
Abstract
Coupling the photo-oxidation of biomass derived substrates with water splitting in a photoelectrochemical (PEC) cell is a broadly discussed approach intended to enhance efficiency of hydrogen generation at the cathode. Here, we report a PEC device employing a nanostructured semitransparent WO3 photoanode that, irradiated with simulated solar light achieves large photocurrents of 6.5 mA cm−2 through oxidation of glucose, a common carbohydrate available in nature that can be obtained by processing waste biomass. The attained photocurrents are in a large part due to the occurrence of the photocurrent doubling, where oxidation of glucose by the photogenerated positive hole is followed by injection by the formed intermediate of an electron into the conduction band of WO3. Selection of an appropriate supporting electrolyte enabled effective reforming of glucose into valuable products: gluconic and glucaric acids, erythrose and arabinose with up to 64% total Faradaic yield attained at ca 15% glucose conversion.
Similar content being viewed by others

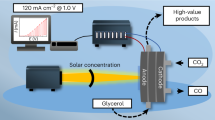

Introduction
The continuing research on photoelectrochemical (PEC) water splitting to allow generation of sustainable hydrogen fuel from water and sunlight is now principally oriented towards improving the stability and performance of the employed photoelectrodes1,2,3. In the most frequently employed PEC cell configuration, an n-type semiconductor (SC) photoanode (where photogenerated positive holes, h+, oxidize water to form O2) is combined with a metallic cathode (where electrons, e-, transferred from the photoanode, reduce protons to form H2). It is to be noted, in this connection that, considering high overpotentials accompanying the oxygen evolution reaction (OER) on conventional (unilluminated) anodes, the SC photoanodes may arguably offer larger advantage than the photocathodes for the performance of a PEC water splitting cell. As regards the photoanodes, the only materials able to photo-oxidize water whilst avoiding photocorrosion (essentially for kinetic reasons) are metallic oxides4,5, and only few of them (Fe2O3, BiVO4, WO3) feature optical absorption spectra that match terrestrial solar spectrum. Since none among those photostable - metallic oxide - semiconductors exhibits band-edge energy levels that match those of hydrogen and oxygen evolution reactions to allow unassisted water splitting, the present efforts focus on minimizing the bias voltage allowing the photoanode to reach plateau (saturation) photocurrent when performing visible light-driven photo-oxidation of water6,7,8,9,10,11. In fact, the moderate operating potential point at which the photocurrent already reaches saturation is a precondition for combining the photoelectrolyzer with a photovoltaic (PV) cell to form a tandem device12,13,14. Actually, a bias voltage of the order of 1 V (at which the semitransparent WO3 photoanode approaches saturation photocurrent) may be provided by several single-junction photovoltaic (PV) cells likely to operate in a tandem device with the PEC cell.
The principal strategy developed for improving the photocurrent-voltage (j-E) behavior of the SC photoanodes, by reducing e--h+ recombination losses, involved deposition on the photoanodes of OER electrocatalysts such as mixed cobalt phosphate-oxide (Co–Pi)15,16 or oxyhydroxides of nickel and iron17,18.
To address this issue, several authors also attempted to combine water splitting with the kinetically easier photo-oxidation of biomass derivatives to replace or complement oxygen evolution on the photoanode. However, an optimal implementation of such concept would require, rather than total mineralization at the photoanode of the used biomass-derived reagent (to form carbon dioxide), its efficient conversion into value-added products. Early studies aimed to enhance hydrogen production at the cathode via photo-reforming at the anode of biomass-derived products used principally nano-crystalline (NC) TiO2 photoanodes19,20,21,22 and ethanol as model compound acting as a hole scavenger. When an alkaline electrolyte (1 M NaOH) was employed by the authors19,20,22 in the anode compartment, the ethanol solutions did not reach total mineralization due, in particular, to the formation of insoluble aldol condensation products - high molecular weight aldehydes22. The latter investigations followed parallel studies in this area that used relatively low concentrations of oxygenate biomass derivatives with suspended in the solutions TiO2 nanoparticles (NPs) acting as photocatalysts23,24.
However, an important advantage of the PEC cell configuration is in the facility of produced gas separation and the possibility of controlling the atmosphere in the anode compartment. Although the photo-oxidation of biomass-derived oxygenates at NC TiO2 photoanodes has been shown to be highly efficient25 and to benefit, in addition, from the photocurrent doubling effect26, the large band gap of the semiconductor (Eg = 3.2 eV), for the anatase form (that is the major component of the widely employed commercial P25 particles) is an evident limitation for the effective use of sunlight as the irradiation source.
The photocurrent doubling effect has also been demonstrated for the photo-oxidation of organic C127 and larger28 molecules at nanostructured (NS) tungsten trioxide photoelectrodes that, due to their lower bandgap energy (Eg = 2.5–2.6 eV), are capable to also absorb light over the visible blue portion of the solar spectrum. Despite the fact that Eg of WO3 is larger than the optimum value of 2-2.2 eV, the NS WO3 photoanodes attained considerable photocurrent efficiencies29,30 due to relatively long hole diffusion length of ca 150 nm31,32 that reduces bulk electron-hole recombination within the NPs that form the film. On the other hand, the efficient photogenerated current collection at the electrode substrate is favored by large (in the range of 6.5-12 cm2 V−1 s−1) electron mobility in the crystalline WO333,34.
A more recent work35, employing thin film WO3 photoanodes formed by sputtering confirmed the occurrence of the photocurrent doubling also in the case of photo-oxidation of a biomass-derived monosaccharide – glucose. Significant information that stemmed from that study was a relatively low extent of glucose conversion into CO2, in particular in more concentrated (0.1 M) solutions, that indicated the dominant formation of intermediate oxidation products that could not been identified. We note in this connection that numerous experiments on photo-reforming glucose and/or lignocellulose using suspensions of TiO2 photocatalysts loaded with noble metal’s (most frequently Pt) NPs36,37,38,39, in which changes in the solution composition were monitored using high-performance liquid chromatographic (HPLC) analyses, showed the formation of a wide variety, some of them high-value, oxidation products. In one of such studies, the TiO2 doping with tungsten allowed the operation of such Pt-TiO2(W) photocatalyst under sunlight irradiation while TiO2 doping with nitrogen favored formation, besides H2 and CO2, of arabinose, erythrose and formic acid as partial glucose oxidation products37.
