
Abstract
The broad presence of azaarene moieties in natural products has promoted the development of new functionalization reactions, giving access to larger libraries of bioactive compounds. The light promoted [2 + 2] photocycloaddition reaction to generate cyclobutanes has been extensively studied in photochemistry. In particular, De Mayo reported the [2 + 2] cycloaddition followed by retroaldol condensation between enols of 1,3-dicarbonyls and double bonds to synthesize 1,5-dicarbonyls. Herein, we describe the [2 + 2] photocycloaddition followed by a ring-opening rearomatization reaction between electron-deficient 2-methylene-azaarenes and double bonds, taking advantage of the ability of these heterocyclic derivatives to form the corresponding pseudo-enamine intermediate. The procedure shows a high functional group tolerance either on the double bond or the heteroarene side and allows the presence of different electron-withdrawing groups. In addition, the wide applicability of this reaction has been demonstrated through the late-stage derivatization of several natural products. Photochemical studies, together with theoretical calculations, support a mechanism involving the photosensitization of the pseudo-enamine intermediate.
Similar content being viewed by others

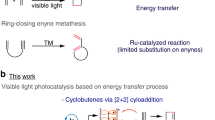
Introduction
Azaarene moieties such as the quinoxaline, pyrazine, quinoline or isoquinoline structures are a ubiquitous theme in natural products with interesting biological properties (Fig. 1)1. These scaffolds are present in a large number of pharmaceutical compounds, such as Timapiprant (an anti-asthmatic and anti-inflammatory agent)2, Papaverine (a smooth muscle relaxant)3,4, Emetine5,6 (an anti-protozoan) and the quinoline derivative I (an atherosclerosis treatment)7. Other azaarenes such as quinoxaline derivatives II with anti-inflammatory properties, and pyrazines such as Terezine A with antifungal characteristics, are also prevalent biological compounds8,9 (Fig. 1). In view of the wide application of electron-deficient azaarenes in drug discovery, the development of new methods to functionalize these structures are of considerable interest, affording new procedures to introduce such core in the skeleton of bioactive molecules and opening the door to the development of new libraries of bioactive compounds.

Compounds bearing different azaarene moieties with interesting properties.
The [2 + 2] photocycloaddition of double bonds to generate cyclobutanes is one of the most employed reactions in photochemical synthesis10,11,12,13,14,15. The inherent strain of cyclobutanes makes them prone to undergo ring-opening reactions. In this context, de Mayo reported that, under UV irradiation, the enol of 1,3-dicarbonyl compounds involved in a [2 + 2] photocycloaddition with olefins to form a non-isolable cyclobutanol intermediate, which evolved to afford 1,5-diketones (X = O, EWG = COR) after retro-aldol condensation (Fig. 2a)16,17. The synthetic potential of this reaction was fully demonstrated in the rapid formation of complex molecular structures and natural product synthesis18,19,20,21,22,23,24. More recently, a visible light-mediated photocatalytic version of the reaction has been reported via photosensitization of styrene using the [Ir{dF(CF3)2ppy}2(bpy)]PF6 complex25. This approach solved the selectivity problem associated with undesired secondary reactions linked to the use of UV-light as the irradiation source. Regarding the scope of both the classical de Mayo photochemical reaction26 and the visible-light photocatalytic approach, the common restraint is that the first [2 + 2] cycloaddition step only takes place with enols or preformed enamines, thus limiting the scope of substrates that can participate in this reactivity.

a De Mayo reaction, b This work: [2 + 2] cycloaddition & ring-opening rearomatization.
We envisioned that other pseudo-enamine species with a heteroarene core in the structure could undergo a [2 + 2] cycloaddition in the presence of an olefin, thus giving access to the derivatization of compounds with pharmacological or biological interest (see imine-enamine equilibrium, Fig. 2b). Therefore, in this work we present the [2 + 2] cycloaddition reaction between β-electron withdrawing substituted azaarene derivatives and different double bonds followed by a ring-opening rearomatization of the cyclobutane intermediate. DFT calculations together with fluorescence quenching studies provide evidences of which species play a key role in the mechanistic proposal.
