
Abstract
Atomic scale studies of the anchoring of catalytically active complexes to surfaces may provide valuable insights for the design of new catalytically active hybrid systems. In this work, the self-assembly of 1D, 2D and 3D structures of the complex fac-Re(bpy)(CO)3Cl (bpy = 2,2′-bipyridine), a CO2 reduction catalyst, on the Ag(001) surface are studied by a combination of low-temperature scanning tunneling microscopy and density functional theory calculations. Infrared and sum frequency generation spectroscopy confirm that the complex remains chemically intact under sublimation. Deposition of the complexes onto the silver surface at 300 K leads to strong local variations in the resulting surface coverage on the nanometer scale, indicating that in the initial phase of deposition a large fraction of the molecules is desorbing from the surface. Low coverage regions show a decoration of step edges aligned along the crystal’s symmetry axes <110>. These crystallographic directions are found to be of major importance to the binding of the complexes to the surface. Moreover, the interaction between the molecules and the substrate promotes the restructuring of surface steps along these directions. Well-aligned and decorated steps are found to act as nucleation point for monolayer growth (2D) before 3D growth starts.
Similar content being viewed by others


Introduction
In order to reach CO2 neutrality, catalytically active molecules that convert CO2 to valuable products have been in the focus of research for decades1. Besides optimizing the catalytic activity2, one of the goals is to develop and to design new catalytically active hybrid systems, e.g. molecules adsorbed on a surface to combine the advantages of molecular systems, e.g. in selectivity, with the advantages of heterogeneous systems3,4,5,6. Anchoring the molecules to a surface can result in a well-defined geometry/orientation of the molecules to a liquid or a gas phase eventually providing a defined catalytically active interface. A large-scale ordered structure can be realized by molecular self-assembly7, which also provides access to a well-defined playground for tailoring the molecular reactivity within a specific local environment8.
Self-organization of molecular clusters, also fundamental in the fields of molecular electronics and sensing technology, has attracted a lot of attention9,10,11. In this context, the role of surface defects can be very contradictory. For example, on the one hand, surface steps acting as grain boundaries12 can prevent long-range order across different substrate terraces. On the other hand, steps have shown potential to be used as selective adsorption sites13 as well as to induce rotational alignment of molecules14,15. And, applying multi-step preparation techniques, e.g. thermally induced polymerization causing an alignment of the step edges, the prepared step edges can be used to steer molecular layer growth16,17. To find suitable combinations of surfaces and molecules, one has (a) to establish appropriate preparation methods, (b) to prove chemical stability of the molecule on the surface as well as (c) to understand the interaction of the molecules with the surface and its defects in order to finally control the growth of 1D (molecular wires), 2D (monolayers) and 3D molecular structures15,18.
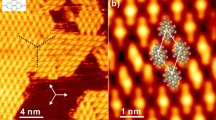
In this study, we have investigated the growth of fac-Re(bpy)(CO)3Cl (bpy = 2,2′-bipyridine), notably a three-dimensional complex, on the noble metal surface Ag(001) using scanning tunneling microscopy (STM). The complex fac-Re(bpy)(CO)3Cl (bpy = 2,2′-bipyridine), a 3D depiction is shown in Fig. 1a, is a well-known photo- and electrochemical CO2-reduction catalyst19. Several groups have already shown interest in tuning the molecule’s structure20 and in mimicking its properties in larger complexes to be anchored on certain surfaces4,21,22,23. In addition, studies of the electro-reduction products of a related complex on graphite24 and the covalent binding of a different related complex to TiO225 were reported.

a The structure of the complex as a ball-and-stick model in top and side view44,45. The colors of the balls represent atoms of different elements: black = C, white = H, red = O, blue = N, green = Cl, turquoise = Re. b High resolution topography showing self-assembled molecular structures on a stepped Ag(001) surface, (Iset = 50 pA, Ubias = 0.4 V and T = 80 K). The coverage of the individual terraces is inhomogeneous. Molecular chains are found at monoatomic step edges oriented along the crystal direction [110] of the silver substrate (indicated by the white arrow). c Top: Ex-situ IR spectra of fac-Re(bpy)(CO)3Cl powder as KBr pellets directly after the synthesis (light blue), after sublimation of the complex at T = 200 °C and with p < 10–3 mbar (orange), and after sublimation onto silver wool (red). The shown wavenumber regime with the indicated CO vibrational modes is used to monitor the chemical stability of the complex46. Bottom: IRpump pulse induced change in SFG response of the molecular monolayer covered crystal surface at 300 K. One can clearly identify CO stretching bands at 1920 and 2025 cm−1. The inset shows the measurement scheme.
We report strong evidence that the growth at room temperature predominantly starts at well-oriented steps. Furthermore, molecular-induced surface restructuring promotes the growth of self-assembled molecular structures. Density functional theory (DFT) provides the basis for a comprehensive understanding of growth.
