
Abstract
Diastereodivergent heterocycle synthesis has been recognized as an important tool for drug discovery in recent years, yet strategies based on nickelacycle formation have not been established. Here, we report a NHC-Ni catalyzed highly 1,3- and 1,4-diastereodivergent heterocycle synthesis from enyne, which is achieved by manipulating the enyne N-substituent (allowing switching of selectivity from up to 2:98 to 98:2). The key to success is the efficient diastereodivergent formation of a nickelacyclopentene, with broad enyne scope at mild conditions, which subsequently provides reductive hydroalkenylation, acylation and silylation products on demand. Diastereoisomers which are sterically hard to distinguish or difficult to access by conventional routes are now accessible easily, including those with very similar 4°, contiguous and skipped stereocenters.
Similar content being viewed by others



Introduction
Heterocycles bearing multiple stereocenters are common structural motifs in many biologically active molecules1. Given that different diastereomers may exhibit diverse bioactivities, synthesis of individual diastereomers is of great importance. Multi-substituted hydropyran and piperidine skeletons with exocyclic olefin are key core structures or precursors in many biologically active natural products and drug molecules (Fig. 1)2,3,4,5,6,7. Over the years, various innovative strategies and elegant synthetic methods have been developed for (dia)stereodivergent heterocycle synthesis8,9,10,11,12,13. Substituents in 1,2-, 1,3- or 1,4-diastereodivergent relationships could be built by tailor-made ring closure methods efficiently, which are controlled often by selective facial attack either on prochiral sp2 center or on radical acceptors (Fig. 2a)14,15. Some were controlled efficiently by solvent assistance and steric repulsion among substituent Rs on ring closure, or by redesign of reaction pathways or catalysts16,17,18,19. Yet, some are limited severely by the choice of nucleophiles and polarized tether structures. Undesired steric repulsion among substituents (mismatch) as well as the high demands of functional groups often limited the scope and the efficiency of the process.

Examples of bioactive multi-substituted heterocycles involved exocyclic olefin precursors.

a By condition modification. b By cyclic template. c By steric substituents. d By hetero-substituted enynes.
Transition metal-mediated metallacyclopentene formation by using hetero-substituted enyne or related variant is a very powerful technique in catalytic heterocycle synthesis and is a common intermediate for various multi-component coupling20,21,22,23,24. Highly 1,3-syn-diastereoselective synthesis has been achieved in a few examples according to the steric demands of enyne substituents and metallacycle formation (Fig. 2b)25,26,27, however, other possible combinations were explored rarely (e.g., 1,3- 1,4-diastereodivergent, and in higher substituted cases). Moreover, unlike a few examples noted in intramolecular hydroalkenylation of diene (Fig. 2c)28,29, the scope is mostly limited to those equipped with cyclic template, some requires Thorpe-Ingold effect assistance, and no efficient diastereodivergent example has been developed. Developing a novel and general diastereodivergent strategy which is not relied primarily on minimizing undesired steric repulsion among Rs is therefore useful, especially for those sterically less accessible and higher substituted products.
Here, a NHC-Ni catalyzed highly diastereodivergent heterocycle synthetic method is thus developed (Fig. 2d). That is based on conformational cooperation in the nickelacyclopentene formation step, directed by the choice of N-substituents on the heteroenynes, and promoted by NHC-Ni catalyst π-electronic effect. By trapping the NHC-nickelacyclopentenes with alcohol, silane and aldehyde, 1,3- and 1,4-diastereodivergent reductive hydroalkenylation30,31,32, silylation and acylation products are obtained26,33.
