
Abstract
The combination of graphene with metal nanoparticles can produce enhanced catalytic properties because of synergistic effects, and has been used to develop highly active catalysts for different applications. However, the mechanism of the synergistic effect between graphene and metal is poorly understood. Here we demonstrate that graphene-coated nickel foam shows a significant catalytic effect on electrodeless metal (gold, platinum, silver, and copper) deposition without any external reducing agent. This is attributed to the formation of an interface dipole layer, induced by the interaction between graphene and nickel. The interface dipole layer catalytic mechanism accelerates metal reduction reaction and explains the simultaneous formation of nickel hydroxide. The nickel hydroxide-wrapped silver hybrid self-assembly developed on the graphene-coated nickel foam serves as an efficient binder-free electrochemical sensor owing to its hierarchical structure.
Similar content being viewed by others
Introduction
Graphene combined with metal nanoparticles or other compounds is widely recognized to be a viable strategy to assemble high activity catalysts and active composites for energy conversion1,2,3,4,5,6. Properties such as high conductivity, transparency, 2D morphology, and high stability in acidic and alkaline solutions make graphene an excellent electron transfer medium on the interface of graphene/active materials as catalysts7,8,9. Among them, metal nanoparticle/graphene (MNP/G) composites have been attracting more interest because of remarkably enhanced catalytic property, which is usually ascribed to a synergistic effect from the interface of graphene and active metal nanoparticles5,10,11,12. The interface properties of graphene-adsorbed metals have been studied by using first principle theories, which predict the existence of graphene-metal chemical bond and interface diploe layer (IDL)13,14,15,16. However, experimentally, there is still no answer to what the synergistic effect comes from besides the high conductivity of graphene as an apparent reason.
In general, reducing agents and electrodeposition methods have been employed to in situ reduce metal ions (Mx+) such as gold ion (Au3+), platinum ion (Pt4+), silver ion (Ag+), and copper ion (Cu2+) to MNP on the graphene to form MNP/G composites17,18,19. Auto-deposition of MNP (Ag or Au) was also found to occur on the graphene-coated copper substrate via the principle of galvanic displacement reaction20,21,22. Following this principle, some metal ions can be reduced by nickel via electrodeless deposition under the condition that the reduction potential of the metals must be higher than that of nickel (Ni2+/Ni, −0.257 V vs. SHE), which is the necessary condition to trigger the displacement reactions. Therefore, Cu2+ (Cu2+/Cu, 0.342 V vs. SHE), Ag+ (Ag+/Ag, 0.799 V vs. SHE), Pt4+ ([PtCl6]2−/[PtCl4]2−, 0.68 V vs. SHE; [PtCl4]2−/Pt, 0.755 V vs. SHE) and Au3+ ([AuCl4]−/Au, 1.002 V vs. SHE) can be reduced to Cu, Ag, Pt, and Au metals by nickel, respectively23. However, no work has been done using a composite of both Ni and graphene as a reactive scaffold.
Besides, it is reported that nickel hydroxide/M (Ni(OH)2/M) hybrids have unique synergistic effect as catalysts for hydrogen evolution reaction24,25,26. Therefore, the self-assembly of Ni(OH)2/M on graphene-coated nickel foam (GNF) is a promising way to develop advanced binder-free catalytic electrodes for various applications. Loading active materials on nickel foam (NF) has been extensively investigated to create a hierarchical-structured electrode because of high conductivity and porous structure of the NF27,28,29,30,31. Furthermore, Ni(OH)2 has been investigated as effective material for glucose sensing but with a limitation of poor conductivity32,33,34. Direct combination of active materials with the conductive scaffold as a binder-free electrode is an effective method to eliminate the resistance from binder35,36,37,38. To the best of our knowledge, there is no report on using GNF as an autocatalytic reaction scaffold to in situ prepare binder-free electrode.
Here, we study metal (Au, Pt, Ag, and Cu) deposition on GNF and NF to uncover the IDL catalytic mechanism, experimentally confirming the interfacial interaction between graphene and nickel. The IDL mechanism may lead to graphene/metal-based catalyst design principles. The Ni(OH)2-wrapped Ag electrode self-assembly, prepared using this deposition method shows promise for glucose sensing. In this study, graphene is grown on NF by chemical vapor deposition (CVD), and GNF composite is then used for MNP (Au, Pt, Ag, and Cu) deposition. Unlike bare NF, GNF can greatly speed up the electrodeless reduction of Mx+ ions on the surface of graphene. Furthermore, the MNP deposition and Ni(OH)2 nanosheet assembly simultaneously occur on the GNF. The high catalytic effect of metal deposition on GNF is ascribed to the formation of IDL induced by strong interactions between graphene and nickel. The IDL catalytic mechanism explains the catalytic function of GNF for the reduction of Mx+ and can be extrapolated to answer the above-mentioned synergetic effect on the interface of graphene and metal nanoparticles. This discovery can facilitate the understanding of catalytic mechanism of graphene-based materials applied to photocatalysis, oxygen reduction and other reductive conversions, and be the useful reference for the design of highly active catalysts. In particular, the Ni(OH)2-wrapped Ag hybrid generated on GNF (Ag@Ni(OH)2-GNF) is used as a binder-free electrode for glucose sensing. The Ag@Ni(OH)2-GNF electrode possesses desirable structural properties including porous Ni(OH)2 shells for fast molecule diffusion in the electrolyte, highly conductive silver cores for current collection, and large surface area GNF scaffold for combined function of active material loading and current collection. These attributes render Ag@Ni(OH)2-GNF electrode an excellent binder-free sensitive platform for chemical or biological species detection.