An interesting approach, intended to avoid the use of noble metal catalysts required to reduce protons into H2 in the case of photo-reforming of lignocellulose at TiO2 photocatalysts39, resulted in the development of a photocatalytic system based on n-type SC cadmium sulfide quantum dots40. In fact, CdS (Eg = 2.4 eV) that absorbs the blue portion of the visible spectrum features the conduction band (CB) level at a potential sufficiently negative (ca −0.5 V vs SHE) to favor H2 evolution in the absence of co-catalysts. To conduct photo-reforming of α-cellulose, the authors used a strongly alkaline (10 M KOH) solution, the conditions under which the surface of suspended CdS quantum dots became passivated by a CdOx overlayer40. However, despite efficient sunlight-driven H2 evolution, due to the complex chemistry involved in the synthesis of CdS quantum dots and the well-known toxicity of cadmium compounds, the latter work may be rather considered as an interesting proof of concept.
Among few examples in which upgrading of biomass-derived compounds was performed in a PEC cell, and not in a photocatalytic system featuring suspension of SC particles41, is the oxidation on a BiVO4 - based photoanode of an important intermediate in biomass conversion 5-hydroxymethylfurfural (HMF) into 2,5-difurancarboxylic acid (FDCA) - a monomer used for the synthesis of several polymers42. The advantage of the employed in that case PEC configuration is that hydrogen was co-generated in a separate cathode cell compartment.
Herein we describe a series of experiments performed in a PEC cell featuring NS WO3 photoanodes, in which we aimed to optimize the photocurrents generated through photo-oxidation of glucose, selected as the target compound, in order to enhance the efficiency of hydrogen generation at the cathode. Let us to recall that glucose is a common carbohydrate available in nature that can be obtained through the hydrolysis of cellulose and hemicellulose present in waste biomass. We demonstrate that a significant replacement of OER with the photo-oxidation of glucose results in a large increase of the photocurrents attained at the NS WO3 photoanode due to the more favorable reaction kinetics and, especially, due to the occurrence of the photocurrent doubling. We probed the photocurrent efficiency as well as stability of the photoanode to also find out the most appropriate electrolyte composition, which allows the formation of the largest amount of valuable organic products – gluconic and glucuronic acids, arabinose and erythrose.
Results
Preparation and structural characterization of WO3 film electrodes
We synthesized the NS WO3 films by a sol-gel method involving layer-by-layer deposition on the substrate of a colloidal solution of freshly prepared tungstic acid, complemented by the addition of polyethylene glycol (PEG 300), followed by high temperature (550-600 °C) annealing in oxygen. Conductive fluorine-doped tin oxide (FTO)-coated glass sheets served as electrode substrates. As confirmed by repeated PEC experiments performed with the same electrodes, the use of the sequential layer-by-layer precursor deposition/annealing procedure is essential for building structurally stable, relatively thick (up to 3 μm thick) WO3 films with good adherence to the substrate. A single standard application of the precursor produced typically a ~0.4 μm thick WO3 film. The films used in this work were formed by three-to-eight consecutive applications of the precursor solution, each followed by the high-temperature annealing in O2 for 30 min. See Methods for further details.
As shown by the cross-sectional scanning electron microscopic (SEM) image of a ~1.2 μm thick WO3 film in Fig. 1a, the employed sol-gel method produces a mesoporous film with some pores extending apparently down to the FTO substrate. The significant porosity is even better perceived on the SEM image of a thinner 0.4 μm thick WO3 film (Supplementary Fig. S1). We consider the formation of such a porous film being the result of burning the PEG, present in the precursor, occurring during the high-temperature annealing in oxygen. We also presume that the local heating involved in the latter process allows to establish crystalline interparticle contacts43 and contributes to the stability of the formed mesoporous film. The top-down surface SEM image in Fig. 1c shows that the film consists of a network of small individual, ~20–40 nm in size, NPs as well as of multiparticle agglomerates.

a and b, Cross-sectional SEM images of ~ 1.2 μm (a) and ~ 3 μm (b) thick WO3 films. c, top-down SEM image of the film in (a). d, X-ray diffraction pattern of WO3 film in (a). e, Raman spectrum of WO3 film in (a).
The annealing at a temperature of 550 °C or above it results in the formation of WO3 films with the well-defined monoclinic crystal structure. X-ray diffraction (XRD) pattern (Fig. 1d) exhibits three main sharp peaks (002), (020), (200) with three other less intense (004), (040) and (041) peaks - consistent with the preferential orientation of the WO3 crystallites parallel to the substrate plane (COD - Crystallography Open Database # 96-210-6383). Also the Raman spectrum of the formed film (Fig. 1e) shows the principal features characteristic of the monoclinic structure of WO3 with prominent bands at around 715 and 805 cm−1.
Photoelectrochemical characterization
Concomitant with the use of WO3 film as a photoanode is the choice of supporting electrolyte - the most appropriate to perform the PEC oxidation of the chosen organic compound. As initially reported44 and subsequently confirmed in a series of works45,46,47, the WO3 photoanodes have a pronounced tendency to favor oxidation of anions of the acidic electrolytes, the reactions that in some cases (H2SO4, HClO4) are accompanied by the formation of the peroxide species that, over prolonged water splitting experiments, causes progressive electrode deactivation. As demonstrated by Raman spectroscopic analyses of the H2SO4 electrolytes, submitted to several hours long photoelectrolysis, the peroxodisulfates (S2O82-) become in that case the main product formed at the WO3 photoanode44.
However, an important exception is the excellent photostability of the WO3 electrodes in electrolytes, including chlorides with the dominant chlorine evolution occurring at the photoanode44,48. Another exception is the behavior of WO3 photoanodes in methanesulfonic acid (CH3SO3H) electrolyte in which stable oxygen evolution photocurrents are observed48,49 that we assign to high reactivity with water of the initially formed, CH3SO3· radical species50. The photo-oxidation products of the anions of acidic supporting electrolytes, including the corresponding radical species, are expected to play a role of intermediates in the PEC reactions involving organic substrates especially when they are present at low concentrations28.