Results
Stability of the complex during the sample preparation procedure
Molecules containing CO ligands are known to easily lose CO ligands during heating procedures or upon irradiation26. Moreover, ligand dissociation upon adsorption of the three-dimensional complex on the flat surface cannot be excluded a priori. Thus, we tested the thermal stability of the complex by sublimation at temperatures of 200 °C using ex-situ infrared (IR) spectroscopy. In Fig. 1c the IR spectra are shown directly after synthesis (light blue), and after sublimation (orange). The characteristic CO vibrational bands stay invariant under sublimation indicating that the complex is unaffected by the heating procedure and stays chemically intact. In a second step, we tested the stability of the complex once it has been deposited on silver wool (details of the preparation can be found in Supplementary Note 1) serving as a multi-faceted model surface.
The IR spectrum of the washed-out and dried material is shown in Fig. 1c in red. Due to the small amount of complexes sublimed, the IR absorbance is rather low. For comparison, a spectrum of a blank sample is given in blue. The IR data show the characteristic high-energy CO-vibrations at 2020 cm−1. If the chloride ion dissociates, we would expect the formation of either [Re(bpy)(CO)3(solvent)]+ or [Re(bpy)(CO)3]2 after dissolution, which exhibit characteristic IR bands at ~2050 and ~1950 cm−1, respectively27. Neither of these bands is present in the IR spectra, indicating that the molecules stay intact upon deposition on randomly oriented silver.
The lower panel of Fig. 1c shows the normalized IRpump induced sum-frequency generation (SFG) spectrum of a monolayer fac-Re(bpy)(CO)3Cl on a Ag(100) surface. The spectrum shows two features at 1920 ± 5 and 2025 ± 3 cm−1 which agree well with the IR spectrum of the carbonyl stretching bands of the solid material (upper panel in Fig. 1c). The conventional SFG spectrum of the A’(1) band, as described in Supplementary Note 2, gives a center frequency of 2033 ± 6 cm−1 consistent with Fig. 1c, too. These data support that the complex stays intact within the self-assembled molecular monolayers on the oriented silver surface.
Figure 1b shows a high-resolution topography mapping several silver terraces covered by a sub-monolayer of fac-Re(bpy)(CO)3Cl. While the complexes are found to form well-ordered structures, the coverage of the individual terraces is surprisingly inhomogeneous. In contrast to all the other terraces visible, the center terrace is only partially covered with complexes, although the incoming molecular flux is homogeneous on the given length scales. Topographies acquired on surfaces with small molecular coverage (Fig. 1b) show that even large molecular areas can coexist with Ag terraces, of which only the steps are decorated by molecular structures. However, only very rarely do we observe single isolated molecules and never molecular clusters on the free Ag surface.
While IR and SFG already strongly support the fact that the surface is covered by chemically intact molecules, STM can be used to search for local variations. Careful investigating of the molecular layers with small bias voltages and small tunnel currents revealed that none of the acquired data show any indication of different species being present on the surface. In other words, we do not have evidence for any chemical modification, e.g. ligand dissociation, of the complex on the surface after deposition.
Growth of molecular clusters on Ag(001)
One central finding of this study is that the occupied step edges show a clear preferential orientation, namely the crystal direction [110] of the silver substrate, as can be seen in Fig. 1b. This stands in direct contrast to the pristine silver surface that does not show any preferential alignment of the steps (Supplementary Fig. 2).
Figure 2a shows an example, in which small clusters at step edges are separated by randomly oriented unoccupied step edge regions. Crystallographic analysis of the occupied sections reveals a preferred step orientation, the direction \([1\bar 10]\), equivalent to [110]. Also, in the case of fully decorated step edges, the local preferential orientation can be seen as is illustrated by white lines in Fig. 2b. Many of the aligned segments have a length of a few nanometers.

a Topography of several steps edges that are barely covered by molecules. The occupied segments are aligned along the [110] direction of the substrate. b Topography shows several silver steps that are fully decorated by molecules. The step edges exhibit a predominant orientation along the [110] or the geometrically equivalent \([1\bar 10]\) crystal direction of the silver substrate. The step edge regions being aligned along these directions are indicated by white lines for one step edge. c Topography shows steps perfectly aligned along the substrate direction [110] that are fully decorated by molecules and show a periodic pattern. Tunneling parameters of all topographies were Iset = 50 pA, Ubias = 2 V and T = 80 K.
Figure 2c shows that these ordered segments may extend to several tens of nanometers in this way forming long-range ordered 1D molecular wires. For a surface that, after deposition, was stored at room temperature for several days we even find ordered 1D wires as long as 100 nm (cf. Supplementary Fig. 3).
In the large-scale topography in Fig. 3a the transition from 1D growth at the steps to 2D growth on the terraces is shown. All the step edges are decorated by molecules. While on the center terrace full monolayer coverage is found, two-thirds of the image shows unoccupied silver. Small 2D monolayer areas are found at the positions indicated by the light and dark blue arrows. All 2D areas adjoin step edges that are well-aligned along the crystal direction [110] for several nanometers. In addition, a monolayer grows exclusively on the upper part of the steps, i.e. no growth is observed on the bottom of a step. This is visible, e.g., for the monolayer in the lower left-hand corner of Fig. 3a, dark blue arrow, that solemnly attaches to the lower flanking step of the silver terrace.