Results
Optimization for N-substituents directed diastereodivergent reductive hydroalkenylation
Our study commenced with a set of simple 1,7-heteroenynes 1a–f having a racemic stereocenter and terminal propargyl-Z for a study of N-substituents on nickelacyclopentene selective formation (Table 1, entry 1–6, Eq. (1), Z = O, NH or NMs). The condition employed is as simple as an NHC-Ni(0) catalyzed enyne cycloaddition reported in the literature with shorter tethers24, except 1-phenylethanol is used as terminal reductant to complete the desired catalytic cycle by reductive hydroalkenylation under mild condition (Supplementary Table 1). To our delight, a NHC-Ni(0) catalyzed reductive hydroalkenylation of enyne 1 was observed. High yield and high reactivity were obtained by using a catalytic amount of IPr-Ni(0) in toluene. This is useful because selective nickelacyclopentene formation is less efficient for those terminal enynes due to highly competitive and undesired [2 + 2 + 2] oligomerization or dimerization. Yet, the most remarkable result is that a highly diastereodivergent piperidine synthesis was achieved by this rarely employed strategy. Also, the role of R1 size in 1,3-syn-:anti-selectivity was found not so crucial (R1 = Ph c.f. Me). A high 1,3-syn-:anti-selectivity (3:3′) was observed by using 1c-d (with propargyl-Z = NH). On the contrary, a high 1,3-anti-selectivity (4′) was noted in 1e-f (with propargyl-Z = NMs). Oxa-enyne 1a-b followed the same reactivity and selectivity pattern as 1c-d did. Moreover, a highly efficient diastereodivergent 1,4-stereotransfer was observed by using enyne 2 with different allyl-Z as substrate (Table 2, Eq. (2), Z = O, NH or NMs, R1 = Ph or Me). Heterocycles 5′ and 6 were obtained as major products (Table 2, entry 1–6). Similarly, the preferred product stereo-configuration (syn-:anti-selectivity) was found correlated to the selected choice of Z. Yet, the preferred stereo-configuration directed by allyl-Z was found different from the propargyl-Z cases (Tables 1 and 2, e.g., allyl-NMs favored 1,4-syn-, propargyl-NMs favored 1,3-anti-).
At first, one might consider the steric difference in Z is sufficient to explain the above diastereodivergent synthesis. Yet, very similar efficiency among enynes having different sizes of R1 in both Eqs. (1) and (2) caught our attention. That made us suspected it was not just directed by simple steric interaction, so Z with different steric and electronic property was evaluated next (Supplementary Table 2). First, using sterically bulkier Z than NMs were found not helpful in favoring anti-selectivity further but led to a drop in selectivity (Table 1, N-Ms vs -Ts and -Bn, entry 5, 7 and 8). This is unusual since a larger Z like NTs or NBn is expected to favor a stronger steric repulsion among R1 and Z for more anti-product. Second, using a bulky N-Bn could favor the syn-product as N-H did (Table 1, entry 3, 8). These 2 sets of results are in sharp contrast to the former rationale that based on steric effect increment on Z from NH to NMs alone. Furthermore, Z = NTf did not provide the desired reactivity. Altogether, the diastereodivergent synthesis as noted above was attributed mainly to the use of Z with different electronic property. Yet, the size of Z is still critical in terms of diastereodivergent efficiency, in which using small Z could avoid competing steric interactions. Overall, the diastereodivergent synthesis efficiency and preference did not follow the order of the Z sizes to change gradually (Size: N-H < Ms < Bn~Ts, while the Syn-:Anti-ratio ranged from 96:4 to 6:94 in order of N-H > Bn > Ts > Ms). Similar phenomenon was noted in Table 2, Eq. (2), entry 3 and 5.
Ligand effect on diastereoselectivity
The above results prompted us to screen ligands with different steric and electronic properties by using 1a, 1c and 1e as well as 2a, 2c and 2e (Tables 1, 2, Supplementary Table 3, 4). In general, using bulky NHC is one of the keys to obtain desired reactivity. Indeed, non-selective oligomerization and dimerization was noted in IMes and PCy3 (Table 1, entry 5). More interestingly, the NHC electronic effect has a strong impact on the diastereodivergent efficiency in both equations and IPr performed much better than SIPr in general (Table 1, entry 1, 5; Table 2, entry 1, 5), in which dramatic drops were observed when using Z = NMs and SIPr. This result suggested that the use of NHC with lower π-accepting ability is highly desirable for higher diastereodivergent efficiency, given that IPr and SIPr are very similar in size and σ-donating power34,35. Also, it showed that the above is not entirely a Z controlled process.