In order to compare the effect of different supporting electrolytes upon glucose photo-oxidation efficiency, we used a three-electrode cell configuration with a Pt cathode separated from the WO3 photoanode by a Nafion membrane and an Ag/AgCl reference electrode (see Methods). For those preliminary screening tests we employed relatively thin (~1.2 μm thick) WO3 film photoanodes. The initially used electrolytes consisted of moderately concentrated 0.1 M solutions of sodium chloride, perchlorate, sulfate and methanesulfonate acidified to pH 2 that remains in the range of thermodynamic WO3 stability extending up to pH 4–5. In Fig. 2 are represented linear sweep voltammograms (j-E) recorded for WO3 photoelectrodes in contact with supporting electrolytes alone (Fig. 2a) and, subsequently, after addition to the anodic compartment of 0.1 mol L−1 of glucose (Fig. 2d). In both cases the photoelectrodes were irradiated with simulated solar light (AM 1.5 G of 100 mW cm−2 intensity). The voltametric scans recorded in supporting electrolytes showed photocurrent onset potentials at ~0.15 V versus Ag/AgCl (corresponding to 0.47 V vs reversible hydrogen electrode (RHE)) and reached the plateaus of around 2.2 mA cm−2, at ~0.8 V vs Ag/AgCl. The largest plateau photocurrent was observed in a CH3SO3Na electrolyte and a slightly lower in NaHSO4, apparently indicative of the formation of peroxodisulfate species on the WO3 surface. It is to be noted that these photocurrents are limited by the restrained optical thickness of the WO3 photoanodes that we used in those screening test.

a Photocurrent densities vs imposed potential (j-E) plots recorded under simulated AM 1.5 G irradiation and b, The corresponding incident photon-to-current efficiency (IPCE) spectra measured at 1.23 V vs RHE for a ~1.2 μm thick film WO3 electrode in a series of 0.1 M supporting electrolytes acidified to pH 2. c The photo-oxidized species formed on the WO3 photoanode in the employed electrolytes. d Effect of the addition of 0.1 mol L−1 of glucose upon j-E plots recorded for the electrolytes listed in (a). e, The corresponding IPCE spectra.
Addition to the supporting electrolytes of 0.1 mol L−1 of glucose led to a strong increase of the recorded photocurrents (Fig. 2d) consistent with the occurrence at the WO3 electrode of the photocurrent doubling26,27. We wish to mention that the use, in this context, of the term photocurrent doubling implies, in fact, an important contribution of the mechanism where oxidation of glucose by the photogenerated positive hole, h+, is followed by the injection, by the formed reaction intermediate, of an electron into the CB of WO3. The largest increase of the photocurrents was observed in the solutions containing acidic sulfate and chloride electrolytes to achieve plateaus of ~4.2 mA cm−2. As shown in Fig. 2d, the photocurrents set in at about 0.1–0.15 V more negative potentials suggesting that the presence of glucose also significantly diminished the extent of charge carrier e- - h+ recombination. The corresponding dark currents measured in the supporting electrolytes and with added 0.1 mol L−1 of glucose are represented in Supplementary Fig. S2. It is to be noted, in this connection, that our choice of 0.1 M glucose concentration used in photo-reforming experiments, came from preliminary measurements (Supplementary Fig. S3) that showed that further increase of glucose concentrations in the anolyte did not bring any significant advantage in terms of the achieved photocurrents.
Performed in parallel, measurements of incident photon-to-current conversion efficiency (IPCE) spectra of the WO3 photoelectrodes in the four supporting electrolytes revealed the spectra extending up to 480 nm with maxima close to 80% occurring in the 360–380 nm wavelength range (Fig. 2b). As already mentioned, because of the indirect optical transition in WO348, the figures in the IPCE spectra are strongly dependent upon the SC film thickness, with the maximum that (vide infra) becomes shifted up to 400 nm for a ~3 μm thick WO3 film.
The extent of the photocurrent doubling resulting from the presence of glucose in the anolyte is directly illustrated by the IPCE plots (Fig. 2e) with the enhancement, with respect to the supporting electrolytes, close to factor 2. Like in the case of j-E plots (Fig. 2d), a larger increase of the IPCEs was observed in the solutions containing sulfates or chlorides, where peroxodisulfates (S2O82-)44,47 or hypochlorous acid (HClO)48 may be formed, respectively.
An important insight into the nature of the products formed on a WO3 electrode during photo-oxidation of C1 organic molecules in a H2SO4 supporting electrolyte was provided51 by potentiodynamic on-line differential electrochemical mass spectrometry (DEMS) measurements employing continuous flow system. That study showed that although e.g., 0.01 M formic acid is photo-oxidized preferentially on the WO3 electrode this does not totally suppress the O2 formation that still accounts for about 10% of current efficiency.
In comparison, larger (0.1 M) concentration of glucose employed in our experiments might suggest, at least at the initial stage of the photoreaction, less important contribution of radicals formed by oxidation of anions of the supporting electrolyte (such as HSO4· in the case of 0.1 M NaHSO4) to the observed photocurrents. Consequently, the dominant pathway in the 2e- oxidation of glucose to form gluconic acid might involve a direct transfer of a photogenerated positive hole to the adsorbed molecule of glucose (Fig. 3a) followed by injection by the reactive intermediate of an electron into the CB of WO3 to form gluconic acid (Fig. 3b).

Sketch a represents a positive hole transfer to the adsorbed molecule of glucose followed by injection by the reactive intermediate of an electron into the CB of WO3, shown in sketch b, to form gluconic acid.
However, assuming that further PEC reaction steps follow the desorption-re-adsorption mechanism51, the concentration of the intermediate (i.e., gluconic acid) leading to the formation of arabinose (corresponding to the transfer of 2e-) and of that of erythrose (transfer of additional 2e-) must be substantially lower. For this reason, it appears plausible that further stages of photoreaction (leading to arabinose and erythrose) follow an indirect pathway involving as intermediates the oxidation products of anions of the supporting electrolyte (cf. Fig. 2c).
Identification of the glucose photo-oxidation products
To check the photostability of the WO3 photoanodes and, at the same time, probe glucose photo-oxidation products we conducted prolonged PEC experiments using a cell depicted in Fig. 4a. As shown in Fig. 4b, the photoanodes sustained a 20 h long photo-oxidation of 0.1 M solutions of glucose (present in the anolytes) in the four tested supporting electrolytes (i.e., chlorides, acidic sulfates, perchlorates and methanesulfonates) with a relatively small decline of the photocurrent that we assign to accumulation of the formed products within the pores inside the WO3 film. We also wish to notice, in this connection, that in contrast with the PEC behavior of the WO3 electrode in acidic sulfates electrolyte alone, characterized by a progressive photoanode deactivation associated with the formation of the surface peroxo species44,47, this did not occur when an equivalent amount of glucose was present in the anolyte. This observation might suggest that a part of the generated HSO4· radical species, instead of forming peroxodisulfates, rapidly reacted with glucose.