Results
Graphene-nickel interface-induced catalytic effect on electrodeless metal deposition
The GNF is discovered to have remarkable catalytic function on the metal replacement reaction (Ni-Mx+, M: Au, Pt, Ag, and Cu). Figure 1 shows the optical photographs of NF and GNF before and after electrodeless metal deposition on the NF and GNF substrates in 0.09 mM HAuCl4 (pH = 4.2), 0.09 mM H2PtCl6 (pH = 4.5), 0.9 mM AgNO3 (pH = 5.3), and 0.9 mM CuSO4 (pH = 4.6) solution for 1 h, respectively. Mx+ is reduced to metal nanoparticles on the surface of the NF or GNF. There is not much color change for NF substrates, and the color of the NF becomes gradually brighter from Au to Cu (sample B in Fig. 1a–d) because of the gradually decreasing oxidizing ability from Au3+ to Cu2+. The slight color change of the NF indicates the low deposition rates of Au, Pt, Ag, and Cu on the bare NF. By comparison, however, the GNF substrate shows different reaction phenomenon because of the intervention of graphene on the nickel surface, with which the metal ions are reduced drastically by the underlying nickel, and the color changes from initial gray to black concomitantly (sample D in Fig. 1a–d) without any extra reducing agent or electrochemical reducing conditions.

Optical photographs of NF and GNF before and after metal deposition in different electrolytes. a–d Comparison of NF and GNF before and after 1 h deposition in a 0.09 mM HAuCl4 (pH = 4.2), b 0.09 mM H2PtCl6 (pH = 4.5), c 0.9 mM AgNO3 (pH = 5.3), and d 0.9 mM CuSO4 (pH = 4.6), respectively
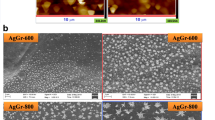
Figure 2 shows surface morphology of GNF and NF after metal deposition in 0.09 mM HAuCl4 (pH = 4.2), 0.09 mM H2PtCl6 (pH = 4.5), 0.9 mM AgNO3 (pH = 5.3), and 0.9 mM CuSO4 (pH = 4.6) for 1 h, respectively. Unexpectedly, a large amount of Ni(OH)2 is generated simultaneously with Au, Pt, and Ag deposition on GNF (Fig. 2a, b, e, f, i, j), while only a few metal nanoparticles are observed on NF (Fig. 2c, d, g, h, k, l). TEM images show that Au, Pt, and Ag are wrapped by Ni(OH)2 sheets to generate MNP@Ni(OH)2 hierarchical structures (Supplementary Fig. 1), which is well confirmed by energy dispersive spectroscope (EDS) (see Supplementary Figs. 2 and 3). Interestingly, however, Ni(OH)2 sheets are not observed for Cu deposition on both GNF and NF (Fig. 2m, n, o, p) and Supplementary Fig. 4).

SEM images of different metals deposition on GNF and NF. a, e, i, and m images of Au, Pt, Ag, and Cu deposition on GNF in 0.09 mM HAuCl4 (pH = 4.2), 0.09 mM H2PtCl6 (pH = 4.5), 0.9 mM AgNO3 (pH = 5.3), and 0.9 mM CuSO4 (pH = 4.6) for 1 h, respectively (b, f, j and n are corresponding high resolution images). c, g, k, and o images of Au, Pt, Ag, and Cu deposition on NF in the same electrolytes above for 1 h, respectively (d, h, l, and p are corresponding high resolution images). Scale bar is 5 µm for a, e, i, and m; 1 µm for b, f, j, k, and o; 0.5 µm for c and g; and 0.2 µm for d, h, l, n, and p
In order to clearly observe the surface morphology progression, 0.0125 mM HAuCl4 (pH = 5.0), 0.025 mM H2PtCl6 (pH = 4.7), 0.45 mM AgNO3 (pH = 5.5), and 0.6 mM CuSO4 (pH = 4.8) solutions were prepared according to their decreasing oxidizing ability and used to investigate metal deposition on GNF (see Fig. 3). In the initial 1 h, GNF is covered by both metals (Au, Pt, and Ag) and Ni(OH)2 sheets (Fig. 3a, d, g) which show morphologies similar to that in Fig. 2a, e, i. Interestingly, 2 h later, it was observed that Ni(OH)2 sheets fully disappear in the cases of Au and Pt, and only Au and Pt nanoparticles are clearly visible on the GNF (Fig. 3b, e), and continue to grow to form a thicker layer after 4 h (Fig. 3c, f). However, Ni(OH)2 nanosheets still largely remain on silver flakes (Fig. 3h, i). In case of copper, only cubic box-like Cu nanoparticles grow individually on the GNF (Fig. 3k, l), and Ni(OH)2 sheets are not observed during the whole deposition process. The above phenomena indicate typical catalytic effect of GNF on metal deposition.