Substrate scope of diastereodivergent reductive hydroalkenylation
The fine-tuned reductive hydroalkenylation reactivity, which was brought by propargyl-/allyl-Z, 2° alcohol and NHC-Ni cooperation, also came with a broad scope of 1 and 2 (Fig. 3). It provided a general access to functionalized hydroalkenylation products (3 and 6, 4′ and 5′) in one step by simply using an in situ generated catalyst from over-the-counter sources. Enynes with aryl and alkyl substitutions (Set 1), with internal alkenes and alkynes (Set 2 and 3), with nearby stereocenter interference at 2-position (Set 4), with gem-disubstituted alkenes (Set 5) and with a longer chain length (Set 6) are all good substrates for this reductive hydroalkenylation (Eqs. (1) and (2)). Only a few cases required a slow addition of enyne, protected NH and employed IPrMe as NHC. Those changes were used to compete with the undesired oligomerization. Overall, this method allows us to synthesis heterocycle derivatives with higher substituted olefins, longer side chains, 1,2,3-contagious and 1,2,4-skipped stereocenters, new quaternary centers, and 7-member rings.

See Methods section for hydroalkenylation procedure; see Tables 1 and 2 for Eqs. (1) and (2). Product syn:anti-selectivity was determined by NMR, yield of desired product in relative configuration is in parenthesis. a By GCMS. b 2 h addition, to suppress oligomerization. c 20 mol% catalyst. d IPrMe was used instead of IPr. e enyne: alcohol = 1:1.5, to avoid undesired enyne direct reduction. f 3 equiv. of NaBH4, to avoid acetophenone insertion (from the 1-phenylethanol).
The robust reactivity also came along with moderate to excellent 1,3- and 1,4-diastereodivergent efficiency (e.g., from up to 2:98 to 98:2). Several notable results deserve further comment. First, the diastereodivergent synthesis efficiency is excellent even in cases with substituents as small as Me. It highlighted that the basis of this switching is not primarily on steric repulsion (Set 1, Eqs. (1) and (2)). Second, the method can still perform even when interfered by an extra 2-substituent at syn/anti-relative configuration (Set 4). This indicated that such gauche interactions are not competitive enough to the desired preference directed by our strategy (e.g., Z = NH, 95:5 vs 90:10 in Eq. (1); <5:95 in Eq. (2)). Hence, assortments of densely substituted stereoisomers are now easily accessible by this reductive hydroalkenylation. Third, stereo-defined 4° centers only having marginal steric differences are hard to make selectively (Set 5, e.g., Me vs Et). Our approach offers a possible entry to build those very similar products as well as isomers. Overall, it complements to a few shortcomings by the traditional steric approaches, the cyclic templates, the rearrangement of side chains or the stoichiometric organometallic reagents19,36,37,38,39.
Discussion
The precise mechanism for our desired reactivity and the diastereodivergent strategy are still under investigation, yet some insights were obtained from the studies that used CD3OH, ArCHO and TESH instead of 1-phenylethanol (Fig. 4). First, using CD3OH in the reductive hydroalkenylation of 1c and 1e yielded the corresponding D1-3c and D1-4′e (Fig. 4a). The stereo-defined D1-olefin obtained and high D-incorporation suggest a nickela-cyclopentene mechanism rather than Ni-H insertion to enyne at this stage40. Second, by using ArCHO or TESH as enyne reaction partners in place of alcohol, several sets of highly efficient diastereodivergent acylations or silylations were achieved, respectively (Fig. 4b, c)24,26. The excellent diastereodivergent synthesis efficiency and trend noted at here are comparable with the above reductive hydroalkenylation (Tables 1, 2, Fig. 3). This strongly suggested the strategy is related to a common diastereodivergent nickelacyclopentene formation step, and not by an in situ generated Ni(II)H from NHC-Ni(0) and 2° alcohol. Thus, the diastereodivergent synthesis could be a Z directed NHC-nickelacyclopentene formation with a suitable choice of NHC, not on choices of partners41,42,43. Synthetically, the exocyclic-olefin also links up many useful organic transformations (Fig. 5), which broaden the impacts and the applications of this hydroalkenylation.

See Supplementary Methods. a D-Labeling experiment of reductive hydroalkenylaition. b Diastereodivergent acylation; NHC = IPr, Enyne: (p-anisyl)CHO = 1:1.5. c Diastereodivergent silylation; NHC = IPrCl, Enyne: TESH = 1:8. d 50 mol% catalyst, NHC = IPr, 91–94% D-labeled in the products. e 10 mol% catalyst, Syn-:Anti-ratio by GCMS (Supplementary Method E). f by NMR.
