
Abstract
Nanoscale open spaces formed by partial overlap of two-dimensional nanosheets in clays, abundantly and ubiquitously available, possess reactive molecular sites such as nanosheet edges in their interior. Here, the capture and storage of CO2 molecules in open spaces within saponite clay are explored by solid-state nuclear magnetic resonance coupled with open space analysis using positronium. CO2 physisorption occurs on the nanosheet surfaces inside the open spaces under ambient conditions. Thereby, CO2 molecules are activated by picking off weakly-bound oxygen from octahedral sites at the nanosheet edges and carbonate species are stabilized on the nanosheet surfaces. This instantaneous mineral carbonation and CO2 physisorption occurs in the absence of an energy-consumption process or chemical solution enhancement. This finding is of potential significance for CO2 capture and storage and presents an approach of environmentally friendly recycling of low contaminated soil in Fukushima.
Similar content being viewed by others
Introduction
Owing to the significant climate change caused by the steady increase of CO2 in the atmosphere along with industrial activity1, the technology of CO2 capture and storage with respect to less-energy intensiveness, cost effectiveness, and environmental friendliness has been long-awaited. In power plants, electricity is generated by burning fossil fuels such as coal and natural gas, which could be a large point source yielding ~26% of global CO2 emission through the combustion process2. An approach to reduce CO2 emission is the efficient capture of CO2 and its storage in a stable manner before release to the atmosphere. There are three pathways for CO2 capture: pre-combustion capture, oxyfuel combustion, and post-combustion capture3. Pre-combustion capture is the decarbonation process before combustion, in which primary fuels are converted into a mixture of hydrogen and CO2 using gasification or reforming. In oxyfuel combustion, fuels are burned with an oxygen-enriched gas mixture instead of air, resulting in flue gases that mainly comprise CO2 and H2O. Post-combustion capture removes diluted CO2 from the flue gases produced by the combustion of fuel in the air, which could be applied to most conventional power plants owing to its adaptable operation.
Post-combustion technology for CO2 capture is based on the fundamental process of mainly chemical and physical absorption with absorbents of high CO2 solubility, and physical adsorption employing solid sorbents3. Chemical absorption is the CO2 capture process involving the reaction of CO2 with chemical solvents in aqueous solution, whereas CO2 is absorbed into solvents by applying pressure to promote physical absorption. In the standard environment of power plants, flue gases contain 12–14 vol.% of CO2 and are emitted under atmospheric conditions, therefore, they require treatment at elevated pressure to promote CO2 removal4. Physical adsorption utilizes the physisorption of CO2 molecules onto the surface of an adsorbent via the quadrupole interaction in addition to the size effect, thus requiring microporous materials with a high specific surface area5. At present, physical adsorption of CO2 is still in the early research stage together with the subject as to the thermal and chemical stability of the sorbed CO2, mechanical strength as well as production cost.
Another important factor of CO2 capture and storage is the long-term sequestration of CO2, where CO2 storage in geological reservoirs has been often considered6. In this approach, CO2 is injected into a deep geological formation to be physically confined below an impermeable or very low permeability caprock, such as a shale, allowing for a sequence of possible trapping mechanisms7. A fraction of injected CO2 is fixed as thermodynamically stable mineral carbonates, as e.g., CaCO3 or MgCO3 in the geological formation via the reaction with alkaline minerals there8. The formation of stable carbonates in the deep underground geology, known as in situ mineral carbonation9, could be suitable for long-term storage of CO210,11. On the contrary, ex situ mineral carbonation is the above-ground process involving the reaction of CO2 with alkaline earth metals extracted from basic rock12. Ex situ carbonation is generally conducted using acidic solutions at high temperature/pressure to accelerate the reaction between CO2 and materials, resulting in an energy-intensive process that generates a number of liquid wastes12,13.