Results and discussion
Optimization of the model reaction
Taking into account that quinolines are electron-deficient heteroarenes and can form pseudo-enamine species, we chose 1a as model substrate for the [2 + 2] cycloaddition reaction. Therefore we studied the reaction between the quinoline derivative 1a and styrene 2a in the presence of the iridium complex 3a (ET = 62 kcal mol−1)27 as photocatalyst in CH3CN and irradiating the mixture with blue LED (455 nm) for 17 h (entry 1, Table 1). To our delight, the product 4a was obtained with a 38% yield, thus corroborating the initial hypothesis and opening the door to the development of this transformation with electron-deficient azaarenes. As expected, the presence of DBU as the base enhanced the reactivity increasing the formation of 4a to an 83% yield (entry 2, Table 1). Other photocatalysts such us Ru(bpy)3Cl2 (3b, ET = 46 kcal mol−1)28 or the Fukuzumi´s catalyst (3e, ET = 44 kcal mol−1)29 with lower triplet energies did not show any reactivity, while Ir(ppy)3 (3c, ET = 55 kcal mol−1)30 or 4CzIPN (3d, ET = 60 kcal mol−1)31 afforded the final product although in a lower yield than 3a (entries 3–6, Table 1). Carrying out the reaction in THF, 4a was isolated with a 98% yield (entry 7, Table 1). It should be highlighted that the reaction proceeded in a regioselective manner since the other plausible product coming from the enolization of the ester [CH2-CO2Me → CH = C(OH)OMe] was not observed (See Supplementary Note 5). In addition, the photocatalytic nature of the reaction was confirmed when the experiment was performed without light or a photocatalyst, without conversion for both cases (entries 8 and 9, Table 1).
Substrate scope
With the optimized conditions established (entry 7, Table 1), a variety of heteroarenes, different EWGs and double bonds were examined in the reaction (Fig. 3). The presence of halogens in different positions of the quinoline ring did not affect the reactivity (4b-4d), and excellent yields were obtained in all cases. The methodology also allowed the presence of electron-withdrawing groups (CO2H, 1e) or electron-donating substituents at the aryl moiety (MeO, 1f). Next, other electron-withdrawing substituents in the β-position to the azaarene were studied (EWGs row, Fig. 3), obtaining good to excellent results for the quinoline derivatives with amide (4g), nitrile (4h), sulfone (4i) and arylketone (4j). Moreover, even in the case of the more enolizable arylketone 4j, we observed exclusively one regioisomer. Furthermore, the reaction is not exclusive for 2-substituted quinolines but 4-substituted quinolines, isoquinolines, quinoxalines, and pyrazines also underwent this reactivity, affording the final products 4k-4o in moderate to good yields (azaarenes row).

aReaction conditions: All the reactions were carried out with 0.1 mmol 1, 0.5 mmol 2, under N2, 25 °C, 455 nm LED irradiation (22 W m−2), over 17 h and the following conditions: A: 0.002 mmol Ir[dFCF3ppy]2(bpy)PF6 3a, 0.05 mmol DBU, THF (1 mL), except in the cases indicated with conditions B: 0.002 mmol Ir(ppy)3 3c, 0.05 mmol DBU, THF (1 mL) or conditions C: 0.002 mmol Ir(ppy)3 3c, 0.05 mmol K2CO3, CH3CN (1 mL); bReaction carried out over 30 h. c4 s was isolated as an inseparable mixture 5.2:1 of the adduct and the Michael addition product. dLower yield is obtained due to the formation of the competing Michael adduct.