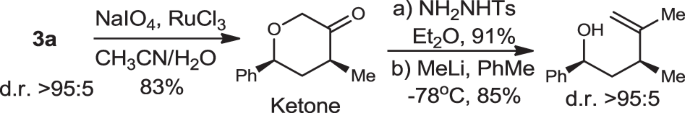
(i) Heterocyclic ketone, and (ii) Acyclic alkenol (Supplementary Methods).
In summary, an efficient 1,3- and 1,4-diastereodivergent heterocycle synthesis strategy was established by tuning property of Z on enynes under NHC-Ni catalysis (switch from up to 2:98 to 98:2). It was demonstrated by a reductive hydroalkenylation of enynes using 2° alcohol as reductant. Highly competing side reactions were suppressed and broad scope was achieved simultaneously. Unlike a number of former diastereodivergent approaches that based on facial selection at the latter stage of the catalytic cycle, here the diastereodivergent efficiency is governed by Z directed nickelacyclopentene forming step at the start (a 3 x bonds formation event, including 1 x C–C and 2 x Ni–C bond). Moreover, that can be promoted by choice of NHC ligand (IPr vs SIPr). That feature makes the preferred diastereodivergent outcome highly predicable and tunable, which in turn makes it less dependent on steric interactions among external reaction partners and substituents on the tether. Products packed densely with diastereocenters which is difficult to be made by conventional steric approaches (either sterically unfavorable or hardly distinguishable, e.g., preparation of 1,2,3-contagious and skipped stereocenters, and new 4° center with Me vs Et) are now easily accessible expediently, rather than just the sterically favorable ones. Finally, the diastereodivergent results obtained in acylations and silylations imply a great potential of this work in joining many other transformations and cross-coupling reactions that based on nickelacyclopentenes and related species for broader scope. Exploration along this line is now underway.
Methods
Preparation of hetero-substituted enynes: See Supplementary Methods.
Determination of diastereoselectivity: See Supplementary Methods, Supplementary Figs. 4–10.
Acylation procedure: See Supplementary Methods.
Sylilation procedure: See Supplementary Methods.
Products characterization: See Supplementary Data 1.
Standard reductive hydroalkenylation procedure: Ni(cod)2 and IPr (0.05 mmol each) were dissolved in toluene (2 mL) and stirred at 30 °C in glovebox for 1 hr. An enyne and 1-phenylethanol (0.5: 1.5 mmol) toluene solution (1 mL) were added dropwise to the above catalyst in 0.5 h, and was stirred at 30 °C for 3 h. After work up, the yield, structure and selectivity were determined by 1H NMR, NOESY, isolation and GCMS (average of two runs). Mesylation of NH products were carried out for direct comparison of the isomers when necessary (Supplementary Method A).
Data availability
The authors declare that all the other data supporting the findings of this study are available within this paper, its Supplementary Information file and Supplementary Data 1.
References
Majumdar, K. C. & Chattopadhyay, S. K. Heterocycles in Natural Product Synthesis (Wiley-VCH Verlag, 2011).
Hayden, M. R., Leavitt, B. R., Yasothan, U. & Kirkpatrick, P. Tetrabenazine. Nat. Rev. Drug. Discov. 8, 17–18 (2009).
Seneca. in Alkaloids - Secrets of Life (ed. Tadeusz Aniszewski) 61–139 (Elsevier, 2007).
Fujii, T. & Ohba, M. In The Alkaloids: Chemistry and Biology Vol. 51 (ed. Geoffrey A. Cordell) 271–321 (Academic Press, 1998).
Ishihara, J. et al. Total synthesis of (−)-Verrucarol1. J. Org. Chem. 63, 2679–2688 (1998).
Wang, X. et al. A radical cascade enabling collective syntheses of natural products. Chem 2, 803–816 (2017).
Zhang, W., Bah, J., Wohlfarth, A. & Franzen, J. A stereodivergent strategy for the preparation of corynantheine and ipecac alkaloids, their epimers, and analogues: efficient total synthesis of (−)-dihydrocorynantheol, (−)-corynantheol, (−)-protoemetinol, (−)-corynantheal, (−)-protoemetine, and related natural and nonnatural compounds. Chem. Eur. J. 17, 13814–13824 (2011).
