryls. Acc. Chem. Res. 53, 45–61 (2020).
Zhang, L. & Hu, X. Nickel catalysis enables convergent paired electrolysis for direct arylation of benzylic C–H bonds. Chem. Sci. 11, 10786–10791 (2020).
Hou, Z. W., Mao, Z. Y., Song, J. & Xu, H. C. Electrochemical synthesis of polycyclic N- heteroaromatics through cascade radical cyclization of diynes. ACS Catal. 7, 5810–5813 (2017).
Kingston, C. et al. A survival guide for the “electro-curious”. Acc. Chem. Res. 53, 72–83 (2020).
https://www.ika.com/en/Products-Lab-Eq/Electrochemistry-Kit-csp-516/ElectraSyn-20- pro-Package-cpdt-40003261/
Pelliccioli, V. et al. Enantioselective synthesis of dithia[5]helicenes and their postsynthetic functionalization to access dithia[9]helicenes. Angew. Chem. Int. Ed. 61, e202114577 (2022).
Wang, Q., Zhang, W. W., Zheng, C., Gu, Q. & You, S. L. Enantioselective synthesis ofazoniahelicenes by Rh-catalyzed C–H annulation with alkynes. J. Am. Chem. Soc. 143, 114–120 (2020).
Stará, I. G. & Starý, I. Helically chiral aromatics: The synthesis of helicenes by [2+2+2] cycloisomerization of π-electron systems. Acc. Chem. Res. 53, 144–158 (2020).
Yubuta, A. et al. Enantioselective synthesis of triple helicenes by cross-cyclotrimerization of a helicenyl aryne and alkynes via dynamic kinetic resolution. J. Am. Chem. Soc. 142, 10025–10033 (2020).
Dhbaibi, K., Favereau, L. & Crassous, J. Enantioenriched helicenes and helicenoids containing main-group elements (B, Si, N, P). Chem. Rev. 119, 8846–8953 (2019).
Acknowledgements
This study is dedicated to Professor Masakatsu Shibasaki on the occasion of his 75th birthday. The work was funded by grants from JSPS KAKENHI Early Career Scientists (No. JP20K15281), Transformative Research Areas (A) 21A204 Digitalization-driven Transformative Organic Synthesis (Digi-TOS) from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT), and the Japan Society for the Promotion of Science (JSPS), JST CREST (No. JPMJCR20R1), Masuyakinen Basic Research Foundation, and Hoansha Foundation. We acknowledge the technical staff of the Comprehensive Analysis Center of SANKEN, Osaka. University (Japan). We sincerely appreciate Prof. Masahiro Miura and Mr. Shotaro Nakamura for the CPL analysis of the heterodehydrohelicenes.
Author information
Authors and Affiliations
Contributions
M.I.K., M. S. H.S.: Performing the experiments, analysing data, and writing the paper. S. M. and K. M.: Designing the experiments. H. S. and S. T.: Supervising the project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Chemistry thanks Takayuki Tanaka and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Khalid, M.I., Salem, M.S.H., Sako, M. et al. Electrochemical synthesis of heterodehydro[7]helicenes. Commun Chem 5, 166 (2022). https://doi.org/10.1038/s42004-022-00780-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42004-022-00780-7
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
