
Understanding the biogeochemistry of radionuclides in the environment is essential for effective isolation of nuclear waste in repositories, management of contaminated sites, ensuring long-term protection of our ecosystems, and limiting impacts on human health. Here the authors discuss the extreme complexity of this multidimensional chemistry problem, highlighting the outstanding open questions for the next generations of environmental radiochemists.
Anthropogenic radionuclides are created during nuclear explosions, irradiation in nuclear reactors, particle or X-ray bombardment of materials, and α, β, γ, and ε decay or even fission (spontaneous or neutron induced) of parent radioisotopes. Natural radionuclides are produced by stellar nucleosynthesis and cosmic-ray spallation processes. However, with the exception of the uranium series (notably 238/234U and 226Ra), natural radioactive isotopes do not pose a significant health concern or environmental perturbation. Anthropogenic radionuclides are produced under extreme pressures, temperatures, and radiation fields, as well as non-equilibrium physicochemical conditions, and this may subsequently influence the surrounding geology and the radionuclides’ fate in the environment. For example, radionuclides deposited in the immediate aftermath of an underground nuclear detonation undergo temperatures that exceed 1,000,000 K, vaporizing ~70 tons and melting another 700 tons of rock for every kiloton of yield1. Fortunately, the vast majority of the radionuclides produced by anthropogenic activities (electricity generation, medicine, research, etc.) are short-lived (half-lives <10 years) and the associated waste can be managed by leveraging the natural fast decay of the isotopes. The main concern shared by past, present, and future generations will be the longer-lived (>10 years) radionuclides (lanthanide fission products, 3H, 90Sr, 94Nb, 99Tc, 137Cs, 226Ra, 233/234/235/236/238U, 237Np, 239/240/241/242/244Pu, 241/243Am, 244/245/246/247Cm, and so on), which will ultimately decay to harmless isotopes too—but only after hundreds or thousands of years. Thus, their potential to adversely impact human health and our environment must be understood at the millennial timescale.
Radionuclide contamination: a global, complex, and long-lasting issue
Identifying processes controlling radionuclide migration and predicting their behavior at environmentally relevant timescales (e.g., hundreds or thousands of years) represents one of the most unique challenges that face scientists and society. The problem is inherently interdisciplinary, and involves constraints from geochemistry, biochemistry, mineralogy, inorganic chemistry, material, and nuclear sciences, all of which can be influenced by environmental changes and human intervention. Our understanding of the fate and transport of radionuclides in the environment is limited by the inherent complexity of the problem and challenges associated with radiochemistry, including:
-
Long-lived radionuclides (half-lives >10 years) are, by nature, highly hazardous and controlled substances. A restricted availability of some radioisotopes, combined with strict safety protocols for material handling, has limited the number of institutions and scientists willing to embark on environmental radiochemistry research.
-
The sites of radionuclide contamination are chemically and geologically diverse, from the atmosphere to the ocean, from surface soils to the deep subsurface, from deserts to the polar regions, resulting in a wide range of biogeochemical conditions influencing their chemical behavior (Fig. 1)2.
Fig. 1: Global view of radiologically impacted sites and Pu inventories at selected sites. 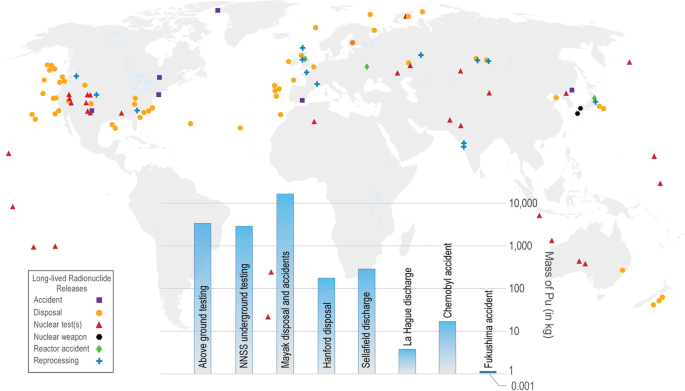
Notes: Data taken from Geckeis et al.2 and not intended to be comprehensive. Nuclear power generation sites and uranium mining-related sites not included (inset: histogram of Pu inventories at selected sites that are the result of nuclear tests, fuel reprocessing, disposal, discharge, and accidents).