Saponite, a silicate clay mineral abundantly and ubiquitously available in nature, is structured through stacks of 2D nanosheets with thicknesses of a few nm, which are the minimum structural unit. The 2D nanosheets have a variety of sizes and cannot be perfectly stacked, resulting in partial overlapping as schematically illustrated in Fig. 1a, which has been observed in images of field-emission type scanning electron microscopy14. This results in the formation of nanoscale open spaces (see Fig. 1b), which have been identified by positronium (Ps) annihilation spectroscopy together with molecular dynamics simulation as is detailed later15,16,17. Naturally, there exist local molecular sites in the interior of above open spaces such as nanosheet edges that are chemically active owing to the presence of unpaired electrons at ionically bound octahedron18. In the present work, CO2 adsorption in the open spaces originated from overlapped nanosheets in saponite clay minerals is explored by solid-state nuclear magnetic resonance (NMR) spectroscopy, open space analysis using positronium (Ps), and Fourier transform infrared (FT-IR) spectroscopy coupled with conventional chemical techniques. Besides the Na-type saponite, the Cs type is studied to find out an approach of environment-friendly recycling for low contaminated soil in Fukushima. The emergence mechanism of instantaneous mineral carbonation together with the physisorption of CO2 gas molecules, feasible in the absence of energy-consumption process as well as chemical solution enhancement, is highlighted as a future strategy of CO2 capture and storage.

a Partial overlap viewed from the direction perpendicular to the surface of 2D nanosheets marked by solid squares. b Cross-section view. Note that the nanoscale open space is locally formed as marked by a solid circle. Red, gray, and green atoms correspond to oxygen, carbon, and magnesium, respectively.
Results
CO2 adsorption on 2D nanosheet surfaces
Chemical analysis by inductively coupled plasma (ICP) spectroscopy indicated that the Cs-type saponite contained ~0.04 mmol/g Cs (see ccesium in Table 1). According to a recently developed analytical method using the data of elution test, 133Cs magic-angle spinning (MAS) NMR, and radiocesium interception potential, the local molecular structures, as e.g., nanosheet surface, nanosheet edge, and oncoming hexagonal cavity, have been shown to act as Cs adsorption sites18,19. In this analysis, 69% of the loaded Cs are found to physisorb on the surface of the 2D nanosheet, which amounts to a concentration of ~0.03 mmol/g. The carbon, hydrogen, and nitrogen (CHN) elemental analysis revealed that the C contents, ccarbon, in the Na- and Cs-type saponite samples after CO2 loading are ~0.02 and ~0.18 wt.%, respectively (see Table 1). In light of the fact that the samples are isolated from the air during CO2 loading, the C contents detected in the CHN analysis are solely associated with CO2. Therefore, CO2 molecules are sorbed to the Na- and Cs-type samples at concentrations of ~0.02 and ~0.15 mmol/g, respectively (see cCO2 in Table 1). The concentration of CO2 higher than that of Cs on the nanosheet surface implies the adsorption of several CO2 molecules at the Cs cation sites, which will be discussed more in detail later.
Ps lifetime spectroscopy prior to CO2-loading reveals two kinds of open spaces for both the Na- and Cs-type saponite samples. The similar sizes of small and large open spaces with R1 ~ 3 Å and R2 ~ 9 Å are obtained for the Na- and Cs-type samples (see Table 2). Our former studies revealed that the above two open spaces commonly observed for both the Na- and Cs-type saponite samples are caused by overlapped nanosheets: the small and large open spaces are the consequence of one- and two-nanosheet insertion into the interlayer spaces15. The relative intensity of large open space I2 for the Cs-type sample is ~25%, much higher than that of the Na-type sample, though the intensities I1 of small open spaces at 5% are similar to each other. The high intensity I2 for the Cs-type sample indicating the large amount of 9 Å open space is resultant from insufficient self-assembly toward densification, which originates from interlayer Cs cations with low hydration degree16,17.
Figure 2 shows the 133Cs MAS NMR spectra obtained for the Cs-type saponite before (black curve) and after CO2 loading (red curve), and their difference spectrum (blue curve). The dominant signal at ~−134 ppm observed for the sample prior to CO2 loading correspond to the Cs+ interlayer cations physisorbed on the surfaces of the 2D nanosheets, whereas the additional broad signal at ~−15 ppm originates from Cs2O compounds at nanosheet edges18,19,20. The dominant peak arising from the Cs+ interlayer cations is slightly shifted and becomes broader upon CO2 loading, which can be seen in the difference spectrum as well. This demonstrates that the CO2 molecules adsorb at the Cs+ cation sites on the nanosheet surfaces at a concentration of ~0.03 mmol/g, as estimated above.