a Topography (T = 80 K) showing several step edges being fully covered by molecules. Molecular monolayer growth is found for step edges being aligned along with the directions [110], indicated by light and dark blue arrows, and along [210] (dotted white line), cf. Supplementary Fig. 10. b Topography (T = 8 K) of a region with perfectly aligned step edges that show almost complete monolayer coverage. c Topography (T = 8 K) of 3D cluster growth. The molecular islands growing on top of the monolayer adopt the geometric orientation along [110]. The inset shows a high-resolution topography (6 × 6 nm²) of a molecular island showing a gear-rail-like structure on top of the island. The tunneling parameters for all topographies were Iset = 50 pA and Ubias = 2 V.
Figure 3b shows a large-scale topography of a nearly perfectly ordered monolayer. Such a complete monolayer occupation of the surface is mainly found in regions, in which the step edges are close to perfectly aligned along the silver crystal’s high symmetry axes 〈110〉.
For higher surface coverage, molecular islands are found on top of the monolayer. This can be seen in Fig. 3c. Moreover, for the growth of three-dimensional structures, the same 〈110〉 crystal directions have been found to have a major impact on structure growth. The islands clearly adopt the underlying monolayer geometry and, thus, are aligned along with the directions 〈110〉. In the high-resolution images in the inset of Fig. 3c the islands appear to have a base plateau of about 3 nm width. On top of this, a gear rail-like structure can be seen.
DFT calculations of a single complex on Ag(001)
Starting the DFT analysis with a single fac-Re(bpy)(CO)3Cl molecule on a flat Ag(001) surface, we have considered two fundamentally different configurations: One in which the Cl atom faces the Ag(001) surface and the other one with the opposing CO group towards the Ag(001) surface. The DFT results show that the configuration with the Cl atom towards the Ag(001) surface has a binding energy of −1.86 eV, which is significantly lower than that with the CO group towards the Ag(001) surface, −1.38 eV (Fig. 4a, b). Comparing the experimental findings, e.g. Fig. 5d, with the simulated constant current STM topography of a single fac-Re(bpy)(CO)3Cl molecule in Fig. 4a supports this result. Consequently, analyzing more complex molecular structures, we have focused on configurations with the Cl atom facing the Ag(001) surface.

a Ball-and-stick model in top view and side view with chloride ion facing the surface as well as the simulated constant current topography, Ubias = 2 V. b Ball-and-stick model in top view and side view of a molecule with CO group facing the surface. c–f Four different geometric configurations of a single complex attached to a monoatomic silver step edge that is aligned along the crystal direction [110] in top view and side view. Below the belonging binding energies, Eb, are shown.

a, b Simulation of the molecular monolayer on the terrace and step edge in top view, side view, and the simulated STM images, Ubias = 2 V. c High-resolution topography of the molecular monolayer on the flat silver surface. While the primitive unit cell is depicted in red, the conventional unit cell that resembles the underlying geometry by taking into account the quadratic structure of the silver lattice is shown in orange. d High-resolution topography giving access to the orientation of the molecules within the primitive unit cell, framed in red. For comparison, the conventional unit cell is indicated in orange. e A simple model showing the orientation of the molecules within the conventional unit cell as identified on basis of STM data. The primitive and the conventional unit cell are framed in red and orange, respectively. The tunneling parameters for all topographies were T = 80 K, Iset = 50 pA, and Ubias = 1 V.
In the next step, triggered by the experimental findings, we have investigated a single fac-Re(bpy)(CO)3Cl molecule attached to a monatomic [110] step edge. Figure 4c–f shows different configurations with binding energy being greatly decreased up to Eb = −3.35 eV with respect to the free surface. Careful exploration of the three energetically most favorable fac-Re(bpy)(CO)3Cl configurations on the step edge shows that the respective binding energy differs only by about ~0.1 eV (Fig. 4c–e). In Fig. 4f the local environment of the Cl ligand is comparable to the local environment of the molecule on the undisturbed surface, shown in Fig. 4a. Both configurations are attributed very similarly but higher binding energies compared to the configurations Fig. 4c–e. Hence, we attribute the strong decrease in the binding energy to the interaction between the Cl ligand and the step edge. In general, the binding of the chloride ion to the silver surface is accompanied by significant charge transfer. At the step edge, additional charge transfer occurs between the molecule and the nearby step atoms leading to a stronger chemical bond compared to the molecule on the free surface. The charge transfer becomes evident, e.g., by the Bader charges calculated for the chloride ion changing from −0.62e (the molecule in vacuum) over −0.56e (molecule on the surface) to −0.54e (molecule at step), cf. Supplementary Fig. 4. In contrast, the interaction of the molecule’s dipole with the slightly charged step edges cannot explain the decrease of the binding energy. The complex has a dipole along the axis of the Re–Cl bond of about P = −0.276 e Å or −1.32 Da with respect to the Re atom and the metal atoms close to the step edge show only little charging of about 0.01e per atom. Also, the binding of the bipyridine to the surface plays only a minor role, as the rotational orientation of the molecule with respect to the step edge does not affect the binding energy.