Morphology changes of different metals deposited on GNF with variation of time. a–l SEM images of Au (a, b, c), Pt (d, e, f), Ag (g, h, i), and Cu (j, k, l,) depositions on GNF in 0.0125 mM HAuCl4 (pH = 5.0), 0.025 mM H2PtCl6 (pH = 4.7), 0.45 mM AgNO3 (pH = 5.5), and 0.6 mM CuSO4 (pH = 4.8) for different time, respectively (scale bar, 1 µm)
Reaction mechanism
Graphene, a zero-gap semiconductor, is reported to have strong electron communication with metal when it is chemically adsorbed on the metal surface14. The interaction between graphene and metal substrates has been theoretically investigated39,40,41, which intrinsically originates from three kinds of interaction: dispersive force (van der Waals interaction), Pauli repulsion, and donation/back-donation bonding42. The charge transfer between transition metal and graphene leads to the change of work function of graphene, resulting in the Fermi level of graphene shifting near the Dirac point up or down43. Julian Gebhardt’s study also elucidates that graphene on Ni(111) can be n-doped or p-doped depending on different interface distance13.
In this experiment, strong charge interaction occurs on the interface of graphene and nickel because graphene tightly adsorbs on the nickel surface (CVD method). Figure 4a shows ultraviolet photoelectron spectroscopy (UPS) plots of graphene foam (GF), NF and GNF, from which the work functions of GF, NF, and GNF are obtained in Fig. 4b44. It is observed that work function of GNF is 0.57 eV lower than that of GF, indicating the electrons in nickel move to graphene and the Fermi level in Dirac point of graphene shifts up. In other words, graphene on nickel surface is n-doped, which causes a Fermi energy difference ∆EF1. Moreover, the work function of GNF is 0.35 eV lower than that of bare NF, resulting in the generation of the ∆EF2 as shown in Fig. 4b, which may be due to a small amount of NiO on NF or may have other more complex reasons because chemisorption strongly perturbs the electronic structure of graphene. Moreover, graphene is chemically bonded with nickel surface through strong interaction, where pz orbitals of graphene have hybridization with 3d orbitals of nickel. Charge transfer between two atoms (metal and carbon) results in the formation of a single dipole because of polarization, and a large number of dipoles allow the formation of an IDL (also called electric double layer) with an associated electrostatic potential difference, ∆U13,14. The diploe layer in the interface consists of electropositive nickel surface and electronegative graphene surface (Fig. 4c).

Work function analysis and IDL model-based catalytic reaction. a Ultraviolet photoelectron spectroscopy plots of GF, NF, and GNF. b Work functions of GF, NF, and GNF calculated from (a). c Single dipole and IDL model. d IDL catalytic mechanism of GNF on the reduction of Mx+ and simultaneous generation of Ni(OH)2. Blue circles in (a) show the places where Fermi edge and cutoff edge are obtained, and Ф is energy width of UPS spectrum. ∆U is electrostatic potential difference between the dipole layers
X-ray photoelectron spectroscopy (XPS) on C 1s discloses that graphene has strong electrical interaction with nickel atoms (Fig. 5). It is noticed that C 1s peak of GNF slightly shifts to lower energy (0.3 eV less than that of GF), which is probably due to the innermost electronegative graphene layer. Moreover, graphene is chemically bonded with nickel surface through Ni-C bond, which corresponds to a typical peak at 283.2 eV in the C 1s XPS spectrum of the GNF45,46.

The C 1s XPS spectra of GNF and GF. a The black and purple dashed lines show the C 1s spectra of GNF and b GF have peaks at 284.4 and 284.7 eV, respectively. The red dashed line shows peak position of Ni-C bond at 283.2 eV. The black line with a bubble labeled as “original curve” represents the curve obtained from original XPS data (the black line is obscured by the bubbles)
From another point of view, the smaller work function of GNF may indicate that the electrons in the dipole (graphene side) are more active or have less constraint than those in bare NF, which is confirmed by the superior catalytic effect of GNF on the metal deposition. Surprisingly, Ni(OH)2 is also observed on GNF in the Au, Pt, and Ag deposition reactions except for Cu, and it is no doubt that the OH− comes from electrolytes. To figure out the catalytic mechanism, the hydrolysis reactions for different electrolytes and probable metal deposition reaction mechanism on GNF are written as below.
In the reaction (5), Y represents [AuCl3(OH)]−, [PtCl5(OH)]2−, AgOH, or Cu(OH)2.
In HAuCl4 and H2PtCl6 solution, it is difficult to form Au3+ and Pt4+ from [AuCl4]− and [PtCl6]2−, respectively because of their high coordination ability. Instead, in the diluted HAuCl4 and H2PtCl6 solution with pH around 4.5 or higher, [AuCl3(OH)]− and [PtCl5(OH)]2− complexes are easily generated because of hydrolysis (reactions (1) and (2))47,48. Ag+ and Cu2+ have weak hydrolysis equilibriums to generate compounds AgOH (Ksp (solubility product) = 2.0 × 10−8) and Cu(OH)2 (Ksp = 2.2 × 10−20), respectively as shown in reactions (3) and (4). Y complex contains metal ion Mx+ and OH−, and Mx+ is fast reduced to MNP by taking electrons from n-doped graphene layer and OH− is simultaneously released, temporarily leading to a high concentration of OH− near the MNP surface. Ni2+ ions come out from gaps and holes among graphene domains and react with OH−, resulting in simultaneous generation of Ni(OH)2 sheets outside the deposited MNP (as shown in reaction (5) and Fig. 4d). Even though the degree of hydrolysis may be low, fast catalytic reduction of Mx+ on the GNF increases the degree of hydrolysis (hydrolysis equilibrium moves toward the right) to produce more Y complexes. H+ is also concomitantly generated during the above process (reactions (1–4)), which may cause Ni(OH)2 to dissolve in the electrolyte (reaction (6)). However, during the metal deposition, the acidity of electrolyte does not change significantly, and thus does not lead to fast dissolution of the large amount of aggregated Ni(OH)2 for Ag deposition case. However, exceptionally, the formation of Ni(OH)2 is difficult for Cu deposition because Cu2+ is very weak oxidizing agent and fast generation of high concentration OH− is impossible in low reaction rate.