Later, the reaction between the quinoline 1a and a variety of styrene derivatives 2 was studied (olefin row, Fig. 3). The presence of electron-donating substituents (4p-4r) and electron-withdrawing ones (4s) did not have a large influence on the final results, whereas the use of halogens (4t, 4u) and ortho substituents (4u and 4v) were also compatible. Pleasantly, the reaction also allowed the synthesis of quaternary centres, starting from the α-methyl styrene and giving the 4w quinoline derivative with a 70% yield. In addition, when β-methyl styrene derivatives were used, two new chiral centres were obtained with a complete diastereoselectivity for both cases (anti-4x, and anti-4y). The 2,3-dimethylbuta-1,3-diene previously employed in photocatalytic [2 + 2] cycloadditions32, afforded the synthesis of the dialkylic quaternary centre product 4z with a 70% yield. The reaction also took place with double bonds bearing electron-poor and electron-rich heteroarenes. Therefore, the reaction worked with the pyridine derivative (4aa) and the thiophene (4ab), with moderate to good yields. Interestingly, the indol derivative 4ac, which is present in the Timapiprant core (an oral CRTH2 antagonist which has been proved to be an anti-asthmatic)33 was isolated with a good yield. Some limitations were also observed such as the use of cyclohexene, free acidic protons at the aryl moiety in the double bond, or the use of stilbene (see limitations row).
In the field of drug discovery, late-stage functionalization (LSF) has become an excellent tool for the development of libraries of bioactive compounds, starting from lead structures and avoiding the de novo synthesis34. LSF facilitates the development of structure-activity relationships, the optimization of on-target potency and the improvement of the physical properties of the lead compounds. This is evidenced by the large number of methods for the functionalization of peptides35,36,37,38,39,40 or the number of methodologies that have been recently developed for the functionalization of complex bioactive molecules41,42,43,44,45,46.
Therefore, to prove the utility of our methodology, we introduced electron-deficient azaarenes in the structure of complex natural products in one step (Fig. 4). Thus, the hormone estrone was derivatized to 5a with an 85% yield, as well as the amino acid Tyrosine derivative, that afforded 5b with an excellent 89% yield. Finally, δ-Tocopherol (vitamin E), which is employed as a food preservative for its antioxidant properties, was transformed to 5c with an excellent 92% yield.

Reaction conditions: 0.1 mmol of 1a, 0.5 mmol of the corresponding styryl derivative, 0.5 equivalents of DBU and 1.0 mL of THF, under N2, 25 °C, 455 nm LED irradiation (22 W m−2) over 17 h. a Functionalization of Estrone derivative, b Functionalization of Tyrosine derivative, c Functionalization of Tocopherol derivative.
Mechanistic proposal
Elucidation of the reaction mechanism was further addressed (Fig. 5). Based on a previous report25, after photoexcitation of the photocatalyst 3 to its excited state 3*, energy transfer from 3* to 2a or the enamine 1a′ (that is almost isoenergetic to the quinoline 1a, see Supplementary Data 1) takes place to afford either 3(1a′)* or 3(2a)* (Fig. 5a). From the triplet excited state both 3(1a′)* or 3(2a)* undergo a [2 + 2] cycloaddition with 2a or 1a′ respectively, yielding a common intermediate I. With regard to the experimental triplet energies described in the literature (ET 2a = 60.8 kcal mol−1 47; ET 3a = 62.0 kcal mol−1 27; ET 3c = 57.6 kcal mol−1 30), styrene 2a can be photosensitized by 3a, but not by 3c, whose triplet energy is lower than the triplet energy of 2a. However, considering that the reaction with photocatalyst 3c afforded 4a with an 80% yield (entry 3, Table 1), photosensitization of 1a′ seems to be a probable mechanistic pathway.

a Mechanistic proposal, b Fluorescence quenching studies of 3a and 3c.
Fluorescence quenching studies of 3a and 3c are consistent with the previous data, finding an efficient interaction of both 1a′ and 2a with 3a*, while in the case of photocatalyst 3c, only the quinoline 1a′ was able to quench the excited state (see Fig. 5b and Supplementary Note 5). Therefore, our results indicate that the most probable mechanistic pathway is the photosensitization of 1a′.