-
Radionuclide concentrations of interest span ~15 orders of magnitude, from concentrated fuel rods or solid particles to femtomolar regulatory limits for drinking water standards (e.g., US Environmental Protection Agency Maximum Contaminant Levels for drinking water of 6.4 × 10–16, 1.7 × 10–14, and 3.7 × 10–15 mol/L for 90Sr, 137Cs, and 238Pu, respectively). The variation in concentration and initial form can control the chemical behavior of the radionuclides.
-
The behavior of radionuclides involves timescales that are orders of magnitude longer than the typical laboratory experiments (thousands of years versus a few years at best), questioning if short-term experiments (days to weeks) capture the long-term environmental behavior of radionuclides.
-
The radionuclides of concern are spread across the periodic table, spanning from light elements that have corresponding stable isotopes in nature (tritium, strontium, etc.) to heavy atoms that are normally not present on earth (technetium, neptunium, plutonium, americium, etc.). Hence, the behavior of nuclear waste is intrinsically difficult to predict due to the wide range of elements they contain, covering almost the entire speciation realm (gas, solids, soluble cations, anions, oxo-species, etc.).
-
Environmental systems are inherently complex and the associated radiochemistry may involve solubilized ions, precipitates, aqueous chelators, radicals, mineral phases, colloids, microorganisms, redox changes, and anthropogenic materials (e.g., fuel cladding, waste canister, repository filler, co-contaminants)3.

Mobility of long-lived radionuclides in the environment: progress and emergent behavior
Mobility of plutonium
The Comprehensive Nuclear Test Ban Treaty (CTBT) was adopted by the United Nations General Assembly, <25 years ago, on September 10, 1996. While the CTBT had little effect on the accumulation of radioactive waste generated by civilian nuclear activities, it did significantly reduce the direct release of radionuclides into the environment. For example, >6000 kg of Pu was released to the environment during underground and atmospheric nuclear testing2. It is estimated that an amount of the same order of magnitude was released globally as a result of poor environmental management of civilian and military activities (Fig. 1). Moreover, an additional ~70,000 kg of Pu are added to the non-released inventory annually for electricity production, yielding a global inventory of 2,630,000 kg Pu as of 20142. In other words, we have taken an almost irreversible path where long-lived radionuclides will exist in our environment for generations to come and must be studied and managed accordingly.
A major breakthrough in the field of environmental radiochemistry was the discovery that sparingly soluble metals can migrate rapidly over several kilometers from their initial source. Kersting et al.4 demonstrated—post CTBT—that the migration of radioisotopes, including actinides, over kilometer distances at the Nevada National Security Site (USA) is due to the association between radionuclides and mobile clay and zeolite colloids in suspension in groundwater. Similarly, Novikov et al.5 later confirmed long-range (<4 km) colloid-facilitated transport of Pu at Mayak (Russia) due to its association with mobile iron oxide colloids. In a very different scenario, trace concentrations of U and Pu have been detected over a large area (>50 km radial distance) stemming from the Fukushima Daiichi Nuclear Power Plant reactor meltdown accident, 2011 (Japan)6. In this case, recent measurements, using advanced microscopy techniques and forensic analysis tools (scanning transmission electron microscopy (TEM), high-resolution TEM, high-angle annular dark-field scanning TEM, scanning TEM energy-dispersive X-ray mapping, secondary ion mass spectrometry, synchrotron micro-focus X-ray fluorescence mapping, and X-ray absorption near-edge spectroscopy) showed that U and Pu are associated with cesium-rich microparticles5. These three colloidal/microparticle-facilitated migration mechanisms illustrate the chemical complexity of radionuclide transport.