As reduction agents, graphene and CNT can reduce some metal ions such as Au3+, Pt4+, Pd2+, Ir3+ and so on44,49,50, which indicates that n-doped graphene should be much stronger reduction agent than bare graphene. Therefore, the n-doped graphene present in GNF works as a highly efficient catalyst for electron transfer in those reactions compared to bare NF. Along with consumption of electrons in the n-doped graphene layer (δ− side), the electrons provided by the oxidation of underlying nickel move continuously from nickel to graphene to keep charge balance or equilibrium in the IDL (Fig. 4c). Figuratively speaking, the electrostatic potential difference (∆U) between the IDL can maintain electron transfer from nickel to graphene, driving the redox reaction at a high rate.
The unique phenomena in Figs. 2 and 3 can be explained based on the above catalytic mechanism. Figure 2 shows metal (Au, Pt, Ag, and Cu) deposition on GNF and NF for 1 h. HAuCl4, H2PtCl6, and AgNO3 are the stronger oxidizing agents than CuSO4. Therefore, fast consumption of [AuCl3(OH)]− and [PtCl5(OH)]2− complexes leads to high concentration of OH− near the graphene surface and high concentration of Ni2+ near the nickel surface, and then Ni2+ ions combine with OH− to generate Ni(OH)2 sheets. In the case of Au and Pt, large amount of Ni(OH)2 can be observed in the beginning (Fig. 2a, e, Fig. 3a, d) in the acidic HAuCl4 and H2PtCl6 solution probably because of high catalytic reaction rate. However, when the deposition time becomes much longer (Fig. 3b, c, e, f), the Ni(OH)2 gradually dissolves in the moderately high acidic solution as shown in reaction (6). Interestingly, Ni(OH)2 nanosheets are still observed on Ag plates even after 4 h, which is ascribed to the weakly acidic AgNO3 solution (pH ~ 5.5), where Ni(OH)2 is difficult to dissolve (Fig. 3i). Besides, the generated H+ may be not enough to change the acidity of AgNO3 solution significantly (after 1 h reaction, the pH of AgNO3 solution still maintains around 5).
The deposition of Cu is an exception. Ni(OH)2 is not observed in both deposition conditions (Fig. 2m, n) and Fig. 3j–l). Cu2+ shows a very weak hydrolysis reaction rate and thus leads to low concentration of Y group (Cu(OH)2) in the reaction (4). Furthermore, the Cu(OH)2 has an extremely small solubility product and thus fails to generate high concentration of OH− near the GNF surface. In addition, Cu2+ is the weakest oxidizing agent among those metal ions studied in this work, resulting in the much slower reduction of Cu2+. Therefore, Ni(OH)2 does not form because only a small amount of OH− can release from Cu(OH)2. Eventually, the experimental phenomena in Figs. 2 and 3 are explained by the IDL catalytic mechanism.
Density functional theory calculations
DFT calculation was employed to estimate the states of electron transfer on the interface between nickel and graphene (detailed method can be found in Supplementary Method). Calculation models are designed according to graphene domains of different layers chemically adsorbed on NF. In order to guarantee a scientific simulation, two models (Supplementary Fig. 5), where nickel crystal surface (111) is covered by one-layer graphene (Model I) or five-layer graphene (Model II) are considered, respectively. As indicates in Model I of Fig. 6a, it is found that some regions (red color) show increased electron density in the graphene layer, indicting electrons transferred from nickel to graphene. Interestingly, the electron transfer also largely happens in the first layer in the Model II (Fig. 6b–f), and decreases quickly with the increase of graphene layers and has almost no influence for the fourth or much outer layer. The IDL model is visible and well confirmed by the charge distribution on the interface (see Fig. 6g, h). It is observed that the poor electron region and rich electron region for both models are located on the nickel surface and the nearby graphene surface, respectively. Therefore, the number of graphene layers on nickel surface almost has no effect on the formation of IDL. However, the electrons can move easily outside irrespective of single-layer graphene or multilayer graphene. In fact, the free-standing graphene foam (GF) is obtained after nickel is removed by HCl, which contains graphene domains or nanosheets. The number of graphene can be distinguished quantitatively from full width at half-maximum (FWHM) of the 2D band and the intensity of G band because band structures of graphene layers are closely related to the G and 2D bands51,52. Raman spectra indicates that the GF consists of composites of single-layer and multilayer graphene (Supplementary Fig. 6a and b), and the multilayer graphene is found to be dominant in GF as shown in XRD in Supplementary Fig. 7.