Further mechanistic insights were obtained by performing DFT calculations at the M062X/6–311 G** level of theory including solvation effects using the SMD model (solvent CH3CN ε = 37.5) (Fig. 6 and Supplementary Table 1). According to the experimental results, the S0 → T1 gap, considering the free energy of optimized geometries in both singlet and triplet spin states, is significantly lower for 1a than for 2a (ET calculated 1a′ = 48.8 kcal mol−1; ET calculated 2a = 54.9 kcal mol−1) (Comparison between experimental and theoretical data for 2a indicates that our calculations slightly underestimate the energy values of S0→T1 gaps by approximately 10%). Therefore, once the photosensitization of 1a′ takes place, [2 + 2] cycloaddition between 3(1a′)* and 2a proceeds. The pathway found consisted of a stepwise addition that produces firstly the 1,4-biradical intermediate I, with the unpaired electrons in the most stable positions (benzylic and α to the N). This initial stage of the cycloaddition is both thermodynamically and kinetically very favourable (ΔG‡ = 7.2 kcal mol−1 and ΔG = −17.2 kcal mol−1, Fig. 6). Such a biradical was calculated as both a triplet spin state and an open shell singlet spin state, giving comparable thermodynamic stabilities (ΔG = 1.6 kcal mol−1).

DFT calculations of the reaction mechanism.
Exploration of the further evolution of the open-shell singlet spin state revealed that the intermediate I was not very stable because its evolution was even more favourable than its formation. Therefore, the process from I, in the open-shell spin state, to the cyclobutane intermediate II presents a kinetic barrier of ΔG‡ = 6.2 kcal mol−1 and its thermodynamics correspond to ΔG = −21.9 kcal mol−1. Under the reaction conditions, cyclobutane II was not experimentally observed. Instead, a ring-opening rearomatization from II accounts for the product 4a was found. Such a ring-opening process takes place through a kinetic barrier of 17.5 kcal mol−1, being the enolic product isoenergetic to intermediate II. However, the thermodynamics of this process is widely compensated by keto-enol tautomerism, with the keto form which is 26.6 kcal mol−1 more favourable. Owing to the kinetic barriers calculated, it is inferred that the cyclobutane opening is the actual rate-determining step. In order to corroborate the mechanistic proposal, we performed the reaction with styrene-D8, obtaining 4a-D with a CD-CD2 fragment (see top-right, Fig. 6).
Conclusions
In conclusion, we have developed a [2 + 2] cycloaddition and ring-opening rearomatization between electron-deficient methylene azaarenes and double bonds. The reaction gives good results with quinolines, isoquinolines, quinoxalines and pyrazines bearing different electron-withdrawing substituents in the β-position. The process is highly functional group tolerant, and it was performed efficiently with a variety of styrenes, bearing either electron donating or withdrawing groups, as well as heteroaryl or alkyl-substituted double bonds. Moreover, its applicability could be demonstrated by late-stage functionalization of different natural products. Finally, the fluorescence quenching studies together with the theoretical calculations supported a mechanism based on the photosensitization of the pseudo-enamine of the azaarene, instead of the alkene derivative.
Methods
General information
For more details, see Supplementary Methods.
Detailed optimization of the reaction conditions
For more details, see Supplementary Note 1 and Supplementary Table 1.
Synthesis and characterization
See Supplementary Notes 2–4 and Supplementary Figs 4–99 for NMR spectra.
Mechanistic investigation
For more details, see Supplementary Note 5 and Supplementary Data 1.
General procedures for the photocatalytic reaction
General procedure A: A dry vial equipped with a magnetic stir bar was charged with 3a (2.0 mg, 2 μmol, 0.02 equiv.), the corresponding azaarene derivative 1 (0.1 mmol, 1.0 equiv.), DBU (7.6 mg, 0.05 mmol, 0.5 equiv.), alkene 2 (0.5 mmol, 5.0 equiv.) and 1.0 mL of THF (0.1 M). Degasification of the reaction mixture was performed via freeze-pump-thaw cycling (3 × 10 min under vacuum). Then, the reaction mixture was irradiated and stirred in the photoreactor setup at 455 nm for 17 h (unless otherwise stated). The reaction mixture was concentrated under reduced pressure and purified by flash column chromatography (silica gel) to provide the product.
General procedure B: A dry vial equipped with a magnetic stir bar was charged with 3c (1.4 mg, 2 μmol, 0.02 equiv.), the corresponding azaarene derivative 1 (0.1 mmol, 1.0 equiv.), DBU (7.6 mg, 0.05 mmol, 0.5 equiv.), alkene 2 (0.5 mmol, 5.0 equiv.) and 1.0 mL of THF (0.1 M). Degasification of the reaction mixture was performed via freeze-pump-thaw cycling (3 × 10 min under vacuum). Then, the reaction mixture was irradiated and stirred in the photoreactor setup at 455 nm for 17 h. The reaction mixture was concentrated under reduced pressure and purified by flash column chromatography (silica gel) to provide the product.