Bihani, M. & Zhao, J. C. G. Advances in asymmetric diastereodivergent catalysis. Adv. Synth. Catal. 359, 534–575 (2017).
Krautwald, S. & Carreira, E. M. Stereodivergence in asymmetric catalysis. J. Am. Chem. Soc. 139, 5627–5639 (2017).
Beletskaya, I. P., Najera, C. & Yus, M. Stereodivergent catalysis. Chem. Rev. 118, 5080–5200 (2018).
Singha, S., Serrano, E., Mondal, S., Daniliuc, C. G. & Glorius, F. Diastereodivergent synthesis of enantioenriched α,β-disubstituted γ-butyrolactones via cooperative N-heterocyclic carbene and Ir catalysis. Nat. Catal. 3, 48–54 (2020).
Wu, X., Chen, Z., Bai, Y.-B. & Dong, V. M. Diastereodivergent construction of bicyclic gamma-lactones via enantioselective ketone hydroacylation. J. Am. Chem. Soc. 138, 12013–12016 (2016).
Zhu, J. et al. Remote ester groups switch selectivity: diastereodivergent synthesis of tetracyclic spiroindolines. J. Am. Chem. Soc. 136, 6900–6903 (2014).
France, S. P. et al. One-pot cascade synthesis of mono- and disubstituted piperidines and pyrrolidines using carboxylic acid reductase (CAR), ω-Transaminase (ω-TA), and imine reductase (IRED) biocatalysts. ACS Catal. 6, 3753–3759 (2016).
Zhao, Y., Hu, Y., Wang, H., Li, X. & Wan, B. Transition-metal controlled diastereodivergent radical cyclization/azidation cascade of 1,7-enynes. J. Org. Chem. 81, 4412–4420 (2016).
Morgen, M., Bretzke, S., Li, P. & Menche, D. Stereodivergent synthesis of 1,3-syn- and -anti-tetrahydropyrimidinones. Org. Lett. 12, 4494–4497 (2010).
Zhong, C., Wang, Y., O’Herin, C. & Young, D. W. Synthesis of substituted morpholines using stereodivergent Aza-Michael reactions catalyzed by Bronsted acids. ACS Catal. 3, 643–646 (2013).
Williams, J. T., Bahia, P. S. & Snaith, J. S. Synthesis of 3,4-disubstituted piperidines by carbonyl ene and prins cyclizations: a switch in diastereoselectivity between Lewis and Bronsted acid catalysts. Org. Lett. 4, 3727–3730 (2002).
Seel, S. et al. Highly diastereoselective arylations of substituted piperidines. J. Am. Chem. Soc. 133, 4774–4777 (2011).
Gandeepan, P. & Cheng, C.-H. Cobalt catalysis involving π components in organic synthesis. Acc. Chem. Res. 48, 1194–1206 (2015).
O’Rourke, N. F., Kier, M. J. & Micalizio, G. C. Metallacycle-mediated cross-coupling in natural product synthesis. Tetrahedron 72, 7093–7123 (2016).
Jackson, E. P. et al. Mechanistic basis for regioselection and regiodivergence in nickel-catalyzed reductive couplings. Acc. Chem. Res. 48, 1736–1745 (2015).
Ohashi, M., Hoshimoto, Y. & Ogoshi, S. Aza-nickelacycle key intermediate in nickel(0)-catalyzed transformation reactions. Dalton T 44, 12060–12073 (2015).
Thakur, A. & Louie, J. Advances in nickel-catalyzed cycloaddition reactions to construct carbocycles and heterocycles. Acc. Chem. Res. 48, 2354–2365 (2015).
Tang, X.-Q. & Montgomery, J. Nickel-catalyzed preparation of bicyclic heterocycles: total synthesis of (+)-Allopumiliotoxin 267A, (+)-Allopumiliotoxin 339A, and (+)-Allopumiliotoxin 339B. J. Am. Chem. Soc. 122, 6950–6954 (2000).
Tekavec, T. N. & Louie, J. Nickel-catalyzed cycloadditions of unsaturated hydrocarbons, aldehydes, and ketones. J. Org. Chem. 73, 2641–2648 (2008).
Mizuno, K., Ikeda, M., Toda, S. & Otsuji, Y. Regioselective double vicinal carbon-carbon bond forming reactions of electron-deficient alkenes by use of allylic stannanes and organoiodo compounds. J. Am. Chem. Soc. 110, 1288–1290 (1988).