133Cs MAS NMR spectra obtained for the Cs-type saponite before (black curve) and after (red curve) CO2 loading, and their difference spectrum (blue curve). The inset is schematic illustration of Cs+ cations (purple) located on the surface of tetrahedral sheet consisting of oxygen (red) and carbon (gray) atoms.
Physisorption and chemisorption of CO2 molecules
Figure 3a shows the 13C MAS NMR spectra obtained for (I) Cs-type saponite 13C-enriched CO2 (13CO2) unloaded, (II) Na-type saponite 13CO2 loaded, (III) Cs-type saponite 13CO2 loaded, and (IV) Cs-type saponite 13CO2 loaded and subsequently heat treated at 200 °C for 2 h in the N2 atmosphere. Before loading 13CO2, no signal is observed in the 13C MAS NMR spectrum of the unloaded Cs-type saponite. Similarly to that, no signal appears in the NMR spectrum of unloaded Na-type saponite (data not shown here). Upon 13CO2 loading, an intense peak and broad hump arising from 13CO2 adsorption appeared at around the chemical shifts of 125 and 170 ppm (see (II) in Fig. 3a). This together with the above result of 133Cs MAS NMR indicates that CO2 adsorption occurs at Na+ cation sites on the surface of the 2D nanosheet. The dominant and additional low-field signals become intense for the Cs-type saponite (see (III) in Fig. 3a), providing the important information that the Cs-type saponite captures CO2 molecules more efficiently than that in the Na-type one. After heat treatment at 200 °C for 2 h, the dominant peak disappeared, whereas the broad hump at ~170 ppm remained (see (IV) in Fig. 2a), demonstrating that the intense and small signals are ascribed to physisorption (dominant) and chemisorption (secondary) on the nanosheet surfaces, respectively. It is reasonably inferred from the study of a ternary catalyst composed of copper, zinc oxide, and alumina21 that the broad signals at ~170 ppm originate from carbonate species, such as Cs2CO3.

a 13C MAS NMR spectra obtained for (I) Cs-type saponite 13CO2 unloaded, (II) Na-type saponite 13CO2 loaded, (III) Cs-type saponite 13CO2 loaded, and (IV) Cs-type saponite 13CO2 loaded and then heat treated at 200 °C for 2 h in the N2 atmosphere. b Schematic illustration of CO2 physisorption and optimized structure of Cs2CO3 as chemisorption on the nanosheet surface. Red, gray, green, and purple atoms correspond to oxygen, carbon, magnesium, and cesium, respectively.
It is of interest that both the physisorption and chemisorption for CO2 molecules occur at the alkali metal cations located on the surface of the 2D nanosheet. It is most probable that the CO2 physisorption on the nanosheet surface is caused by the quadrupole interaction between the alkali metal cations and CO2 molecules. Molecular orbital calculation, on the one hand, predicts the resultant carbonate species Cs2CO3 from CO2 chemisorption, in which the CO2 molecule stabilizes by bridging with two alkali cations on the nanosheet surface (Fig. 3b). The adsorption energy for the optimized structure calculated with the SCIGRESS program (Fujitsu Ltd. Japan) is −97.8 kcal/mol, which is similar to that of CO2 chemisorption onto the graphitic surface with two-bond conformation22. The concentrations of CO2 physisorption, cPhysCO2 and CO2 chemisorption, cChemiCO2 for the Na- and Cs-type samples obtained from the total amount of CO2 and the ratio of two peak intensities in 13C MAS NMR spectra are listed in Table 1. It is noted that ~13% of the loaded CO2 is instantaneously chemisorbed as the carbonate species in both samples without employing strong acid solution at ambient pressure and temperature. As the concentration of Cs+ cations physisorbed on the surface of the 2D nanosheet is ~0.03 mmol/g (see Table 1), the cesium carbonate formed with two Cs+ cations on the nanosheet surface has the concentration of ~0.02 mmol/g. This is consistent with the concentration of CO2 chemisorption, cChemiCO2 (see Table 1), signifying that Cs+ cations on the nanosheet surface dominantly take part in the CO2 chemisorption. On the other hand, the concentration of CO2 physisorption, cPhysCO2 is higher than that of Cs+ cations on the nanosheet surface. Presumably, a few CO2 molecules are physisorbed at Cs+ cations with the distance of several angstroms (see Fig. 3b).