We conclude that the strong affinity between the fac-Re(bpy)(CO)3Cl molecule and the 〈110〉 step edge is the driving force to explain the experimental observation that the formation of fac-Re(bpy)(CO)3Cl clusters always starts at 〈110〉 steps (Fig. 2a).
On the one hand, the DFT analysis of a single fac-Re(bpy)(CO)3Cl molecule attached to a step does not provide a preferential configuration for the complex relative to the surface, as evident by the hardly varying binding energies in Fig. 4c–e. But on the other hand, the experimental data clearly indicate that only one ordered cluster structure is found at the steps, for (i) isolated clusters, for (ii) extended structures fully decorating step edges and for (iii) completely covered surface areas.
The structure of 1D and 2D molecular clusters
To refine the model, the structures of quasi-one-dimensional chains as well as of monolayers were evaluated on basis of experimental data, the detailed discussion is given in Supplementary Note 3. We find that the chain structure is relatively robust and thus not affected by the attachment of a monolayer on either of the adjacent terraces. On the basis of this analysis, we conclude that the configuration in Fig. 4e is the one used to build up the 1D molecular nanowires and thereby also the starting point for the 2D monolayers.
The molecular arrangement within a monolayer on the flat silver surface was analyzed on the basis of high-resolution topographies as shown in Fig. 5c, d. Based on the simulated constant current topography in Fig. 4a we attribute every topographic maximum to be belonging to one molecule and, in a first step, propose a simple model for the underlying molecular structure as shown in Fig. 5e. The obvious periodicity of the layer originates in a column-wise rotation of the complex, as is indicated by the colored arrows in Fig. 5e, repeating itself after four molecules.
Taking into account that the chloride ion is facing the surface (cf. Fig. 4), we propose a possible molecular arrangement on the Ag(001) surface and for a monolayer being attached to an occupied step edge, as shown in Fig. 5a, b. In the monolayer, each unit of four molecules is separated from the next by a gap of a few rows of uncovered silver (between the red and green molecules in Fig. 5e). The interaction of neighboring molecules is likely to be governed by van der Waals forces. The neighboring complexes do not form chemical bonds and the interaction energy E of neighboring dipoles (distance of r ~ 0.5 nm) is firstly repulsive and secondly also rather small, \(E = 2P^2/\left( {4\pi \varepsilon _0r^3} \right)\sim 0.02\,{{{{{{{\mathrm{eV}}}}}}}}\), with the vacuum permittivity ε0.
While we are aware that alternative models can be envisioned, the simulated STM images are consistent with the experimental results, supporting the rationality of the proposed atomic configuration for the molecular monolayer.
Growth model
The key findings of our study is a one-dimensional faceting of decorated step edges which in turn is the cornerstone of the growth of highly oriented monolayers. Combining the experimental with DFT results, we propose the following growth model for fac-Re(bpy)(CO)3Cl on Ag(001) at 300 K substrate temperature.
Nucleation of molecular clusters
We would like to start by mentioning that the overall coverage of the surface does not represent the amount of evaporated and deposited molecules. As on many terraces, full monolayer coverage is found, obviously, enough molecules have been deposited to achieve monolayer coverage. The impact of diffusion of the molecules on the surface as a possible origin of the observed inhomogeneity is discussed in detail in Supplementary Note 4. But from the strong variations of the local surface coverage, we conclude that desorption of the molecules from the surface plays an important role in suppressing nucleation of small clusters on the free surface. Instead, the experimental, as well as the DFT results, provide strong evidence for nucleation to start at steps.
In addition, even for low coverage but also for completely covered surfaces we observe a clear preferential step orientation of the occupied segments along 〈110〉 (e.g. Figs. 2a and 3b). Regarding the highly inhomogeneous coverage on the atomic scale as well as the experimental observation that already small isolated clusters are found at the preferential step edge configuration running along the 〈110〉 direction, it is most likely that nucleation happens at such specific sites, i.e. the growth starts with molecules diffusing to steps and attaching at segments having the preferred local orientation along the crystal direction [110]. The minimum observed number of molecules in a cluster is four implying that several molecules are necessary to form a stable nucleus.
Molecule-step edge interaction
One of the central experimental findings of this study is that the overwhelming majority of the covered step edges show a perfect alignment along the directions 〈110〉. In contrast, on the uncovered silver surface, we do not find any comparable alignment of the step edges, (cf. Supplementary Fig. 2). This alignment is not only found on the local scale, as it is found for steps that are deviating from the directions 〈110〉 (cf. Fig. 2b). It is also found on larger scales, for example, shown in Fig. 2c. For a covered sample that was held at room temperature for several days, we even find ordered molecular structures at quasi perfectly aligned step edges with up to a hundred nanometers length (cf. Supplementary Fig. 3).