Density functional theory calculation of graphene-coated nickel foam. a–f Redistribution of the electron densities of single-layer graphene in the Model I (a) and each graphene layer from the first to the fifth layer (b–f) in the Model II. g, h Redistribution of the electron densities in the cross section (IDL formation) of Model I (g) and Model II (h). The differential charge density is defined as the difference in the electron density with and without the nickel substrate. The red and blue regions are regions of increased and decreased electron density, respectively (the gradient ruler represents electron density, e/Å3)
Structure characterization of Ag@Ni(OH)2-GNF monolith electrode
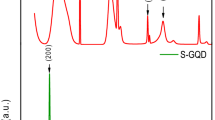
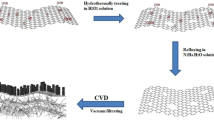
The self-assembled Ni(OH)2-wrapped Ag is grown on GNF by a simple treatment with silver nitrate solution, as schematically depicted in Fig. 7a. Structurally, it’s believed that the inner Ag conductive skeleton greatly benefits the collection of the electrons generated from Ni(OH)2 shell. The obtained Ag@Ni(OH)2 nanohybrids are shown in Fig. 7b. The individual irregular Ag nanoplates stack together and are wrapped by porous Ni(OH)2 nanosheets to form flower-like structure (Fig. 7c). The Ag cores encapsulated by Ni(OH)2 nanosheets are clearly observed from the TEM image in Fig. 7d. The mixture of Ag and Ni(OH)2 composite is confirmed by X-ray diffraction pattern (Supplementary Fig. 7). All the diffraction peaks can be well indexed to Ni(OH)2 and Ag (JCPDS No. 14-0117 and JCPDS No. 04-0783, respectively) except for one weak peak at 26.5° which is ascribed to a small amount of graphene peeled off from nickel foam during high power sonication (Ag@Ni(OH)2 nanohybrids were separated from nickel foam by sonication).

Mechanism of Ag@Ni(OH)2 electrode on glucose sensing on and its morphology. a Schematic illustration of the fabrication processes of Ag@Ni(OH)2 directly on the GNF and the hierarchical structure of the electrode for glucose sensing. b, c SEM images of Ag@Ni(OH)2-GNF prepared with 0.45 mM AgNO3 after 4 h at different magnifications. d TEM image of Ag wrapped by Ni(OH)2. Scale bar is 2 µm for b, and 1 µm for c and d
As shown in Supplementary Fig. 8, the calculated BET (Brunauer–Emmett–Teller) surface area of Ag@Ni(OH)2-GNF sample is determined to be 59.9 m2 g−1, which contains both small mesopores (~ 4 nm) and larger mesopores (~ 15 nm), contributing a pore volume of 0.28 cm3 g−1. Furthermore, elemental analyses show that the hybrid structure consists of Ag core and porous Ni(OH)2 shell (Supplementary Fig. 9). The Ag signal is consistent well with the inner dark region of the composite (Supplementary Fig. 9a and b). The Ni and O signals originating from Ni(OH)2 are distributed over the whole region (Supplementary Fig. 9c and d). Moreover, the formation process of hierarchical flower-like Ag@Ni(OH)2 hybrid and its optimal deposition time are discussed in Supplementary Note 1 (Supplementary Figs. 10 and 11).
Application of Ag@Ni(OH)2-GNF electrode in non-enzymatic glucose sensing
Ni(OH)2 has been reported as active non-enzymatic glucose sensor based on the following detection mechanism53.
The cyclic voltammetric (CV) curve shows three anodic peaks at 0.29 (A1), 0.35 (A2), and 0.52 V (A3) at a scan rate of 2 mV s−1 with absence of glucose in a 0.1 M NaOH solution (Fig. 8a). The two smaller oxidation peaks A1 and A2 are assigned to the formation of Ag(I)-oxygen containing species, and a strong broad reduction peak located at 0.15 V (C2) is ascribed to the reduction of Ag(I)-oxygen to Ag54. A pair of redox peaks C1 and A3 is assigned to the Ni3+/Ni2+ redox couple (reaction (7))55. Glucose can be oxidized to glucolactone by Ni3+ (reaction (8)), and a corresponding increased anodic current is observed (Fig. 8b). However, it is found there is very small current response for pure GNF in the present and absent of glucose (insert in Fig. 8b).

Electrochemical performance of Ag@Ni(OH)2-GNF electrode for glucose sensing. a CV profiles of the Ag@Ni(OH)2-GNF at various scan rates (2, 4, 6, 8, and 10 mV s−1) in 0.1 M NaOH electrolyte. b CV curves of Ag@Ni(OH)2-GNF and GNF (inset) in the absence and presence of 0.25 mM glucose (scan rate: 10 mV s−1). c Typical steady-state current-time response of the Ag@Ni(OH)2-GNF electrode to the successive injection of glucose into 0.1 M NaOH electrolyte (inset is the magnified region from 600 to 820 s as depicted by the blue dashed square, and the successive additions of glucose are depicted by the red arrows). d Amperometric response of the Ag@Ni(OH)2-GNF hybrid electrode to the stepwise addition of 0.2 mM glucose, ascorbic acid, fructose, dopamine, and uric acid, followed by the addition of 0.4 mM glucose at a fixed potential of 0.55 V (The successive additions of different reagents are depicted by the pink arrows). (Ag/AgCl electrode is used as a reference electrode in a and b)
With continuously increasing concentration of glucose, the oxidation peak current densities also gradually increase (Supplementary Fig. 12a) and are linearly proportional to the concentration of glucose in the range of 0.05 mM to 0.225 mM (Supplementary Fig. 12b). Reaction (7) shows the oxidation of Ni(OH)2, which can be disassembled into two parts (one is Ni(OH)2 → NiOOH + H+ + e− within the solid phase, the other H+ + OH− → H2O in the interface of electrode/electrolyte)53. Slow movement of electrons and hindered diffusion of protons will decrease the sensitivity and the reproducibility of sensors. However, porous nanosheet structure of Ag@Ni(OH)2-GNF hybrid can provide sufficient reactive interfaces between Ni(OH)2 and electrolyte, and the coordinated inner silver nanoplates are good electron conductors, which facilitate the electron transfer from the Ni(OH)2 to GNF substrate, contributing to high current response and high sensitivity.