General procedure C: A dry vial equipped with a magnetic stir bar was charged with 3c (1.4 mg, 2 μmol, 0.02 equiv.), the corresponding azaarene derivative 1 (0.1 mmol, 1.0 equiv.), K2CO3 (0.1 mmol, 1.0 equiv.), alkene 2 (0.5 mmol, 5.0 equiv.) and 1.0 mL of MeCN (0.1 M). Degasification of the reaction mixture was performed via freeze-pump-thaw cycling (3 × 10 min under vacuum). Then, the reaction mixture was irradiated and stirred in the photoreactor setup at 455 nm for 17 h. The reaction mixture was concentrated under reduced pressure and purified by flash column chromatography (silica gel) to provide the product.
Data availability
Supplementary Information and relevant data are also available from the authors.
References
Michael, J. P. Quinoline, quinazoline and acridonealkaloids. Nat. Prod. Rep. 25, 166–187 (2008).
Pettipher, R. et al. Heightened response of eosinophilic asthmatic patients to the CRTH2 antagonist OC000459. Allergy 69, 1223–1232 (2014).
Liu, J. K. & Couldwell, W. T. Intra-arterial papaverine infusions for the treatment of cerebral vasospasm induced by aneurysmal subarachnoid hemorrhage. Neurocrit. Care 2, 124–132 (2005).
Takeuchi, K., Sakamoto, S., Nagayoshi, Y., Nishizawa, H. & Matsubara, J. Reactivity of the human internal thoracic artery to vasodilators in coronary artery bypass grafting. Eur. J. Cardiothorac. Surg. 26, 956–959 (2004).
Pailer, M. & Porschinski, K. The constitution of emetine. IV Ipecac alkaliods. Monatsh. Chem. 80, 94–100 (1949).
Battersby, A. R. & Openshaw, H. T. Studies of the structure of emetine. Part IV. Elucidation of the structure of emetine. J. Chem. Soc. 3207–3213 (1949).
Kaila, N. et al. 2-(4-Chlorobenzyl)-3-hydroxy-7,8,9,10-tetrahydrobenzo[H]quinoline-4-carboxylic Acid (PSI-697): identification of a clinical candidate from the quinoline salicylic acid series of P-selectin antagonists. J. Med. Chem. 50, 40–64 (2007).
Badrinarayanan, S. & Sperry, J. Pyrazinealkaloids via dimerization of amino acid-derived α-amino aldehydes: biomimetic synthesis of 2,5-diisopropylpyrazine, 2,5-bis(3-indolylmethyl)pyrazine and actinopolymorphol C. Org. Biomol. Chem. 10, 2126 (2012).
Smits, R. A. et al. Fragment based design of new H4 receptor-ligands with anti-inflammatory properties in vivo. J. Med. Chem. 51, 2457–2467 (2008).
Schuster, D. I., Lem, G. & Kaprinidis, N. A. New insights into an old mechanism: [2 + 2] photocycloaddition of enones to alkenes. Chem. Rev. 93, 3–22 (1993).
Poplata, S., Tröster, A., Zou, Y.-Q. & Bach, T. Recent advances in the synthesis of cyclobutanes by olefin [2 + 2] photocycloaddition reactions. Chem. Rev. 116, 9748–9815 (2016).
Huang, X. et al. Meggers, E. Direct Visible-Light-Excited Asymmetric Lewis Acid Catalysis of Intermolecular [2 + 2] Photocycloadditions. J. Am. Chem. Soc. 139, 9120–9123 (2017).
Hu, N. et al. Meggers, E. Catalytic Asymmetric Dearomatization by Visible‐Light‐Activated [2 + 2] Photocycloaddition. Angew. Chem. Int. Ed. 57, 6242–6246 (2018).