As the samples are at room temperature during sublimation, not only the molecules but also Ag atoms are highly mobile. While it was reported that the most efficient mass transport channel causing fluctuations of silver steps on clean surfaces is the exchange of atoms between step and surface28, other findings suggest the most efficient mass transport channel on clean metal surfaces to run alongside step edges29,30. In any case, it is clear that there are diffusion-driven mechanisms allowing a rearrangement of steps on metal surfaces. Hence, we conclude that there is a strong link between the molecular cluster growth and the reorientation of the steps by silver atom diffusion.
In the literature, adsorbate-induced restructuring of a surface is referred to as faceting31,32. The significance of faceting to molecular growth has been shown in studies of different dimensional molecular growth, for example in 2D for thin-film growth of C60 on Pd(110)33, or the growth of alkane layers on Cu(100)16,17. Furthermore, faceting is found for lower dimensional growth, e.g. in 1D for the anchoring of molecular chains to a surface18. On the single-molecule scale (0D), e.g., the adsorption of the Lander molecule has been investigated34,35,36. It was found that besides adsorbing only at specific surface sites the molecule promotes the growth of surface structures fitting the molecule’s dimensions. For the present model system, we can show that the interaction between molecules and substrate results in low-dimensional faceting that is an essential prerequisite for the growth of well-ordered molecular structures in 1D, 2D, and 3D. It is fair to say that our data do not provide access to the exact atomistic processes leading to the reorientation of the step edges.
Monolayer growth
Since we have not observed any pronounced molecular islands on the terraces, nucleation on the free surface is negligible. Instead, small monolayers areas attached to molecules decorating step edges (Fig. 3a, indicated by the blue arrows) suggest that the decorated step edges are the nucleation sites for monolayer growth. Besides, all the monolayer nuclei are found at step regions well-aligned along the crystal’s symmetry directions 〈110〉. We conclude that the nucleation of the molecular monolayer dominantly happens at completely occupied, for tens of nanometers-oriented step edges and not at randomly oriented step edges. Secondly, a monolayer exclusively grows on the upper part of the steps, i.e. no growth is observed on the bottom of a step. Thereby, the formation of grain boundaries resulting from growth starting either at the lower or the upper flanking step edge37 is strictly avoided.
These very specific nucleation processes found for both 1D clusters and 2D monolayers explain the inhomogeneous coverage of the surface by molecular clusters. For the pristine surface, appropriate binding sites at steps having 〈110〉 orientation are rather limited and at differing sites molecules will desorb with a higher probability from the surface. This is evident, e.g., by the large difference in binding energies for molecules on the undisturbed surface in contrast to molecules at step edges. And, if the fully decorated step edges are not perfectly aligned along the directions 〈110〉 also the lack of appropriate nucleation sites for the monolayer will increase the likelihood of desorption, which means that defects in the 〈110〉 oriented steps are able to prevent further growth. This is visible, e.g. in Supplementary Fig. 9.
In addition, we find the local step density to be an important growth parameter. In surface regions of high step density, we find an additional form of surface faceting promoted through self-assembled molecular clusters. A topography showing several well-ordered clusters is discussed in Supplementary Note 5. The molecules form a periodic structure along the crystal direction [210]. On a very local scale, the [210] steps consist of short [110] step segments and, hence, offer similar binding sites that fit the molecular dimensions. This, again underlines that the crystal symmetry axis [110] is geometrically preferred by the molecule and shows a complex growth mode that is only dominant in a suitable substrate environment.
Conclusion
In summary, our results present a comprehensive picture of the deposition and self-assembly of thermally stable rhenium complexes on the Ag(001) surface. All steps in the molecular growth of fac-Re(bpy)(CO)3Cl on silver rely on the availability of step edges aligned along 〈110〉. Using large-scale DFT calculations the growth hierarchy can be understood and interpreted in terms of the underlying molecular structures and the corresponding binding energies. Rearrangement of the substrate atoms is involved in the molecular cluster growth affecting the local step orientation. This promotes the formation of well-ordered structures. The resulting long-range ordered 1D molecular wires are found to be the prerequisite for 2D growth resulting in long-range ordered molecular monolayers and finally for growing 3D structures. Our results show how well-designed surface morphology can be used to guide and control molecular self-assembly in 1D, 2D as well as 3D.
Methods
Sample preparation
The Ag(001) surface was cleaned by repeated Ar+-sputtering and annealing cycles resulting in a stepped surface with random step orientations and inhomogeneous terrace widths. Sublimation of the fac-Re(bpy)(CO)3Cl (bpy = 2,2′-bipyridine) complex (see Fig. 1a) onto the clean silver was done under ultra-high vacuum (UHV) conditions (pressure p < 10–9 mbar) with the substrate at room temperature.