Very recent progress in the chemistry of actinides and lanthanides also suggest that other phenomena are important in controlling the biogeochemistry of long-lived radionuclides. A recent study by Morrison et al.7 demonstrated that some ubiquitous organic chelators, such as water-soluble siderophores, can impact the redox chemistry of radionuclides, strongly influencing their behavior in the environment (Fig. 2). While other recent studies8,9 have shown that synthetic multidentate chelators can drive the oxidation state of lanthanides and actinides to +IV, Morrison et al.7 demonstrated that even a small bidentate ligand, like acetohydroxamic acid, reduces Pu(VI) to Pu(V) under environmentally relevant conditions, and that the reduction reaction does not produce Pu(IV) or Pu(III). These results raise many questions about the role of commonly occurring natural chelators in environmental samples that are often organic-rich mixtures containing trace amounts of radionuclides.

Reaction of 100 μM Pu(V) with 200 μM acetohydroxamic acid (AHA) at pH 5.6. a UV–Vis–NIR spectra of Pu(V) control and Pu(V) reacted with AHA showing no reduction to Pu(IV) over 7 days. b Concentrations of AHA monitored using GC-MS after 1 and 7 days determined that AHA is not being consumed while reacting with Pu(V). c Reaction of 100 μM PuO22+ with 200 μM desferrioxamine B (DFOB) at pH 5.5. The UV−Vis–NIR spectra show an immediate reduction of Pu(VI) to Pu(V) that remains stable throughout the experiment. A reference spectra of Pu(IV) DFOB is included showing that the reaction of Pu(VI) with DFOB does not produce any spectral features associated with a Pu(IV) DFOB complex. Adapted with permission from ref. 7. Copyright 2018 American Chemical Society.
Mobility of fission products and early actinides
In the past few years, natural macromolecular systems that are highly selective for the lanthanide (Ln) ions over other salts or co-contaminants have also been identified. Methylotrophic bacteria have been shown10 to specifically scavenge and utilize Ln3+ ions for essential biological functions. Cotruvo et al.11 recently isolated a small protein (Lanmodulin) produced by these bacteria that is even more selective for Ln3+ ions than synthetic ligands (stability ratio Ln/Ca, Ln/Zn, Ln/Cu >1,000,000)12. Based on these recent developments, the existence of biomacromolecule-mediated transport mechanisms for f-element radionuclides at specific field sites should not be ruled out.
The fundamental chemistry of actinide complexes may also be pressure dependent. Recent results from Sperling et al.13 indicate that chemical bonds in some minor actinide complexes become much more covalent at high pressure (1–10 GPa), whereas the equivalent lanthanide species remain almost unaffected. These results suggest that the thermodynamic stability of certain metal contaminants in the subsurface may not conform with behavior observed in the laboratory at atmospheric pressure. Likewise, the influence of elevated subsurface heat is often neglected when modeling the speciation of metal ions in the environment, but two recent studies14,15 have demonstrated that phases like ThO2, usually considered to be insoluble, or nuclear melt glass produced in nuclear tests can release radionuclides at elevated temperature. Nisbet et al.14 found that the solubility of ThO2 reaches the parts per million level in hydrothermal solutions at 175–250 °C—orders of magnitude higher that the reference solubility at room temperature. Similarly, Zavarin et al.15 demonstrated that nuclear melt glass can release radionuclides at moderately elevated temperatures (~200 °C). Nuclear melt glass was hydrothermally altered for 3 years over a range of temperatures (25–200 °C) and the concentrations of 60Co, 137Cs, 152Eu, and Pu in the fluids were ~100 times higher at the highest temperatures (80–200 °C) compared to those at room temperature. Even after 3 years, very little alteration was observed at the lowest temperatures. These studies highlight the necessity to perform long-term laboratory experiments under temperature and pressure conditions expected at field sites.
Advances in actinide spectroscopy
Another recent discovery that challenges our understanding of the mobility of radionuclides is the identification of a new metastable solid-phase containing Pu(V), where PuO2 was formed from soluble Pu(VI) ions under mildly alkaline conditions. These observations would not have been possible without the use of X-ray absorption near-edge spectroscopy at the M4 edge of Pu (compared to the classic but less informative L3 edge used for actinide samples). This new spectroscopic technique pioneered by Kvashnina et al.16 could help to elucidate new reaction mechanisms for radionuclides, but it also opens the question of the existence of previously unnoticed solid phases.