The measurement of amperometric response to concentrations of glucose in 0.1 M NaOH at a fixed potential of 0.55 V (optimal potential in Supplementary Fig. 13 and more discussion in Supplementary Note 2) was performed to evaluate the sensitivity of Ag@Ni(OH)2-GNF (Fig. 8c). Almost 98% of the steady-state current could be achieved within 3 s upon the addition of glucose, revealing a rapid response of the sensor. A stable amperometric response was observed when glucose of different concentrations was successively added into 0.1 M NaOH solution with a time interval of 30 s. Moreover, significantly different amperometric current steps are observed as the glucose concentrations vary. The calibration curve of Ag@Ni(OH)2-GNF electrode reveals excellent linear relationship between the concentration of glucose and the responding current signal at a wide range of glucose concentration from 0.6 μM to 3.5 mM (R2 = 0.9989) (Supplementary Fig. 14). Moreover, a very high sensitivity of 2180 μA cm−2 mM−1 and a low detection limit of 0.3 µM (signal/noise = 3) are achieved, which address excellent performance among the Ni(OH)2-based glucose sensors (Supplementary Table 1). Compared with traditional glassy carbon electrode-supported sensor, the as-prepared GNF-based electrode has three-dimensional (3D) interpenetrating network, which can hold a much higher loading amount of active materials per unit electrode area, and thus is more convenient to be directly used in practical large-area testing systems.
High selectivity is evaluated in the presence of the interfering substances of ascorbic acid (AA), fructose, dopamine, and uric acid (UA), whose concentrations are commonly 30 times lower than that of glucose in human’s blood. The as-synthesized Ag@Ni(OH)2-GNF still shows excellent selectivity for glucose compared to the same concentration of AA, UA, fructose, and dopamine, and reveals 40, 6, 20, and 7 times higher in current density, respectively as shown in Fig. 8d. In addition, the hybrid sensor shows good repeatability with remarkable current increment after the addition of 0.4 mM glucose at existence of the interfering compounds. The high selectivity and sensitivity of Ag@Ni(OH)2-GNF hybrid highly benefit from the unique Ag@Ni(OH)2 core-shell structure which has various mesopores, favoring fast mass diffusion on the solid-liquid interface. Therefore, these excellent various qualities make Ag@Ni(OH)2-GNF a significant structure for sensor or other electrochemical and photochemical electrode applications.
Discussion
Graphene-nickel surface-induced catalytic effect on electrodeless metal deposition and simultaneous formation of M@Ni(OH)2-GNF were discovered and studied here. The catalytic effect originates from the IDL structure on the interface of graphene and nickel, which accelerates the electron transfer during the reactions. The IDL catalytic mechanism is proved experimentally and supports the metal deposition and concomitant formation of Ni(OH)2 on the GNF surface. It is found that the binder-free Ag@Ni(OH)2-GNF composite is easily assembled by this method and has ideal structure for electrochemical reaction, and exhibits good performance as a non-enzymatic glucose sensor in terms of sensitivity, response time, and detection limit. The high electrochemical activity is ascribed to synergistic co-existence of Ag conductive core inside the porous Ni(OH)2 nanosheets. In addition, the catalytic mechanism of the IDL can be a good reference to design new catalysts for other applications such as fuel cell, photocatalysis, and electrochemical reductive conversion.
Methods
Materials
Chloroplatinic acid (H2PtCl6·6H2O), chloroauric acid (HAuCl4·3H2O, 99.9%), silver nitrate (AgNO3, 99.5%), copper sulfate (CuSO4, 97%), sodium hydroxide, glucose, ascorbic acid (AA), fructose, dopamine, and uric acid (UA) were procured from Sigma-Aldrich. The chemicals were used without any further purification. Nickel foam (320 ± 20 g m−2 in areal density and 1.5 mm in thickness) was purchased from Taiyuan Lizhiyuan Battery Material Co., China.
Synthesis of GNF
GNF was synthesized by a low pressure CVD method according to our previous work with a little modification56. In detail, nickel foam was washed with 1 M HCl, acetone and water, respectively, for 15 min per each step to remove organics and other contaminations, and then was placed in a quartz tube of outer diameter 120 mm and inner diameter 115 mm. The nickel foam was then heated to 1000 °C in a horizontal tube furnace under H2 gas (100 sccm) and annealed for 30 min to remove thin surface oxide layer. Subsequently, a mixture gas of methane (50 sccm) and hydrogen (30 sccm) with a total pressure of ~1.6 Torr is introduced into the tube furnace. After 15 min, the methane gas is turned off, and the samples rapidly cool to room temperature under hydrogen (30 sccm).