Scanning tunneling microscopy
The formation of a molecule film of fac-Re(bpy)(CO)3Cl (bpy = 2,2′-bipyridine) on the noble metal surface Ag(001) was studied by STM. After the preparation, the crystal was transferred into a home-built cryogenic STM under UHV (p < 5 × 10–11 mbar). The measurements were performed in constant current mode at temperatures of 80 K as well as of 8 K. Constant current topographies are referred to as topographies in the main text. For better visibility, in the topographic data physically flat areas were fitted to a plane. If useful, data were interpolated, line averages were subtracted, and 3 × 1 median filters were applied.
Synthesis of the complex
For the synthesis of the complex, we followed the procedure as described previously38. To a solution of 2,2´-bipyridine (86.4 mg, 553 μmol, 1.01 eq.) in dry toluene (10 mL) pentacarbonylchloridorhenium (199 mg, 549 μmol, 1.00 eq.) was added and the mixture heated to 100 °C for 6 h. After cooling down to room temperature the solvent was removed under reduced pressure. The yellow residue was washed with diethyl ether (3 × 5 mL) and hexane (3 × 5 mL). Drying in vacuum yielded the desired product (233 mg, 504 μmol, 92%) as a yellow solid. Prior to STM studies the fac-Re(bpy)(CO)3Cl was further purified by sublimation at about 200 °C in HV (p < 10–3 mbar). Once in the preparation chamber, the molecular source was degassed at 150 °C for 3 days under HV (p < 10–6 mbar).
Sum-frequency generation (SFG)
Experimental details to measure vibrational SFG can be found in Supplementary Note 6. In short, tunable 20 ps IR pump and probe pulses, as well as 532 nm laser pulses, were used in order to study the generated SFG signal for pump pulses in the range of the molecule’s characteristic CO absorption bands at roughly 2000 cm−1 on a monolayer of the rhenium complex on the Ag(001) surface. The SFG response (Ipump) is measured at a fixed time delay Δt after the IRpump pulse has excited the system at a certain wavenumber and normalized to the signal without a previous pump pulse (I0). The pulse scheme is depicted as an inset of Fig. 1c. The fac-Re(bpy)(CO)3Cl monolayer covered Ag(001) samples were prepared under UHV conditions, the SFG measurements were carried out under ambient conditions.
Density functional theory
The calculations were performed to explore the geometric configurations of fac-Re(bpy)(CO)3Cl molecules on the Ag(001) surface with and without step edges, the binding energies of a fac-Re(bpy)(CO)3Cl molecule at different sites, and various patterns of fac-Re(bpy)(CO)3Cl molecules on Ag(001) surfaces. All the DFT calculations were carried out with the Vienna ab initio Simulation Package (VASP)39 based on DFT with the van der Waals (vdW) interactions in terms of semi-empirical DFT-D2 correction40. The used exchange-correlation function is the generalized gradient approximation (GGA) of Perdew–Burke–Ernzerhof (PBE)41 and the electronic plane wave interception energy was set to be 400 eV. To avoid interactions between the neighboring periodic images, a vacuum layer larger than 15 Å was used in modeling. In structure optimizations, the energy convergence criteria of 10–5 eV and force convergence criteria of 0.01 eV per Å are used. The binding energies (Eb) of a fac-Re(bpy)(CO)3Cl molecule on Ag(001) surface were calculated as:
where Esystem and Esubstrate are the calculated energies of the whole system and the Ag substrate, respectively. Emolecule is the energy of one molecule and n denotes the number of molecules in one unit cell of the computational model.
As discussed elsewhere42,43, the simulated STM images based on DFT calculations do not correspond to a current value as that in real experiments. Instead, we calculate the 3D charge density distribution and the simulated constant current image corresponds to an iso-value surface.
All the STM images for multiple molecules grown on Ag surfaces are constant height images and were obtained at heights of 6 Å above the metal surface (maximum of z-coordinate).
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request. Besides, the optimized structure of the single complex in a vacuum is provided in Supplementary Data 1.
References
Morris, A. J., Meyer, G. J. & Fujita, E. Molecular approaches to the photocatalytic reduction of carbon dioxide for solar fuels. Acc. Chem. Res. 42, 1983–1994 (2009).
Calle-Vallejo, F. et al. Finding optimal surface sites on heterogeneous catalysts by counting nearest neighbors. Science (80-.). 350, 185–189 (2015).
Qiao, X. et al. Well-defined nanographene–rhenium complex as an efficient electrocatalyst and photocatalyst for selective CO2 reduction. J. Am. Chem. Soc. 139, 3934–3937 (2017).
Zhanaidarova, A., Jones, S. C., Despagnet-Ayoub, E., Pimentel, B. R. & Kubiak, C. P. Re(tBu-bpy)(CO)3Cl supported on multi-walled carbon nanotubes selectively reduces CO2 in water. J. Am. Chem. Soc. 141, 17270–17277 (2019).