a Schematic of the photoelectrochemical cell used in glucose photo-reforming experiments. b The glucose photo-oxidation currents measured along 20 h electrolysis conducted at 1.23 V vs RHE in four different supporting electrolytes, using ~1.2 μm thick film WO3 electrodes irradiated with simulated AM 1.5 G sunlight. The Teflon cell used for those experiments contained 45 mL of solution in the anodic compartment and the illuminated area of the WO3 photoanodes was ~0.7 cm2.
After completion of the 20 h long photoelectrolysis, products of glucose photoconversion present in the anolyte were detected and quantified by gas chromatography with mass spectrometry (GC-MS) analysis. The post photoelectrolysis compounds identified in the selected for analysis solutions of acidic sulfate and chloride electrolytes were: gluconic and glucuronic acids, arabinose and erythrose. Gluconic acid formed by 2e- oxidation of glucose was by far the dominant product with the Faradaic yields (FY) of 16.5% and 14.6% achieved in acidic sulfate and chloride electrolytes, respectively (Supplementary Fig. S4). The FY of three other products remained in the range of 5% with the exception of erythrose produced with 7.7% FY in the sulfate electrolyte. A sample chromatogram in Fig. 5, shows that the photo-oxidized glucose solutions also contained disaccharides that could not be convincingly identified. For comparison we also included a chromatogram of a standard mixture of the principal products detected in the analytes (Supplementary, Fig. S5). Calibration curve was prepared by analyzing a series of standard samples containing compounds that are listed in Supplementary Table S1.

The red trace represents measurement taken before and the blue trace the one performed after 20 h long photoelectrolysis. For the electrolysis conditions see Fig. 4.
An interesting result was also provided by parallel total organic carbon (TOC) analyses (Supplementary, Fig. S6) that showed quite similar amounts of non-purgeable organic carbon (NPOC) species in fresh and in 20 h electrolyzed solutions. This suggests that, under conditions employed in our PEC experiments, the glucose was practically not converted into CO2 or into volatile organic compounds.
In the search for optimal photoelectrolysis conditions allowing to enhance the yield of glucose reforming products, we employed next less acidic supporting electrolytes (NaCl, Na2SO4 and CH3SO3Na) of pH 4. According to reports in the literature, the rate of glucose photo-reforming increases following increasing pH of the photocatalyst (TiO2) suspension36, and we hypothesized that a similar effect might be expected in the electrolytes employed for the PEC glucose oxidation. It is important to mention, in this context, that earlier investigations52 showed that glucose is converted into gluconate ions through hypochlorite oxidation in slightly alkaline solutions with highest yields obtained at pH 11.
The PEC experiments using 0.5 M NaCl electrolyte of pH 4 with 0.1 mol L−1 of glucose (in the anolyte) allowed indeed the formation of substantially larger amounts of erythrose corresponding to FY of more than 27% and of gluconic acid with an about 20% FY. We recall, in this connection, that chlorine and hypochlorous acid are the dominant products generated on the WO3 photoanode during electrolysis of a 0.5 M NaCl solution48. As shown in Fig. 6, the distribution of the glucose photo-oxidation products determined after electrolysis performed in sulfate and methanesulfonate electrolytes of pH 4 was significantly different with, in particular, increased amounts of arabinose and of glucuronic acid, reflecting clearly the effect of anions of the supporting electrolytes. Comparison of the total Faradaic yields of the four analyzed products indicates the largest FY (of around 52%) determined in the 0.5 M NaCl (pH 4)/0.1 M glucose solution. Due to the small surface area of the WO3 photoanode (0.7 cm2) operating in relatively large volume (45 ml) of anolyte (cf. legend of Fig. 4) the extent of glucose conversion along 20 h long photoelectrolysis remained in the range of 15%.

The electrolysis was conducted at 1.23 V vs RHE using WO3 and WO3/TiO2 photoanodes irradiated with simulated AM 1.5 G sunlight of 100 mW cm−2 intensity. The results are represented as Faradaic yields.
To assess the impact of the presence of glucose in the water splitting supporting electrolyte upon the efficiency of hydrogen generation in the PEC cell (see Fig. 4a), we used thicker, ~3 μm thick film photoanodes able to absorb more efficiently the blue portion of the solar spectrum close to the WO3 band edge. The j-E voltammograms recorded for the WO3 photoanode under simulated AM 1.5 G (of 100 mW cm−2 intensity) sunlight in the 0.5 M NaCl pH 4 electrolyte alone (black line) and in the solution containing 0.1 M glucose (blue line) (Fig. 7a) confirm the occurrence of a large extent of the photocurrent doubling resulting from the presence of glucose in the anolyte. At 1 V vs Ag/AgCl (~1.43 V vs RHE) high plateau photocurrents close to 6.5 mA cm−2 were attained. The corresponding IPCE plots (Fig. 7b) provide a clear explanation for the increased photocurrents displayed by the ~3 μm thick WO3 photoanode. Although the IPCEs measured at near UV wavelengths (up to 390 nm) are similar to those observed for a much thinner (1.2 μm thick) film photoanode (Fig. 2b), they are almost twice as large at a 440 nm visible wavelength, due to the substantially larger optical thickness of the WO3 film.

The measurements were performed in a 0.5 M NaCl of pH 4 solution alone and after addition of 0.1 mol L−1 of glucose. a Photocurrent densities vs imposed potential (j-E) plots recorded under simulated AM 1.5 G irradiation. b The corresponding IPCE spectra.
Discussion
In this report, we demonstrated the ability of the semitransparent nanostructured WO3 photoanode to perform photo-reforming of glucose under simulated AM 1.5 G sunlight, with high photocurrent densities exceeding 6 mA cm−2 at 1.23 V vs RHE. Similar bias voltage may be provided by a number of single-junction PV cells enabling them to operate in a tandem device with the water splitting PEC cell. This opens the perspective to perform unassisted solar light-driven water splitting with high solar-to-hydrogen efficiency (STH) of more than 7%.
We note that the high achieved photocurrents are due in part to the preferential (in comparison with OER) photo-oxidation of glucose on WO3, accompanied, in addition, by the occurrence of the photocurrent doubling. In contrast with numerous other PEC and/or photocatalytic water splitting experiments in which biomass-derived compounds served only as sacrificial-kind agents to enhance hydrogen generation, our present work demonstrated photo-reforming glucose into value-added products. The obtained results also revealed the possibility of tuning the distribution of the glucose photo-oxidation products by the selection of anions of the supporting electrolyte and of its pH. In fact, a comparison of the Faradaic yields corresponding to photo-oxidation of glucose in NaCl electrolytes of pH 2 and pH 4 (cf. Supplementary Fig. S4 and Fig. 6 in the full text) shows, in the second case, a substantial increase of FY of both gluconic acid (~20%) and of erythrose (~27%). Considering the FY of the analyzed glucose photo-reforming products (gluconic and glucuronic acids, arabinose and erythrose), the NaCl electrolyte of pH 4 appears as the most effective with the total FY of around 52%. It is important to recall, in this connection, that chlorine and hypochlorous acid are the principal photo-oxidation products at the WO3 photoanode in the 0.5 M NaCl electrolyte48. We also note the close similarity between the distribution of the glucose photo-reforming products identified in CH3SO3Na and Na2SO4 electrolytes of pH 4. Since in the former case, it is the highly reactive CH3SO3· radical (unable to form a dimer)50 that, apparently, mediates photo-oxidation of glucose we hypothesize that in the sulfate supporting electrolyte a similar role is played by the SO4-· radical anion.