Natural long-lived radioisotopes
Naturally occurring radioactive material, particularly the U series isotopes, warrant investigation as they are relatively abundant, some are highly toxic, yet they have received less attention compared to uranium and manmade elements like plutonium. Radium is of particular concern because 226Ra is one of the most radiotoxic natural nuclides (specific activity = 37 GBq/g or 1 Ci/g), it decays to radiotoxic radon gas (222Rn), and its ion, Ra2+, is highly mobile and can replace calcium in bone if ingested. 228Ra (t1/2 = 5.7 years) and 226Ra (t1/2 = 1585 years) are present in the industrial waste from mining and gas operations due to their presence in the 232Th and 238U decay chains, respectively. Matyskin et al.17,18 recently determined the crystal structure of the environmentally relevant radium-barium sulfate, as well as the hydrolysis behavior of Ra2+(aq). An exceptionally high coordination number of 12 (compared to 8–10 for other heavy radionuclides, Fig. 3), long bond metal-oxygen distances (2.85–3.35 Å) and weak hydrolysis behavior were observed. These examples underscore the reality that our current understanding of the properties of many radionuclides is limited, but also highlight that new experimental techniques to probe the solid-state and solution-state chemistry of highly radioactive samples offer hope that our limited understanding will soon be a thing of the past.

Radium is a natural but highly toxic element for which there is little data available. Its chemistry is peculiar with a high coordination number and weak hydrolysis behavior. Extended crystal structure of 226RaSO4 (a), and local coordination environment of Ra2+ in 226RaSO4 (b). Reproduced with permission from ref. 17. Copyright 2017 Elsevier.
Outlook
In the past 20 years, tremendous progress has been made in our understanding of the interactions between radionuclides and the environment. The major open questions that will spark new research in the near future include: the influence of natural organic chelators on the complexation and redox chemistry of metal ions, the role played by macromolecules and microorganisms, the existence of solid phases that control the solubility of radionuclides, and how to perform laboratory experiments that effectively reproduce the behavior of these elements at environmentally relevant (millennial) timescales and physicochemical conditions. Another phenomenon still largely not understood is the formation and/or accumulation of new species (radicals, degradation products, soluble or solid) in the environment induced by the radiation field of nuclear waste (that emit varied radiation: α, β, γ, neutrons, X-rays, etc.) and over a long period of time. Environmental radiochemists will also have to consider the impact of climate change on contaminated field sites, non-nuclear anthropogenic activities, and policy decisions regarding the design of nuclear waste repositories. We hypothesize that these questions can only be answered by stimulating progress in new spectroscopic techniques for radioactive samples, by applying new data science approaches to these complex environmental systems, by encouraging multidisciplinary collaborations, and continuing to motivate the next generation of radiochemists.
In 2021, the Anthropocene Working Group (AWG) plans to submit a proposal to formally adopt a new epoch in geologic time to the International Commission on Stratigraphy, which oversees the official geologic time chart19. Although controversial, the AWG is considering ending the Holocene and designating the new “Anthropoene” epoch that coincides with the start of the atomic age, using the world-wide radionuclide signature from the mid-1950s that signals how significantly humans have altered the planet. If adopted, we are, at present, embarking on a new epoch in geologic time that is marked by the presence of anthropogenic long-lived radionuclides. Understanding the impact of anthropogenic radionuclides at geologic timescales will be central to the definition of this new epoch.
References
Kersting, A. B. & Zavarin, M. in Actinide Nanoparticle Research (eds. Kalmykov, S. N. & Denecke, M. A.) 399–412 (Springer, 2011).
Geckeis, H., Zavarin, M., Salbu, B., Lind, O. C. & Skipperud, L. in Plutonium Handbook (American Nuclear Society, 2019).
Romanchuk, A. Y., Vlasova, I. E. & Kalmykov, S. N. Speciation of uranium and plutonium from nuclear legacy sites to the environment: a mini review. Front. Chem. 8, https://doi.org/10.3389/fchem.2020.00630 (2020).