Discussion
According to a number of earlier works on zeolite materials with CO2 gas, the polarizing power of exchangeable alkali cations is one of the decisive factors for the capacity of CO2 gas adsorption23. The polarizing power of cations is inversely proportional to the ionic radius of alkali metal cations. The capacity of CO2 adsorption owing to the cation-quadrupole interaction thus increases as follows: Cs+<Rb+<K+<Na+<Li+23. This is in sharp contrast with the present observation that the concentration of both the physisorption and chemisorption increase for the Cs-type saponite. It is noted here that the fraction of open space with the size of ~9 Å for the Cs-type saponite probed by Ps is much higher than that of the Na-type one (see relative intensity I2 in Table 2). The open space formed by nanosheet overlap for the Cs-type saponite offers large enough surface area to accommodate CO2 molecules, thus being responsible for both the physisorption and chemisorption for CO2 molecules.
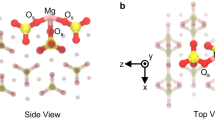
Here, we discuss why the chemisorption of CO2 gas molecules, i.e., ex situ mineral carbonation, instantaneously occurs in saponite samples without acid solution under the ambient condition. This unique adsorption nature is explained by the interior structure of the open nanospace characteristic for 2D materials. Saponite possess a 2:1 layered structure with 2D nanosheets consisting of tetrahedra and distorted octahedra. O and Si atoms are located at the vertices and central site of the tetrahedron, respectively. On the other hand, the O atoms or OH groups sit on the vertices of distorted octahedron, whereas a metallic Mg atom is located at the central site of octahedron (see the inset of Fig. 4). In the case of stevensite, the same family of saponite in aluminosilicate-type 2D materials, ca. 6.7% of the central Mg atoms in the octahedra are absent (see the inset of Fig. 4). An influence of Mg missing in the octahedron on the chemical bond is visible in the wavenumber region of 600–850 cm−1 in the FT-IR spectrum of stevensite. The FT-IR spectrum for the stevensite exhibits intense and small absorption peaks at the wavenumbers of around 650 and 775 cm−1, which can be ascribed to Si-O-Mg and Mg3OH bending vibrations, respectively24. The wavenumbers of the above bending vibration bands for the stevensite are lower than those of saponite, as the missing Mg atom causes lower frequencies in the both bending vibrations. It is noted here that the absorption peaks of Si-O-Mg and Mg3OH bending vibrations are red-shifted for CO2 loaded saponite as well implying atom missing in the octahedron (see Fig. 4). It is reasonably inferred that the above red-shift arise from the disappearance of O atoms from the vertices of the octahedra by the analogy of stevensite as illustrated in the inset of Fig. 4.

FT-IR spectra for CO2 unloaded (black curve) and loaded (red curve) saponite together with stevensite (blue curve). The absorption peaks of Si-O-Mg and Mg3OH bending vibrations are marked by black and gray thick arrows. The schematic illustrations of the disordered octahedra of stevensite, saponite, and CO2-loaded saponite are shown as the insets. Note that Mg and O sites are defective.
Based on the present findings of 133Cs and 13C MAS NMR and FT-IR spectroscopy, we can draw the scenario of instantaneous ex situ mineral carbonation as schematically illustrated in Fig. 5. In saponite, the O atoms are weakly bound to the octahedron by ionic bonding, in contrast to the strong semi-covalent bonding of O and Si atoms in the tetrahedron. CO2 gas molecules thus easily pick up the O atoms from the octahedra at the nanosheet edges in the interior of the above open spaces to be activated as the carbonate ions of CO32− type (see right-hand side of Fig. 5). The CO32− ions are then chemisorbed at the alkali metal cations on the surface of 2D nanosheets (see left-hand side of Fig. 5). The softness of the octahedra is also anticipated from the fact that the decomposition of octahedral sheets by mechanochemical milling proceeds prior to tetrahedral sheets25.