Following earlier reports in the literature36,52 indicating that the rate of glucose oxidation increases with moving the solution pH in alkaline direction, we attempted to perform glucose photo-reforming using less acidic electrolytes. Since the range of thermodynamic stability of crystalline monoclinic WO3 in aqueous solutions extends from strong acids to pH 4.653, in order to minimize the possibility of photo-corrosion we decided to cover the outer surface of the WO3 film with a thin ~0.4 μm thick deposited layer of TiO2 nanoparticles (P25 from Ovonics). Unlike WO3, TiO2 is stable both in acidic as well as in alkaline solutions. Thus formed hybrid WO3/TiO2 electrode was tested in a 0.5 M NaCl (pH 7)/0.1 M glucose solution, irradiated from the rear side (i.e., through the FTO substrate) with simulated 1 sun AM 1.5 G light. Despite slightly diminished light transmission by FTO, the IPCEs attain a maximum of ~160% at 360 nm (Supplementary Fig. S7a) with still observable photoresponse extending up to 480 nm. Interestingly, comparison with the IPCEs recorded in the same configuration for an identical WO3 film electrode without a TiO2 overlayer, shows at 410–460 nm wavelengths a clearly increased values for the hybrid WO3/TiO2 electrode. Since anatase TiO2 (the dominant form present in P25) does not absorb (and largely transmits) wavelengths above 400 nm, we hypothesize that the increased IPCEs may be due to the dispersion/reflection at the WO3 – TiO2 interface of a certain fraction of visible light unabsorbed by WO3. Consequently, the j-E plots measured for the WO3/TiO2 electrode under the rear-side AM 1.5 G (of 100 mW cm−2 intensity) irradiation (Supplementary Fig. S7b) attain high saturation photocurrents of ~ 3.8 mA cm−2 comparable with those observed in the solutions of lower pH under the solution-side illumination (cf. Fig. 2a). The aforementioned conditions were subsequently employed for prolonged 20 h long photoelectrolysis of a 0.5 M NaCl (pH 7)/0.1 M glucose solution, that revealed a large increase of the amount of formed gluconic acid to reach ~34% FY, about 65% larger than the FY determined after experiment conducted in the analogous solution of pH 4 (Fig. 6).
An additional photoelectrolysis employing a 0.5 M NaCl/0.1 M glucose anolyte of pH 7, that involved product analysis also after 5 h and 10 h along the 20 h long PEC experiment, showed regular increase of the amounts of formed products (gluconic acid, glucuronic acid, arabinose and erythrose) over 5 h, 10 h up to 20 h periods (Supplementary Fig. S8).
In summary, we have shown that a PEC device consisting of a nanostructured WO3 photoanode and of a metallic cathode operates in a sustainable way reforming glucose into valuable products and producing hydrogen in a separate cell compartment. By using a WO3 photoanode having larger optical thickness to match the penetration depth of blue light photons, we demonstrated under AM 1.5 G sunlight at 1.23 VRHE photocurrents exceeding 6 mA cm−2. The latter value translates into expected solar-to-hydrogen efficiency above 7% to be attained in the tandem cell. For comparison, we collected together (Supplementary Table S2) the PEC performances of different WO3 photoanodes reported in the papers cited in the present article.
The prolonged photoelectrolysis runs confirmed the absence of any deactivation of the WO3 electrode by the formed products. Our investigation of the photo-reforming process demonstrated the versatility of the WO3 photoanode able to direct the distribution of the glucose oxidation products through the choice of appropriate electrolyte. Further improvements in the selectivity of the photo-reforming process may involve the use of mixtures of supporting electrolytes, i.e., combinations of different anions acting as intermediates in the photo-oxidation reactions on the WO3 electrode.
Methods
Reagents
Sodium tungstate dihydrate, poly(ethylene)glycol (PEG) 300 (used to prepare WO3 films), TiO2 P25 (containing ca. 75% of anatase and 25% of rutile (Ovonics), poly(vinylidene-fluoride), dimethylformamide, methanesulfonic acid (99.5%), perchloric and sulfuric acids, sodium chloride and glucose were purchased from Sigma-Aldrich; hydrochloric acid was purchased from Chempur. The solutions used in electrochemical measurements were prepared with Mili-Q water. The reagents employed to prepare samples for GC-MS and TOC analyses are listed in the Supplementary Methods.
Preparation and structural characterization of the WO3 photoelectrodes
The mesoporous WO3 films used in this work were formed on conductive glass substrates through doctor blade layer-by-layer deposition of a colloidal solution of tungstic acid, containing an organic structure-directing agent – low molecular weight poly(ethylene glycol) PEG 300. The formation of a complex between tungstic acid and the hydrophilic PEG delays the formation of fully crystallized monoclinic tungsten trioxide until heating to ~ 500 °C. Preparation of the precursor solution followed the earlier described original procedure43. Conductive glass plates (Sigma-Aldrich, resistance 7 Ω/square) bearing a 0.9 μm thick overlayer of F-doped SnO2 (FTO) were used as substrates. The WO3 films were formed by three up to eight consecutive applications of the precursor solution each followed by annealing in flowing oxygen at 550 °C for 30 min. The final thickness of such films (determined from the cross-sectional scanning electron microscopic, SEM, images) varied between ~1.2 to about 3 μm.
To obtain hybrid WO3/TiO2 electrodes, we deposited on ~1.2 μm thick WO3@FTO films ca 0.4 μm thick TiO2 overlayers25. The TiO2 film was formed by one application from the prepared suspension (1 g TiO2 powder in 20 ml DMF with 0.4 g poly(vinylidenefluoride)) onto WO3 layer. Then it was dried in air for 30 min., later for solvent evaporation at 100 °C for 40 min and followed by annealing at 500 °C for 1 h in oxygen flow.