Metal electrodeless deposition on NF and GNF
The as-synthesized GNF was cut into a rectangle shape with 5 mm × 20 mm and washed with ethanol for several times before use. Metal (Au, Pt, Ag, and Cu) deposition on GNF and NF was performed as follows. 40 ml 0.09 mM HAuCl4 (pH = 4.2), 0.09 mM H2PtCl6 (pH = 4.5), 0.9 mM AgNO3 (pH = 5.3), and 0.9 mM CuSO4 (pH = 4.6) precursors were prepared. Each solution was filled into four individual glass bottles, respectively and kept in a dark place, and then a piece of GNF was immersed into each solution. One hour later, the GNF samples were washed with ethanol and deionized water for several times, and dried in an oven at 60 °C. As reference, the metal deposition on bare NF (before experiment, NF was washed with acetone, and then annealed in the H2 (100 sccm) at 1000 °C for 30 min to fully remove the impurities and nickel oxide/hydroxide layer on the NF) was also done in the same conditions. Precursors of 0.0125 mM HAuCl4 (pH = 5.0), 0.025 mM H2PtCl6 (pH = 4.7), 0.45 mM AgNO3 (pH = 5.5), and 0.60 mM CuSO4 (pH = 4.8) were also prepared to study the deposition mechanism. For preparation of sensor electrodes, 80 ml 0.45 mM AgNO3 was used to get a thicker layer of Ag@Ni(OH)2 on the GNF.
Materials characterization
Morphologies were examined by scanning electron microscopy (SEM: Hitachi S-4700) at an acceleration voltage of 10 kV, and transmission electron microscopy (TEM; EM 912 Omega, Zeiss) operated at 120 kV. XRD patterns of materials were obtained by using a Rigaku Smartlab diffractometer with Cu Kα radiation and a Ni-filter. The X-ray source was operated at 40 kV and 30 mA. High resolution (HR)-SEM images were obtained by using a Hitachi S-5500 ultrahigh-resolution scanning electron microscope operated at 30 kV. Determination of the specific surface area was carried out by Brunner–Emmett–Teller (BET) N2 adsorption–desorption analysis by using an ASAP 2020 surface area and porosity analyzer. Ultraviolet photoelectron spectroscopy (UPS) of GF, NF, and GNF was characterized by a micro X-ray UV photoelectron spectromicroscopy with photon source: monochromatic Al Kα (1486.6 eV) for XPS and He (21.2 eV) for UPS. The work functions were calculated according to the following equation:
In this formula, W is work function, hν is energy of photon source, and Ф is energy width of UPS spectrum (Ф = Fermi edge − cutoff edge). The values of Fermi edge and cutoff can be obtained by tangential method57.
Electrochemical measurements
Electrochemical measurements of Ag@Ni(OH)2-GNF as a sensor for glucose detection were conducted in a three-electrode cell. Glucose solution with different concentrations was prepared, and 0.1 M NaOH was used as electrolyte for all experiments. The Ag@Ni(OH)2-GNF hybrid works as a working electrode, a Ag/AgCl electrode as a reference electrode, and a carbon rod as a counter electrode. Cycle voltammetry (CV) and linear sweep voltammetry (LSV) measurements were carried out on an electrochemical analyzer (BioLogic VMP3) at different scanning rates (2–10 mV s−1) within a voltage range −0.1–0.6 V (vs. Ag/AgCl). The amperometric response measurements were carried out at a constant potential of 0.55 V in a supporting electrolyte of 0.1 M NaOH solution with successive addition of glucose solution. In detail, the same electrochemical workstation of three-electrode cell with a magnetic bar is used in the experiment. After the background current of the system reached a stable value (a straight horizontal line is observed) under slow and steady stir, glucose solution was added to the cell using a micropipette with a regular interval of 30 s. Then, the resulting current difference was recorded.
DFT calculations
The (111) surfaces of pure Ni has been built with a thickness of six atomic layers. Then, one-layer and five-layer graphene have been laid on the top of Ni surface with the lattice mismatch <1.3% and an interface distance of 0.3512 nm to have Model I and Model II, respectively (see Supplementary Fig. 5). The vacuum space along the Z direction is set to be 25 Å, which is enough to avoid interaction between the two neighboring images. The first-principles calculations in the framework of density functional theory, including structural and electronic performances, were carried out based on the Cambridge Sequential Total Energy Package known as CASTEP58. The exchange-correlation functional under the generalized gradient approximation (GGA)59 with norm-conserving pseudopotentials and Perdew–Burke–Ernzerhof functional were adopted to describe the electron–electron interaction60,61. An energy cutoff of 780 eV was used, and a k-point sampling set of 12 × 12 × 1 was tested to be converged. A force tolerance of 0.01 eV Å−1, energy tolerance of 5.0 × 10−7 eV per atom and maximum displacement of 5.0 × 10−4 Å were considered. Each atom in the storage model is allowed to relax to the minimum in the enthalpy without any constraints. During geometry optimization, the three Ni atomic layers in the bottom have been fixed and the top three Ni atomic layers and graphene layers have been relaxed.
Data availability
The data that support the findings of this study is available from the corresponding author upon reasonable request.
References
Huang, C., Li, C. & Shi, G. Graphene based catalysts. Energy Environ. Sci. 5, 8848–8868 (2012).