Scenario of instantaneous ex situ mineral carbonation under ambient conditions. Red, gray, green, and purple atoms correspond to oxygen, carbon, magnesium, and cesium, respectively.
In the process of physical adsorption utilizing the physisorption of CO2 molecules, a solid adsorbent is exposed to the combustion gas containing acid gasses such as sulfur dioxide. The central concern in the research of solid-adsorbent development is hence always a lifetime of adsorbent material relevant to thermal/chemical stability and mechanical strength in addition to the production cost. Clay minerals, such as saponite, have high tolerances for acids and their cheapness should be emphasized more than anything, thus compensating for the shortcomings of current adsorbent materials mentioned above. In light of the fact that the peak of CO2 physisorption in the 13C MAS NMR spectrum completely disappeared upon heat treatment (see Fig. 3), the open space formed by nanosheet overlap could again offer large enough surface area to accommodate CO2 molecules. It is thus expected that the ability of CO2 physisorption reversibly occurs when the sample is heated under mild conditions as 200 °C. In addition to that, the ability of CO2 chemisorption, i.e., ex situ mineral carbonation, instantaneously emerging without chemical solution under the ambient condition is highly beneficial over amine-based systems with respect to less-energy intensiveness, cost effectiveness, and environmental friendliness. As the nuclear accident in Fukushima, the large quantities of Cs-contaminated soil containing clays generated from decontamination activities have been stored throughout prefecture. The reusing and recycling of contaminated soil on the basis of criteria for both environmental and human impacts upon proper optimization is thus awaited in the near future. The Ministry of the Environment of Japan has promoted the recycling of decontamination soil with a radioactivity level below 8000 Bq/kg for specific construction purposes as, e.g., construction of road26. We believe that the present findings on the ability of CO2 adsorption in well-known clays could open up the recycling strategy of low contaminated soil containing clays with respect to the environment-friendly construction materials.
Methods
Materials
Synthetic Na-type saponite Na0.66[Mg5.34Li0.66]Si8O20[OH]4 produced by Kunimine Industries Co. Ltd., Japan, was employed in the present work. Cs loading was conducted by impregnating the Na-type saponite with a 1 M CsCl solution for ion exchanging with Cs. Aqueous solution was completely eliminated from the sample, which is referred to as Cs-type saponite. The chemical element in the Cs-type sample was examined by ICP spectroscopy (ICPS-8100, Shimadu). The sample was first treated at 423 K for 8 h under a vacuum condition of ~10–5 Torr in order to eliminate physisorbed H2O molecules. The dehydrated sample was successively replaced with 1-mbar CO2 atmosphere for CO2 loading without any contact to air and then kept there for 30 min, which was then analyzed for CHN using an elemental analyzer and 133Cs MAS NMR. In parallel to that, the dehydrated sample was exposed to a 13C-enriched CO2 (13CO2) atmosphere at 50-mbar for 30 min, which was subjected to 13C MAS NMR.
Positronium lifetime spectroscopy
The sizes of open nanospaces and their fractions were investigated by Ps annihilation lifetime spectroscopy employing digital oscilloscope (WaveSurfer 10, Teledyne LeCroy). The positron source (22Na), sealed in a thin foil of Kapton, was mounted in a sample-source-sample sandwich configuration. The 1.27 MeV positron birth γ ray from a 22Na source and one of the 511 keV γ rays emitted as a result of positron annihilation in the samples are detected by BaF2 scintillators with 1'' diameter × 1'' thickness coupled with photomultiplier tubes (H3378-51, HAMAMATSU). Positron lifetime spectra were numerically analyzed. A fraction of energetic positrons injected into samples forms the bound state with an electron, Ps. Singlet para-Ps (p-Ps) with the spins of the positron and electron antiparallel and triplet ortho-Ps (o-Ps) with parallel spins are formed at a ratio of 1:3. Hence, three states of positrons: p-Ps, o-Ps, and free positrons exist in samples. The annihilation of p-Ps results in the emission of two γ-ray photons of 511 keV with a lifetime ~125 ps. Free positrons are trapped by negatively charged parts, such as polar elements, and annihilated into two photons with a lifetime ~450 ps. The positron in o-Ps undergoes two-photon annihilation with one of the bound electrons with a lifetime of a few ns after localization in nanospaces. The last process is known as o-Ps pick-off annihilation and provides information on the free volume size R through its lifetime τo–Ps based on the Tao-Eldrup model27,28.