Skubi, K. L. et al. Enantioselective excited-state photoreactions controlled by a chiral hydrogen-bonding iridium sensitizer. J. Am. Chem. Soc. 139, 17186–17192 (2017).
Zheng, J. et al. Enantioselective intermolecular excited-state photoreactions using a chiral ir triplet sensitizer: separating association from energy transfer in asymmetric photocatalysis. J. Am. Chem. Soc. 141, 13625–13634 (2019).
De Mayo, P., Takeshita, H. & Sattar, A. B. M. A. The photochemical synthesis of 1,5-diketones and their cyclisation: a new annulation process. Proc. Chem. Soc. 119 (1962).
De Mayo, P. & Takeshita, H. Photochemical Synthesis: 6. The formation of heptandiones from acetylacetone and alkenes. Can. J. Chem. 41, 440 (1963).
Begley, M. J., Mellor, M. & Pattenden, G. New synthetic approaches to fused-ring carbocycles based on intramolecular photocycloadditions of 1,3-dione enol esters. J. Chem. Soc., Perkin Trans. 1, 1905–1912 (1983).
Oppolzer, W. The intramolecular [2 + 2] photoaddition/cyclobutane-fragmentation sequence in organic synthesis. Acc. Chem. Res. 15, 135–141 (1982).
Crimmins, M. T. Synthetic applications of intramolecular enone-olefin photocycloadditions. Chem. Rev. 88, 1453–1473 (1988).
Winkler, J. D., Bowen, C. M. & Liotta, F. [2 + 2] Photocycloaddition/fragmentation strategies for the synthesis of natural and unnatural products. Chem. Rev. 95, 2003–2020 (1995).
Winkler, D., Rouse, M. B., Greaney, M. F., Harrison, S. J. & Jeon, Y. T. The first total synthesis of (±)-ingenol. J. Am. Chem. Soc. 124, 9726–9728 (2002).
Winkler, J. D., Muller, C. L. & Scott, R. D. A new method for the formation of nitrogen-containing ring systems via the intramolecular photocycloaddition of vinylogous amides. A synthesis of mesembrine. J. Am. Chem. Soc. 110, 4831–4834 (1988).
Winkler, J. D., Scott, R. D. & Williard, P. G. Asymmetric induction in the vinylogous amide photocycloaddition reaction. A formal synthesis of vindorosine. J. Am. Chem. Soc. 112, 8971–8975 (1990).
Martinez-Haya, R., Marzo, L. & König, B. Reinventing the De Mayo reaction: synthesis of 1,5-diketones or 1,5-ketoesters via visible light [2 + 2] cycloaddition of β-diketones or β-ketoesters with styrenes. Chem. Commun. 54, 11602–11605 (2018).
Li, J. Name Reactions for Carbocyclic Ring Formations (John Wiley & Sons, Inc., New York, 2010).
Hanss, D., Freys, J. C., Bernardinelli, G. & Wenger, O. S. Cyclometalated iridium(III) complexes as photosensitizers for long‐range electron transfer: occurrence of a coulomb barrier. Eur. J. Inorg. Chem. 32, 4850–4859 (2009).
Prier, C. K., Rankic, D. A. & MacMillan, D. W. C. Chem. Rev. 113, 5322–5363 (2013).
Romero, N. A. & Nicewicz, D. A. Organic photoredox catalysis. Chem. Rev. 116, 10075–10166 (2016).
Flamigni, L., Barbieri, A., Sabatini, C., Ventura, B. & Barigel-leti, F. Photochemistry and photophysics of coordination compounds: iridium. Top. Curr. Chem. 281, 143–203 (2007).
Luo, J. & Zhang, J. Donor–acceptor fluorophores for visible-light-promoted organic synthesis: photoredox/Ni dual catalytic C(sp3)–C(sp2) cross-coupling. ACS Catal. 6, 873–877 (2016).
Blum, T. R., Miller, Z. D., Bates, D. M., Guzei, I. A. & Yoon, T. P. Enantioselective photochemistry through Lewis acid–catalyzed triplet energy transfer. Science 354, 1391–1395 (2016).