SEM imaging was performed using a Carl Zeiss Auriga Cross-Beam workstation. The microscope was equipped with a Gemini electron column with Energy Selective Backscattered (ESB) detector.
X-ray powder diffraction (XRD) measurements were performed on the as-grown samples using a Siemens D500 diffractometer equipped with a high-resolution semiconductor Si:Li detector using Cu Kα radiation (U = 40 kV, I = 30 mA). The powder diffraction patterns were measured in a θ/2θ scanning mode with a step of 0.02° and counting time of 10 s by step.
Raman spectra were acquired with the Alpha 300 M + confocal microscope (Witec) equipped with a motorized stage. For analyses we used a 532 nm laser line delivered to the microscope through a single-mode optical fiber. The laser power focused at the sample did not exceed 7 mW. The backscattered Raman signal was collected through a 50× long working distance objective (NA = 0.42), and passed through a photonic optical beam to a lens-based spectrometer (Witec UHTS 300, f/4 aperture, focal length 300 mm) coupled with a back-illuminated Andor iDUS 401 detector thermoelectrically cooled to −60 °C. The spectra were collected in the range of Raman shifts from 100 to 1000 cm−1 with the use of an 600 mm grating. For each sample Raman mapping was conducted on a 50 × 50 μm2 area with a 1 μm step (totaling to 2500 spectra) and a 0.3 s acquisition time. The spectra were postprocessed (background subtraction and cosmic-ray removal) with the Project FIVE software (Witec). For all samples, the spectra in the mapped area were homogeneous. Consequently, an average over 2500 spectra was made for each sample.
Photoelectrochemical measurements
The photocurrent-potential (j-E) plots and incident photon-to-current conversion efficiencies (IPCEs) were measured in a Teflon cell equipped with a quartz window, by illuminating the WO3 electrode either from the side of the solution/film interface or, in some cases, through the FTO substrate. The simulated AM 1.5 G (of 100 mW cm−2 intensity) illumination was obtained from an Oriel 150 W solar simulator. In those measurements, the exposed WO3 electrode surface area was 0.28 cm2. The measurements were performed in a three-electrode configuration, with a WO3 photoanode a platinum grid counter electrode and a silver/silver chloride (Ag/AgCl) in sat. KCl (E = 0.197 vs SHE) as a reference electrode. The potentials are also quoted versus reversible hydrogen electrode (RHE) in the same solution. To obtain j-E plots, the potential of the WO3 electrode was swept from the open circuit potential of ~0.40 VRHE (in the solutions containing glucose) to 1.6 VRHE at a rate of 10 mV sec−1 using a CH Instrument - CHI 660E electrochemical work-station. The IPCE spectra of the WO3 photoanodes were determined using light from a 150 W xenon lamp passing through a photoelectric spectrometer (Instytut Fotonowy) featuring monochromator with a bandwidth of 10 nm. The absolute light intensity was measured with a model OL 730-5 C UV-enhanced silicon detector Gooch&Housego.
Preliminary long-term photoelectrolyses of the anolyte solutions containing glucose, intended to check the photostability of the photoanode, were carried out in a large volume quartz cell containing 400 mL of solution with anodic and cathodic compartments separated by a Nafion membrane. In those tests, a total surface area of the WO3 films deposited on FTO (~1.6 cm2) was exposed to the electrolytes. Further 20 h-long experiments intended for identifying glucose photo-reforming products were performed in a smaller Teflon cell with ca 45 mL of anolyte and ~0.7 cm2 illuminated WO3 surface area.
Identification of the glucose oxidation products
To identify the products of glucose oxidation GC-MS-QP2010 Ultra gas chromatograph (Shimadzu) interfaced with a single quadrupole mass spectrometer was used. The instrument was equipped with AOC-5000 autosampler (Shimadzu) in the liquid-injection mode featuring a 10 µl syringe. The syringe was washed three times before and after each injection with ethyl acetate and then with hexane. The analytes were separated using a capillary column ZB-5MSPlus (Phenomenex): 30 m x 0.25 mm, 0.25 µm stationary phase film. The column head pressure was 26.7 kPa, the total flow of the carried gas was 9.5 ml/min, column flow 0.68 ml/min (30 cm/sec) and the purge flow was 2 ml/min. The linear velocity flow control mode was used and the split ratio was 10. The temperatures of injector, ion source and the mass spectrometer transfer line were 300 °C. The following temperature program was used: initially 50 °C held for 2 min, then linear increase at the rate of 10 °C/min to 225 °C kept for 10 min, then linear increase at the rate of 15 °C/min to 280 °C, held for 10 min.; the total analysis time was 43 min. Mass spectrometer was equipped with the electron impact (EI) ion source and operated in the scan mode in the mass range 45-500 m/z (scan time 150 ms), the solvent cut was 6 min. For the quantitative analysis of the formed products (more concentrated samples), the glucose peak was removed from the chromatogram by turning off the mass spectrometer between 25.9 and 26.3 min to avoid oversaturating the detector and damaging the ion source. GCMSsolution 2.53 (Shimadzu) program was used for data acquisition and processing.
Total organic carbon (TOC) analysis was quantified with TOC-5050A analyzer (Shimadzu) equipped with the ASI-5000A autosampler (Shimadzu) in order to check the possible formation of volatile glucose oxidation products.
Sample preparation
a) for GC/MS analyses
Aliquots of the reaction solution were evaporated to dryness at 60 °C under a gentle stream of N2 and derivatized prior to the GC/MS analysis.
When the reaction solution contained concentrated mineral acids (H2SO4 or HCl), the samples were neutralized before evaporation to avoid the mineralization of glucose and other saccharides that was observed during preliminary experiments. For the quantitative analysis of glucose, the reaction solution was diluted twenty times with DI water to the total volume of 1 ml; 5 µl of this solution was evaporated to dryness at 60 °C under a gentle stream of nitrogen. For the quantification of the products formed, 2 µl of the reaction solution were dried without any prior dilution.
The dried samples were derivatized according to the described procedure that was optimized for saccharides and uronic acids54.
Briefly, this tree-step derivatization protocol involved mercaptalation with ethanethiol and two silylation steps with BSTFA/TMCS (99:1, v/v).
GC measurement uncertainties for products concentrations were calculated from several injections and are presented in Supplementary Table S3.
b) for Total organic carbon
Firstly, the instrument was calibrated with the standard solutions of glucose in DI water with concentrations between 4 and 36 mgTOC × L−1; the squared linear coefficient of determination for the glucose calibration curve (R2) = 0.9994 was obtained.