where R0 = R + ∆R, and ∆R = 0.166 nm is the thickness of homogeneous electron layer in which the positron in o-Ps annihilates. On the one hand, the relative intensity of τo–Ps is assumed to be correlated with the amount of open nanospaces.
Solid-state nuclear magnetic resonance
The 133Cs and 13C MAS NMR experiments were performed using a Bruker Avance III 400WB spectrometer at the resonance frequencies of 52.5 MHz and 100.6 MHz, respectively. 133Cs MAS NMR spectra were recorded with single-pulse excitation of 2.0 µs and the repetition time of 3 s. A sample spinning rate of 22 kHz was used and 24,000 scans were accumulated. The chemical shifts were referenced to a 1.0 M solution of CsCl. 13C MAS NMR measurements were performed via exciting the 13C spins with single-pulses of 2.0 µs and with a repetition time of 20 s thus avoiding relaxation effects by T1. The sample spinning rate was 8 kHz and 320 scans were collected for each spectrum. The chemical shifts of the 13C nuclei in the adsorbed organic species were determined with respect to tetramethylsilane as the external reference with an accuracy ±1 ppm.
FT-IR spectroscopy
Attenuated total reflection (ATR) FT-IR spectra were measured using a Nicolet iS5 FT-IR spectrometer (Thermo Fisher Scientific Inc.) using the ATR device with a diamond crystal plate. All the FT-IR spectra were measured at room temperature with the resolution of 4 cm−1. The measurements were repeated 100 times and the final spectra were obtained by averaging them. OMNIC 8.2 software was used to display absorbance spectra by converting ATR data, where the absorption band in the range of wavenumbers from 600 to 850 cm−1 is focused.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Peters, G. P. et al. Carbon dioxide emissions continue to grow amidst slowly emerging climate policies. Nat. Clim. Change 10, 3–6 (2020).
IPCC, 2007: Climate Change 2007: Synthesis Report. Contribution of Working Groups I, II and III to the Fourth Assessment Report of the Intergovernmental Panel on Climate Change. Pachauri, R. K. & Reisinger, A. eds. IPCC, Geneva, Switzerland, pp. 104 (2007).
Oexmann, J., Kather, A., Linnenberg, S. & Liebenthal, U. Post-combustion CO2 capture: chemical absorption processes in coal-fired steam power plants. Greenh. Gases: Sci. Technol. 2, 80–98 (2012).
Herzog, H. J. Peer reviewed: what future for carbon capture and sequestration? Environ. Sci. Technol. 35, 148A–153AA (2001).
D’Alessandro, D. M., Smit, B. & Long, J. R. Carbon dioxide capture: prospects for new materials. Angew. Chem. Int. Ed. 49, 6058–6082 (2010).
Leung, D. Y. C., Caramanna, G. & Maroto-Valer, M. M. An overview of current status of carbon dioxide capture and storage technologies. Renew. Sust. Energ. Rev. 39, 426–443 (2014).
Smit, B., Reimer, J. A., Oldenburg, C. M. & Bourg, I. C. Introduction to Carbon Capture and Sequestration. (Imperial College Press, London, 2014).
IPCC, 2005: Carbon Dioxide Capture and Storage. Metz, B. et al. eds. Cambridge University Press, UK. pp 431.
Seifritz, W. CO2 disposal by means of silicates. Nature 345, 486 (1990).
Snæbjörnsdóttir, S. O. et al. Carbon dioxide storage through mineral carbonation. Nat. Rev. Earth Environ. 1, 90–102 (2020).
McGrail, B. P. et al. Potential for carbon dioxide sequestration in flood basalts. J. Geophys. Res. 111, B12201 (2006).