Orchanian, N. M., Hong, L. E. & Marinescu, S. C. Immobilized molecular wires on carbon-cloth electrodes facilitate CO2 electrolysis. ACS Catal. 9, 9393–9397 (2019).
Willkomm, J., Bouzidi, S., Bertin, E. & Birss, V. I. Aqueous CO2 reduction by a Re(bipyridine)-polypyrrole film deposited on colloid-imprinted carbon. ACS Catal. 11, 1096–1105 (2021).
Zhuo, M.-P. et al. Hierarchical self-assembly of organic heterostructure nanowires. Nat. Commun. 10, 3839 (2019).
Maksymovych, P., Sorescu, D. C., Jordan, K. D. & Yates, J. T. Collective reactivity of molecular chains self-assembled on a surface. Science (80-) 322, 1664–1667 (2008).
Barth, J. V., Costantini, G. & Kern, K. Engineering atomic and molecular nanostructures at surfaces. Nature 437, 671–679 (2005).
Barth, J. V. Molecular architectonic on metal surfaces. Annu. Rev. Phys. Chem. 58, 375–407 (2007).
Bartels, L. Tailoring molecular layers at metal surfaces. Nat. Chem. 2, 87–95 (2010).
Schiek, M., Balzer, F., Al-Shamery, K., Lü tzen, A. & Rubahn, H. G. Nanoaggregates from thiophene/phenylene co-oligomers. J. Phys. Chem. C 113, 9601–9608 (2009).
Vladimirova, M. et al. Supramolecular self-assembly and selective step decoration on the Au(111) surface. Europhys. Lett. 56, 254–260 (2001).
Frank, E. R., Chen, X. X. & Hamers, R. J. Direct observation of oriented molecular adsorption at step edges: a cryogenic scanning tunneling microscopy study. Surf. Sci. 334, L709–L714 (1995).
Wagner, T., Fritz, D. R., Rudolfová, Z. & Zeppenfeld, P. Role of step edges on the structure formation of α-6T on Ag(441). Surf. Sci. 667, 17–24 (2018).
Wang, L. et al. Tailoring alkane uniaxial self-assembly via polymer modified step edges. J. Phys. Chem. C 123, 28811–28815 (2019).
Zhang, J. et al. Step-edge assisted direct linear alkane coupling. Chemistry 23, 6185–6189 (2017).
Schunack, M. et al. Anchoring of organic molecules to a metal surface: HtBDC on Cu(110). Phys. Rev. Lett. 86, 456–459 (2001).
Hawecker, J., Lehn, J. M. & Ziessel, R. Efficient photochemical reduction of CO2 to CO by visible light irradiation of systems containing Re(bipy)(CO)3X or Ru(bipy) 32+-Co2+ combinations as homogeneous catalysts. J. Chem. Soc. Chem. Commun. 536–538 https://doi.org/10.1039/C39830000536 (1983).
Clark, M. L., Cheung, P. L., Lessio, M., Carter, E. A. & Kubiak, C. P. Kinetic and mechanistic Effects of bipyridine (bpy) substituent, labile ligand, and brønsted acid on electrocatalytic CO2 reduction by Re(bpy) complexes. ACS Catal. 8, 2021–2029 (2018).
Clark, M. L. et al. CO2 Reduction catalysts on gold electrode surfaces influenced by large electric fields. J. Am. Chem. Soc. 140, 17643–17655 (2018).
Paul, L. A. et al. A dinuclear rhenium complex in the electrochemically driven homogeneous and heterogeneous H+/CO2-reduction. Dalt. Trans. 49, 8367–8374 (2020).
Koike, K. et al. Key process of the photocatalytic reduction of CO2 using [Re(4,4‘-X 2 -bipyridine)(CO) 3 PR 3] + (X = CH 3, H, CF 3; PR 3 = phosphorus ligands): dark reaction of the one-electron-reduced complexes with CO2. Organometallics 16, 5724–5729 (1997).
Snyder, S. R., White, H. S., López, S. & Abruńa, H. D. Scanning tunneling microscopy of dimeric and polymeric products of electroreduced [Re(CO)3(4-vinyl, 4’-methyl-2,2’-bipyridine)Cl]. J. Am. Chem. Soc. 112, 1333–1337 (1990).
Anfuso, C. L. et al. Covalent attachment of a rhenium bipyridyl CO2 reduction catalyst to rutile TiO2. J. Am. Chem. Soc. 133, 6922–6925 (2011).
Holleman, A. F., Wiberg, E., Wiberg, N. & Fischer, G. Lehrbuch der anorganischen Chemie (Walter de Gruyter, 2007).
Johnson, F. P. A., George, M. W., Hartl, F. & Turner, J. J. Electrocatalytic reduction of CO2 using the complexes [Re(bpy)(CO)3L]n (n = +1, L = P(OEt)3, CH3CN; n = 0, L = Cl-, Otf-; bpy = 2,2′-bipyridine; Otf- = CF3SO3) as catalyst precursors: Infrared spectroelectrochemical investigation. Organometallics 15, 3374–3387 (1996).