The aliquots of the reaction solution were diluted approx. five hundred times with DI water before the analysis to adjust the total organic carbon (TOC) concentration between 20 and 35 (mg × L−1). Afterward, each sample was filtered with a single-use PTFE syringe filter (pore size 0.22 µm) and 4 ml of the filtered solution was placed in the instrument autosampler. Subsequently, 50 µl of 2 M HCl was added via the autosampler and each sample was sparged with oxygen for 2 min before injection. The injection volume was 21 µl and each sample was injected into the instrument three times.
Data availability
The authors declare that all data supporting the findings of this study are available within the article and its supplementary information files, and from the corresponding author on reasonable request.
References
Hisatomi, T., Kubota, J. & Domen, K. Recent advances in semiconductors for photocatalytic and photoelectrochemical water splitting. Chem. Soc. Rev. 43, 7520–7535 (2014).
Sivula, K. & van de Krol, R. Semiconducting materials for photoelectrochemical energy conversion. Nat. Rev. Mater. 1, 15010 (2016).
Wang, Q. & Domen, K. Particulate Photocatalysts for light-driven water splitting: mechanisms, challenges, and design strategies. Chem. Rev. 120, 919–985 (2020).
Rajeshwar, K. Hydrogen generation at irradiated oxide semiconductor–solution interfaces. J. Appl. Electrochem. 3, 765–787 (2007).
Woodhouse, M. & Parkinson, B. A. Combinatorial approaches for the identification and optimization of oxide semiconductors for efficient solar photoelectrolysis. Chem. Soc. Rev. 38, 197–210 (2009).
Kronawitter, C. X. et al. On the interfacial electronic structure origin of efficiency enhancement in hematite photoanodes. J. Phys. Chem. C. 116, 22780–22785 (2012).
Carvalho, W. M. Jr. & Souza, F. L. Thermal enhancement of water affinity on the surface of undoped hematite photoelectrodes. Sol. Energy Mater. Sol. Cells 144, 395–404 (2016).
Hu, Y. et al. Function and electronic structure of the SnO2 buffer layer between the α-Fe2O3 water oxidation photoelectrode and the transparent conducting oxide current collector. J. Phys. Chem. C. 125, 9158–9168 (2021).
Lee, D. K. & Choi, K.-S. Enhancing long-term photostability of BiVO4 photoanodes for solar water splitting by tuning electrolyte composition. Nat. Energy 3, 53–60 (2018).
Bignozzi, C. A. et al. Nanostructured photoelectrodes based on WO3: applications to photooxidation of aqueous electrolytes. Chem. Soc. Rev. 42, 2228–2246 (2013).
Jelinska, A. et al. Enhanced photocatalytic water splitting on very thin WO3 films activated by high-temperature annealing. ACS Catal. 8, 10573–10580 (2018).
Augustynski, J., Calzaferri, G., Courvoisier, J. & Gratzel, M. Proceedings of the 11th World Hydrogen Energy Conference, ed. T. N. Veziroglu, C.-J. Winter, J. P. Baselt and G. Kreysa, DECHEMA, Frankfurt, Germany, 1996, p. 2378.
Brillet, J. et al. Highly efficient water splitting by a dual-absorber tandem cell. Nat. Photonics 6, 824–828 (2012).
Kawase, Y., Higashi, T., Domen, K. & Takanabe, K. Recent developments in visible-light-absorbing semitransparent photoanodes for tandem cells driving solar water splitting. Adv. Energy Sustainability Res 2, 2100023 (2021).
Zhong, D. K., Choi, S. & Gamelin, D. R. Near-complete suppression of surface recombination in solar photoelectrolysis by “Co-Pi” catalyst-modified W:BiVO4. J. Am. Chem. Soc. 133, 18370–18377 (2011).
Pilli, S. K. et al. Cobalt-phosphate (Co-Pi) catalyst modified Mo-doped BiVO4 photoelectrodes for solar water oxidation. Energy Environ. Sci. 4, 5028–5034 (2011).
Jeon, T. H., Choi, W. & Park, H. Cobalt–phosphate complexes catalyze the photoelectrochemical water oxidation of BiVO4 electrodes. Phys. Chem. Chem. Phys. 13, 21392–21401 (2011).
Kim, T. W. & Choi, K.-S. Nanoporous BiVO4 photoanodes with dual-layer oxygen evolution catalysts for solar water splitting. Science 343, 990–994 (2014).
Antoniadou, M., Bouras, P., Strataki, N. & Lianos, P. Hydrogen and electricity generation by photoelectrochemical decomposition of ethanol over nanocrystalline titania. Int. J. Hydrog. Energy 33, 5045–5051 (2008).
Lianos, P. & Antoniadou, M. Photoelectrochemical oxidation of organic substances over nanocrystalline titania: optimization of the photoelectrochemical cell. Catal. Today 144, 166–171 (2009).
Zhou, B. et al. Photoeletrochemical generation of hydrogen over carbon-doped TiO2 photoanode. Appl. Catal., B 92, 41–49 (2009).
Panagiotopoulou, P., Antoniadou, M., Kondarides, D. I. & Lianos, P. Aldol condensation products during photocatalytic oxidation of ethanol in a photoelectrochemical cell. Appl. Catal., B 100, 124–132 (2010).
Kondarides, D. I., Daskalaki, V. M., Patsoura, A. & Verykios, X. E. Hydrogen production by photo-induced reforming of biomass components and derivatives at ambient conditions. Catal. Lett. 122, 26–32 (2008).
Shimura, K. & Yoshida, H. Heterogeneous photocatalytic hydrogen production from water and biomass derivatives. Energy Environ. Sci. 4, 2467–2481 (2011).
Wahl, A. & Augustynski, J. Charge carrier transport in nanostructured anatase TiO2 films assisted by the self-doping of nanoparticles. J. Phys. Chem. 102, 7820–7828 (1998).
Morrison, S. R. & Freund, T. Chemical role of holes and electrons in ZnO photocatalysis. J. Chem. Phys. 47, 1543–1551 (1967).
Santato, C., Ulmann, M. & Augustynski, J. Photoelectrochemical properties of nanostructured tungsten trioxide films. J. Phys. Chem. B 105, 936–940 (2001).
Solarska, R., Santato, C., Jorand-Sartoretti, C., Ulmann, M. & Augustynski, J. Photoelectrolytic oxidation of organic species at mesoporous tungsten trioxide film electrodes under visible light illumination. J. Appl. Electrochem. 35, 715–721 (2005).