Gerdemann, S. J., O’Connor, W. K., Dahlin, D. C., Penner, L. R. & Rush, H. Ex situ aqueous mineral carbonation. Environ. Sci. Technol. 41, 2587–2593 (2007).
Teir, S., Eloneva, S., Fogelholm, C. J. & Zevenhoven, R. Fixation of carbon dioxide by producing hydromagnesite from serpentinite. Appl. Energy 86, 214–218 (2009).
Sato, K., Fujimoto, K. & Kawamura, K. Enhanced adhesivity of water molecules confined in angstrom-scale open spaces formed by two-dimensional nanosheets. J. Phys. Chem. C. 120, 27509–27514 (2016).
Sato, K., Fujimoto, K., Kawamura, K., Dai, W. & Hunger, M. Rheological mechanism of long-term self-assembly in saponite nanoparticles. J. Phys. Chem. C. 116, 22954–22959 (2012).
Sato, K., Numata, K., Dai, W. & Hunger, M. Long-term self-assembly of smectite nanoparticles influenced by the states of the interlayer cations. Phys. Chem. Chem. Phys. 16, 10959–10964 (2014).
Sato, K., Numata, K., Dai, W. & Hunger, M. Tunable states of interlayer cations in two-dimensional materials. Appl. Phys. Lett. 104, 131901-1-131901-5 (2014).
Sato, K., Fujimoto, K., Dai, W. & Hunger, M. Molecular mechanism of heavily adhesive Cs: why radioactive cs is not decontaminated from soil. J. Phys. Chem. C. 117, 14075–14080 (2013).
Sato, K., Fujimoto, K., Dai, W. & Hunger, M. Quantitative elucidation of Cs adsorption sites in clays: toward sophisticated decontamination of radioactive Cs. J. Phys. Chem. C. 120, 1270–1274 (2016).
Sato, K. & Hunger, M. Molecular studies of Cs adsorption sites in inorganic layered materials: the influence of solution concentration. Phys. Chem. Chem. Phys. 19, 18481–18486 (2017).
Lazo, N. D., Murray, D. K., Kieke, M. L. & Haw, J. F. In situ carbon-13 solid-state NMR study of the Cu/ZnO/Al2O3 methanol synthesis catalyst. J. Am. Chem. Soc. 114, 8552–8559 (1992).
Yangyang, L. & Wilcox, J. CO2 adsorption on carbon models of organic constituents of gas shale and coal. J. Environ. Sci. Technol. 45, 809–814 (2011).
Yang, S. T., Kim, J. & Ahn, W. S. CO2 adsorption over ion-exchanged zeolite beta with alkali and alkaline earth metal ions. Microporous Mesoporous Mater. 135, 90–94 (2010).
Zhang, C. et al. Metal occupancy and its influence on thermal stability of synthetic saponites. Appl. Clay Sci. 135, 282–288 (2017).
Sugiyama, K., Filio, J. M., Saito, F. & Waseda, Y. Structural change of kaolinite and pyrophyllite induced by dry grinding. Mineral. J. 17, 28–41 (1994).
Web Site of Ministry of the Environment, Government of Japan, “Interim Storage Facility”, Available at http://josen.env.go.jp/en/storage/.
Tao, S. J. Positronium annihilation in molecular substances. J. Chem. Phys. 56, 5499–5510 (1972).
Eldrup, M., Lightbody, D. & Sherwood, J. N. The temperature dependence of positron lifetimes in solid pivalic acid. Chem. Phys. 63, 51–58 (1981).
Acknowledgements
This work was partially supported by Grants-in-Aid of the Ministry of Education, Culture, Sports, Science and Technology of Japan (Grant nos. 16K05394, 18K04884, 18KK0382, and 20K14372).
Author information
Authors and Affiliations
Contributions
K.S. and M.H. designed and conducted the present experiments. K.S. and M.H. discussed the results and contributed to writing of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sato, K., Hunger, M. Carbon dioxide adsorption in open nanospaces formed by overlap of saponite clay nanosheets. Commun Chem 3, 91 (2020). https://doi.org/10.1038/s42004-020-00346-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42004-020-00346-5
This article is cited by
-
Thermodynamic transformations of entangled bulky organic monomers with long alkyl chains
Structural Chemistry (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