Pai, W., Bartelt, N. & Reutt-Robey, J. Fluctuation kinetics of an isolated Ag(110) step. Phys. Rev. B - Condens. Matter Mater. Phys. 53, 15991–15996 (1996).
Giesen, M. & Schulze Icking-Konert, G. Equilibrium fluctuations and decay of step bumps on vicinal Cu (111) surfaces. Surf. Sci. 412–413, 645–656 (1998).
Quaas, N., Wenderoth, M. & Ulbrich, R. G. Diffusive atom transport along step edges on Ag(111) at 295 K. Surf. Sci. 550, 57–64 (2004).
Chen, Q. & Richardson, N. V. Surface facetting induced by adsorbates. Prog. Surf. Sci. 73, 59–77 (2003).
Böhringer, M., Berndt, R. & Schneider, W. D. Transition from three-dimensional to two-dimensional faceting of Ag(110) induced by Cu-phthalocyanine. Phys. Rev. B-Condens. Matter Mater. Phys. 55, 1384–1387 (1997).
Weckesser, J. et al. Binding and ordering of C60 on Pd(110): Investigations at the local and mesoscopic scale. J. Chem. Phys. 115, 9001–9009 (2001).
Schunack, M. et al. Adsorption behavior of Lander molecules on Cu(110) studied by scanning tunneling microscopy. J. Chem. Phys. 117, 6259–6265 (2002).
Gross, L. et al. Lander on Cu(2 1 1)-selective adsorption and surface restructuring by a molecular wire. Chem. Phys. Lett. 371, 750–756 (2003).
Rosei, F. Organic molecules acting as templates on metal surfaces. Science (80-.) 296, 328–331 (2002).
Zhang, X. N., Oteyza, D. G., de, Wakayama, Y. & Dosch, H. STM study of di-indenoperylene molecules on Cu(100) surfaces: mobility, stability and epitaxy. Surf. Sci. 603, 3179–3183 (2009).
Bronner, C. & Wenger, O. S. Proton-coupled electron transfer between 4-cyanophenol and photoexcited rhenium(I) complexes with different protonatable sites. Inorg. Chem. 51, 8275–8283 (2012).
Kresse, G. & Hafner, J. Ab initio molecular dynamics for open-shell transition metals. Phys. Rev. B 48, 13115–13118 (1993).
Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 27, 1787–1799 (2006).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Andryushechkin, B. V. et al. Structure of chlorine on Ag(111): evidence of the (3×3) reconstruction. Phys. Rev. B-Condens. Matter Mater. Phys. 81, 1–5 (2010).
Choudhary, K. et al. Computational scanning tunneling microscope image database. Sci. Data 8, 1–9 (2021).
Kurz, P., Probst, B., Spingler, B. & Alberto, R. Ligand variations in [ReX(diimine)(CO)3] complexes: effects on photocatalytic CO2 reduction. Eur. J. Inorg. Chem. 2006, 2966–2974 (2006).
Kurz, P., Probst, B., Spingler, B. & Alberto, R. CCDC 299061 Experimental Crystal Structure Determination https://doi.org/10.5517/ccb164p (2006).
Hayashi, Y., Kita, S., Brunschwig, B. S. & Fujita, E. Involvement of a binuclear species with the Re−C(O)O−Re moiety in CO2 reduction catalyzed by tricarbonyl rhenium(I) complexes with diimine ligands: strikingly slow formation of the Re−Re and Re−C(O)O−Re species from Re(dmb)(CO) 3 S (dmb = 4,4‘-dimethyl-2. J. Am. Chem. Soc. 125, 11976–11987 (2003).
Acknowledgements
We thank all the members of the tunneling group for fruitful discussions and experimental support. We thank Bernhard Spicher and the teams of the electronics and precision mechanics workshops for technical support. This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) 217133147/SFB 1073, projects (C01, C04, C07). And, we acknowledge support from the Open Access Publication Funds of the Göttingen University.
Author information
Authors and Affiliations
Contributions
M.W., I.S., O.B., and L.A.P. conceived the experiments. L.A.P. synthesized the complex and L.A.P. and O.B. performed the IR measurements. L.A.P. and I.S. interpreted the IR data. O.B., T.C., and A.R. performed the STM experiments. O.B. and M.W. analyzed and interpreted the STM data. H.J. and D.S. performed and analyzed the SFG measurements. F.D. and X.D. designed and performed the DFT calculations. I.S., L.A.P., F.D., X.D., D.S., and H.J. provided materials. O.B. and M.W. wrote the manuscript. All authors commented on the manuscript at all stages.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review information
Communications Chemistry thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bunjes, O., Paul, L.A., Dai, X. et al. Ordering a rhenium catalyst on Ag(001) through molecule-surface step interaction. Commun Chem 5, 3 (2022). https://doi.org/10.1038/s42004-021-00617-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42004-021-00617-9
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