Zheng, H. et al. Nanostructured tungsten oxide – properties, synthesis, and applications. Adv. Funct. Mater. 21, 2175–2196 (2011).
Wang, Y., Tian, W., Chen, C., Xu, W. & Li, L. Tungsten trioxide nanostructures for photoelectrochemical water splitting: material engineering and charge carrier dynamic manipulation. Adv. Funct. Mater. 29, 1809036 (2019).
Butler, M. A. Photoelectrolysis and physical properties of the semiconducting electrode WO2. J. Appl. Phys. 48, 1914–1920 (1977).
Santato, C., Ulmann, M. & Augustynski, J. Enhanced visible light conversion efficiency using nanocrystalline WO3 films. Adv. Mater. 13, 511–514 (2001).
Berak, J. M. & Sienko, M. J. Effect of oxygen-deficiency on electrical transport properties of tungsten trioxide crystals. J. Solid State Chem. 2, 109–133 (1970).
Patel, K. J., Panchal, C. J., Kheraj, V. A. & Desai, M. S. Growth, structural, electrical and optical properties of the thermally evaporated tungsten trioxide (WO3) thin films. Mater. Chem. Phys. 114, 475–478 (2009).
Esposito, D. V. et al. Photoelectrochemical reforming of glucose for hydrogen production using a WO3-based tandem cell device. Energy Environ. Sci. 5, 9091–9099 (2012).
Granone, L. I., Sieland, F., Zheng, N., Dillert, R. & Bahnemann, D. W. Photocatalytic conversion of biomass into valuable products: a meaningful approach? Green. Chem. 20, 1169–1192 (2018).
Bellardita, M., Garcia-Lopez, E. I., Marci, G., Nasillo, G. & Palmisano, L. Photocatalytic solar light H2 production by aqueous glucose reforming. Eur. J. Inorg. Chem. 2018, 4522–4532 (2018).
Zhang, G. et al. Simultaneous cellulose conversion and hydrogen production assisted by cellulose decomposition under UV-light photocatalysis. Chem. Commun. 52, 1673–1676 (2016).
Caravaca, A., Jones, W., Hardacre, C. & Bowker, M. H2 production by the photocatalytic reforming of cellulose and raw biomass using Ni, Pd, Pt and Au on titania. Proc. R. Soc. A. 472, 20160054 (2016).
Wakerley, D. W. et al. Solar-driven reforming of lignocellulose to H2 with a CdS/CdOX photocatalyst. Nat. Energy 2, 17021 (2017).
Rao, V. N. et al. Light-driven transformation of biomass into chemicals using photocatalysts – vistas and challenges. J. Environ. Manag. 284, 111983 (2021).
Cha, H. G. & Choi, K.-S. Combined biomass valorization and hydrogen production in a photoelectrochemical cell. Nat. Chem. 7, 328–333 (2015).
Santato, C., Odziemkowski, M., Ulmannn, M. & Augustynski, J. Crystallographically oriented mesoporous WO3 films: synthesis, characterization, and applications. J. Am. Chem. Soc. 123, 10639–10649 (2001).
Augustynski, J., Solarska, R., Hagemann, H. & Santato, C. Nanostructured thin-film tungsten trioxide photoanodes for solar water and sea-water splitting. Proc. Soc. Photo‐Opt. Instrum. Eng. 6340, U 140 (2006).
Seabold, J. A. & Choi, K.-S. Effect of a cobalt-based oxygen evolution catalyst on the stability and the selectivity of photo-oxidation reactions of a WO3 photoanode. Chem. Mater. 21, 1105–1112 (2011).
Mi, Q., Zhanaidarova, A., Brunschwig, B. S., Gray, H. B. & Lewis, N. S. A quantitative assessment of the competition between water and anion oxidation at WO3 photoanodes in acidic aqueous electrolytes. Energy Environ. Sci. 5, 5694–5700 (2012).
Hill, J. C. & Choi, K.-S. Effect of electrolytes on the selectivity and stability of n-type WO3 photoelectrodes for use in solar water oxidation. J. Phys. Chem. C. 116, 7612–7620 (2012).
Jadwiszczak, M., Jakubow-Piotrowska, K., Kedzierzawski, P., Bienkowski, K. & Augustynski, J. Highly efficient sunlight-driven seawater splitting in a photo-electrochemical cell with chlorine evolved at nanostructured WO3 photo-anode and hydrogen stored as hydride within metallic cathode. Adv. Energy Mater. 10, 1903213 (2019).
Solarska, R., Jurczakowski, R. & Augustynski, J. A highly stable, efficient visible-light driven water photoelectrolysis system using a nanocrystalline WO3 photoanode and a methane sulfonic acid electrolyte. Nanoscale 4, 1553–1556 (2012).
Myall, C. J. & Pletcher, D. Electrochemical preparation of bismethylsulphonyl peroxide and its reactions with aromatic hydrocarbons. J. Chem. Soc., Perkin Trans. 1. 10, 953–955 (1975).
Reichert, R., Zambrzycki, C., Jusys, Z. & Behm, R. J. Photo‐electrochemical oxidation of organic C1 molecules over WO3 films in aqueous electrolyte: competition between water oxidation and C1 oxidation. ChemSusChem 8, 3677–3687 (2015).
Whistler, R. L. & Schweiger, R. Preparation of D-Arabinose from D-Glucose with Hypochlorite. J. Am. Chem. Soc. 81, 5190–5192 (1959).
Pourbaix, M. Atlas d’équilibres électrochimiques. (Gauthier-Villars Ed., Paris, 1963).
Colombini, M. P., Andreotti, A., Bonaduce, I., Modugno, F. & Ribechini, E. Analytical Strategies for Characterizing Organic Paint Media Using Gas Chromatography/Mass Spectrometry. Acc. Chem. Res. 43, 715–727 (2010).
Acknowledgements
K.J.-P. and J.A. acknowledge support from the Polish National Science Center (OPUS grant No. UMO- 2019/33/B/ST4/02718).
Author information
Authors and Affiliations
Contributions
J.A. with K.J.-P. conceived the project, K.J.-P. conducted the entire photoelectrochemical experimental research. B.W. performed the GC-MS and TOC analysis. J.A. wrote the paper with the contributions from all co-authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Chemistry thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jakubow-Piotrowska, K., Witkowski, B. & Augustynski, J. Photoelectrocatalytic hydrogen generation coupled with reforming of glucose into valuable chemicals using a nanostructured WO3 photoanode. Commun Chem 5, 125 (2022). https://doi.org/10.1038/s42004-022-00745-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42004-022-00745-w
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