Yanes, D. A. & Mosser-Goldfarb, J. L. Emerging therapies for atopic dermatitis: the prostaglandin/leukotriene pathway. J. Am. Acad. Dermatol. 78, S71–S75 (2018).
Cernak, T., Dykstra, K. D., Tyagarajan, S., Vachalb, P. & Krska, S. W. The medicinal chemist’s toolbox for late stage functionalization of drug-like molecules. Chem. Soc. Rev. 45, 546–576 (2016).
Witt, K. A., Gillespie, T. J., Huber, J. D., Egleton, R. D. & Davis, T. P. Peptide drug modifications to enhance bioavailability and blood-brain barrier permeability. Peptides 22, 2329–2343 (2001).
Adessi, C. & Soto, C. Converting a peptide into a drug: strategies to improve stability and bioavailability. Curr. Med. Chem. 9, 963–978 (2002).
Biron, E. et al. Improving oral bioavailability of peptides by multiple N‐methylation: somatostatin analogues. Angew. Chem. Int. Ed. 47, 2595–2599 (2008).
Vorherr, T. Modifying peptides to enhance permeability. Future Med. Chem. 7, 1009–1021 (2015).
Wang, W., Lorion, M. M., Shah, J., Kapdi, A. R. & Ackermann, L. Late‐stage peptide diversification by position‐selective C−H activation. Angew. Chem. Int. Ed. 57, 14700–14717 (2018).
Leroux, M. et al. Late‐stage functionalization of peptides and cyclopeptides using organozinc reagents. Angew. Chem. Int. Ed. 58, 8231–8234 (2019).
Ye, F. et al. Aryl sulfonium salts for site‐selective late‐stage trifluoromethylation. Angew. Chem. Int. Ed. 131, 14615–14619 (2019).
Fujiwara, Y. & Baran, P. S. in New Horizons of Process Chemistry (Eds Tomioka, K., Shioiri, T. & Sajiki, H.) (Springer, Singapore, 2017).
Lei, Z. et al. β‐Selective aroylation of activated alkenes by photoredox catalysis. Angew. Chem. Int. Ed. 58, 7318–7323 (2019).
Lv, L., Zhu, D. & Li, C.-J. Direct dehydrogenative alkyl Heck-couplings of vinylarenes with umpolung aldehydes catalyzed by nickel. Nat. Commun. 10, 715 (2019).
Fier, P. S., Kim, S. & Maloney, K. M. Reductive cleavage of secondary sulfonamides: converting terminal functional groups into versatile synthetic handles. J. Am. Chem. Soc. 141, 18416–18420 (2019).
Mondal, A., Chen, H., Flämig, L., Wedi, P. & van Gemmeren, M. Sterically controlled late-stage C–H alkynylation of arenes. J. Am. Chem. Soc. 141, 18662–18667 (2019).
Ni, T., Cadwell, R. A. & Melton, L. A. The relaxed and spectroscopic energies of olefin triplets. J. Am. Chem. Soc. 111, 457–464 (2019).
Acknowledgements
We acknowledge the financial support from the Spanish Government (RTI2018–095038-B-I00), CAM_UAM (SI1/PJI/2019-00237), CCC-UAM (computing time), and ERC (ERC-CG, 647550). L.M. wishes to thank CAM for the ‘Atracción de Talento’ fellowship. N.S. wishes to thank ‘Ministerio de Ciencia e Innovación’ for a FPU predoctoral fellowship. The authors also wish to thank the ‘Comunidad de Madrid’ and European Structural Funds for their financial support to FotoArt-CM project (S2018/NMT-4367).
Author information
Authors and Affiliations
Contributions
N.S. carried out the optimization and scope of the reaction and, in collaboration with L.M., carried out the mechanistic investigation. R.M-B. and L.M. carried out the DFT-computational studies. L.M. and J.A. conceived the project and prepared the manuscript that was edited by all other authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Salaverri, N., Mas-Ballesté, R., Marzo, L. et al. Visible light mediated photocatalytic [2 + 2] cycloaddition/ring-opening rearomatization cascade of electron-deficient azaarenes and vinylarenes. Commun Chem 3, 132 (2020). https://doi.org/10.1038/s42004-020-00378-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42004-020-00378-x
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