Yamamoto, Y. Transition-metal-catalyzed cycloisomerizations of α,ω-dienes. Chem. Rev. 112, 4736–4769 (2012).
Li, K., Li, M.-L., Zhang, Q., Zhu, S.-F. & Zhou, Q.-L. Highly enantioselective nickel-catalyzed intramolecular hydroalkenylation of N- and O-tethered 1,6-dienes to form six-membered heterocycles. J. Am. Chem. Soc. 140, 7458–7461 (2018).
Montgomery, J. & Savchenko, A. V. Nickel-catalyzed cyclizations of alkynyl enones with concomitant stereoselective tri- or tetrasubstituted alkene introduction. J. Am. Chem. Soc. 118, 2099–2100 (1996).
Phillips, J. H. & Montgomery, J. Mechanistic insights into nickel-catalyzed cycloisomerizations. Org. Lett. 12, 4556–4559 (2010).
Chen, J., Han, X. & Lu, X. Palladium(II)-catalyzed reductive cyclization of N-tosyl-tethered 1,7-enynes: enantioselective synthesis of 1,2,3,4-tetrahydroquinolines. Org. Lett. 21, 8153–8157 (2019).
Xi, T. & Lu, Z. Cobalt-catalyzed hydrosilylation/cyclization of 1,6-enynes. J. Org. Chem. 81, 8858–8866 (2016).
Huynh, H. V. Electronic properties of N-heterocyclic carbenes and their experimental determination. Chem. Rev. 118, 9457–9492 (2018).
Yong, X., Thurston, R. & Ho, C.-Y. Electronic effects on chiral NHC–transition-metal catalysis. Synthesis 51, 2058–2080 (2019).
Xiao, Y.-C. & Moberg, C. Silaborative carbocyclizations of 1,7-enynes. Diastereoselective preparation of chromane derivatives. Org. Lett. 18, 308–311 (2016).
Yang, C.-M., Mannathan, S. & Cheng, C.-H. Nickel-catalyzed chemo- and stereoselective alkenylative cyclization of 1,6-enynes with alkenyl boronic acids. Chem. Eur. J. 19, 12212–12216 (2013).
Jeganmohan, M. & Cheng, C.-H. Cobalt- and nickel-catalyzed regio- and stereoselective reductive coupling of alkynes, allenes, and alkenes with alkenes. Chem. Eur. J. 14, 10876–10886 (2008).
Kuang, Y., Wang, X., Anthony, D. & Diao, T. Ni-catalyzed two-component reductive dicarbofunctionalization of alkenes via radical cyclization. Chem. Commun. 54, 2558–2561 (2018).
Diccianni, J. B., Heitmann, T. & Diao, T. Nickel-catalyzed reductive cycloisomerization of enynes with CO2. J. Org. Chem. 82, 6895–6903 (2017).
Kirby, A. The Anomeric Effect and Related Stereoelectronic Effects at Oxygen. 149. (Springer-Verlag, Berlin, 1983).
Juaristi, E. & Cuevas, G. Recent studies of the anomeric effect. Tetrahedron 48, 5019–5087 (1992).
Box, V. G. S. The role of lone pair interactions in the chemistry of the monosaccharides - stereo-electronic effects in unsaturated monosaccharides. Heterocycles 32, 795–807 (1991).
Acknowledgements
C.Y.H. thanks R. H. Grubbs (Shenzhen Nobel Prize Scientists Laboratory Project (C17783101), SZ research fund (JCYJ 20170817105041557), Guangdong Provincial Key Laboratory of Catalysis (2020B121201002) and NSFC (21602099). We thank Dr. Elvis W. H. Ng for useful discussion.
Author information
Authors and Affiliations
Contributions
The manuscript was achieved through contributions of all authors. X.Y. designed and conducted the experiments. W.G. participated in the reductive hydroalkenylation section. X.L. contributed to preparation of substrates. C.Y.H. supervised the project.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yong, X., Gao, W., Lin, X. et al. NHC-Ni catalyzed 1,3- and 1,4-diastereodivergent heterocycle synthesis from hetero-substituted enyne. Commun Chem 3, 50 (2020). https://doi.org/10.1038/s42004-020-0299-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42004-020-0299-9
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