Kersting, A. B. et al. Migration of plutonium in ground water at the Nevada Test Site. Nature 397, 56–59 (1999).
Novikov, A. P. et al. Colloid transport of plutonium in the far-field of the Mayak Production Association, Russia. Science 314, 638–641 (2006).
Kurihara, E. et al. Particulate plutonium released from the Fukushima Daiichi meltdowns. Sci. Total Environ. 743, 140539 (2020).
Morrison, K. D., Jiao, Y., Kersting, A. B. & Zavarin, M. Reduction of plutonium(VI) to (V) by hydroxamate compounds at environmentally relevant pH. Environ. Sci. Technol. 52, 6448–6456 (2018).
Deblonde, G. J.-P. et al. Chelation and stabilization of berkelium in oxidation state +IV. Nat. Chem. 9, 843–849 (2017).
Piro, N. A., Robinson, J. R., Walsh, P. J. & Schelter, E. J. The electrochemical behavior of cerium(III/IV) complexes: Thermodynamics, kinetics and applications in synthesis. Coord. Chem. Rev. 260, 21–36 (2014).
Pol, A. et al. Rare earth metals are essential for methanotrophic life in volcanic mudpots. Environ. Microbiol. 16, 255–264 (2014).
Cotruvo, J. A. Jr, Featherston, E. R., Mattocks, J. A., Ho, J. V. & Laremore, T. N. Lanmodulin: a highly selective lanthanide-binding protein from a lanthanide-utilizing bacterium. J. Am. Chem. Soc. 140, 15056–15061 (2018).
Deblonde, G. J.-P. et al. Selective and efficient biomacromolecular extraction of rare-earth elements using lanmodulin. Inorg. Chem. 59, 11855–11867 (2020).
Sperling, J. M. et al. Compression of curium pyrrolidine-dithiocarbamate enhances covalency. Nature 583, 396–399 (2020).
Nisbet, H. et al. Challenging the thorium-immobility paradigm. Sci. Rep. 9, 1–6 (2019).
Zavarin, M. et al. Hydrothermal alteration of nuclear melt glass, colloid formation, and plutonium mobilization at the Nevada National Security Site, U.S.A. Environ. Sci. Technol. 53, 7363–7370 (2019).
Kvashnina, K. O. et al. A novel metastable pentavalent plutonium solid phase on the pathway from aqueous plutonium(VI) to PuO2 nanoparticles. Angew. Chem. Int. Ed. 58, 17558–17562 (2019).
Matyskin, A. V., Ylmen, R., Lagerkvist, P., Ramebäck, H. & Ekberg, C. Crystal structure of radium sulfate: an X-ray powder diffraction and density functional theory study. J. Solid State Chem. 253, 15–20 (2017).
Matyskin, A. V., Brown, P. L. & Ekberg, C. Weak barium and radium hydrolysis using an ion exchange method and its uncertainty assessment. J. Chem. Thermodyn. 128, 362–371 (2019).
Subramanian, M. Anthropocene now: influential panel votes to recognize Earth’s new epoch. Nature https://doi.org/10.1038/d41586-019-01641-5 (2019).
Acknowledgements
Lawrence Livermore National Laboratory is operated by Lawrence Livermore National Security, LLC, for the US Department of Energy, National Nuclear Security Administration under Contract DE-AC52-07NA27344 (LLNL-JRNL-815005).
Author information
Authors and Affiliations
Contributions
G.J.-P.D., A.B.K., and M.Z. carried out literature search, discussed ideas, and contributed to the writing of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Deblonde, G.JP., Kersting, A.B. & Zavarin, M. Open questions on the environmental chemistry of radionuclides. Commun Chem 3, 167 (2020). https://doi.org/10.1038/s42004-020-00418-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42004-020-00418-6
This article is cited by
-
Sources, seasonal cycling, and fate of plutonium in a seasonally stratified and radiologically contaminated pond
Scientific Reports (2023)
-
Effect of Si content, pH, electrolyte and fulvic acid on the stability of Th(IV)-silicate colloids
Journal of Radioanalytical and Nuclear Chemistry (2023)