Chen, D., Tang, L. & Li, J. Graphene-based materials in electrochemistry. Chem. Soc. Rev. 39, 3157–3180 (2010).
Kamat, P. V. Graphene-based nanoassemblies for energy conversion. J. Phys. Chem. Lett. 2, 242–251 (2011).
Yin, H. et al. Three-dimensional graphene/metal oxide nanoparticle hybrids for high-performance capacitive deionization of saline water. Adv. Mater. 25, 6270–6276 (2013).
Zhao, S. et al. Three-dimensional graphene/Pt nanoparticle composites as freestanding anode for enhancing performance of microbial fuel cells. Sci. Adv. 1, e1500372 (2015).
Zhang, C. & Yu, J.-S. Morphology-tuned synthesis of NiCo2O4-coated 3D graphene architectures used as binder-free electrodes for lithium-ion batteries. Chem. Eur. J. 22, 4422–4430 (2016).
Zhang, Y., Tang, Z.-R., Fu, X. & Xu, Y.-J. TiO2−Graphene nanocomposites for gas-phase photocatalytic degradation of volatile aromatic pollutant: is TiO2−graphene truly different from other TiO2−carbon composite materials? ACS Nano 4, 7303–7314 (2010).
Xiang, Q., Yu, J. & Jaroniec, M. Graphene-based semiconductor photocatalysts. Chem. Soc. Rev. 41, 782–796 (2012).
Lightcap, I. V. & Kamat, P. V. Graphitic design: prospects of graphene-based nanocomposites for solar energy conversion, storage, and sensing. Acc. Chem. Res. 46, 2235–2243 (2013).
Chao, Y. et al. CuNi Nanoparticles assembled on graphene for catalytic methanolysis of ammonia borane and hydrogenation of nitro/nitrile compounds. Chem. Mater. 29, 1413–1418 (2017).
Gowthaman, N. S. K., Raj, M. A. & John, S. A. nitrogen-doped graphene as a robust scaffold for the homogeneous deposition of copper nanostructures: a nonenzymatic disposable glucose sensor. ACS Sustain. Chem. Eng. 5, 1648–1658 (2017).
Yoo, E. J. et al. Enhanced electrocatalytic activity of Pt subnanoclusters on graphene nanosheet surface. Nano. Lett. 9, 2255–2259 (2009).
Gebhardt, J., Viñes, F. & Görling, A. Influence of the surface dipole layer and pauli repulsion on band energies and doping in graphene adsorbed on metal surfaces. Phys. Rev. B 86, 195431 (2012).
Khomyakov, P. A., Giovannetti, G., Rusu, P. C., Brink, J. V. D. & Kelly, P. J. First-principles study of the interaction and charge transfer between graphene and metals. Phys. Rev. B 79, 195425 (2009).
Hu, T. & Hong, J. First-principles study of metal adatom adsorption on black phosphorene. J. Phys. Chem. C. 119, 8199–8207 (2015).
Huar, B., Stander, N., Sulpizio, J. A. & Goldhaber-Gordon, D. Evidence of the role of contacts on the observed electron-hole asymmetry in graphene. Phy. Rev. B 78, 121402 (2008).
Zhuo, Q. et al. Facile synthesis of graphene/metal nanoparticle composites via self-catalysis reduction at room temperature. Inorg. Chem. 52, 3141–3147 (2013).
Maiyalagan, T., Dong, X., Chen, P. & Wang, X. Electrodeposited Pt on three-dimensional interconnected graphene as a free-standing electrode for fuel cell application. J. Mater. Chem. 22, 5286–5290 (2012).
Liu, C. et al. Direct electrodeposition of graphene enabling the one-step synthesis of graphene-metal nanocomposite films. Small 7, 1203–1206 (2011).
Liu, X.-W., Mao, J.-J., Liu, P.-D. & Wei, X.-W. Fabrication of metal-graphene hybrid materials by electrodeless deposition. Carbon N. Y. 49, 477–483 (2011).
Li, Z. et al. Graphene buffered galvanic synthesis of graphene–metal hybrids. J. Mater. Chem. 21, 13241–13246 (2011).
Gutés, A. et al. Graphene decoration with metal nanoparticles: towards easy integration for sensing applications. Nanoscale 4, 438–440 (2012).
Grünewald, H. Handbook of Chemistry and Physics. 73th edn, 8-21–8-25 (CRC Press, Cleveland, 1992).
Danilovic, N. et al. Enhancing the alkaline hydrogen evolution reaction activity through the bifunctionality of Ni(OH)2/metal catalysts. Angew. Chem. Int. Ed. 124, 12663–12666 (2012).
Yin, H. et al. Ultrathin platinum nanowires grown on single-layered nickel hydroxide with high hydrogen evolution activity. Nat. Commun. 6, 6430 (2015).
Yin, H. & Tang, Z. Ultrathin two-dimensional layered metal hydroxides: an emerging platform for advanced catalysis, energy conversion and storage. Chem. Soc. Rev. 45, 4873–4891 (2016).
Wan, Y., Zhang, D., Wang, Y. & Hou, B. A 3D-impedimetric immunosensor based on foam Ni for detection of sulfate-reducing bacteria. Electrochem. Commun. 12, 288–291 (2010).
Kung, C.-W., Cheng, Y.-H. & Ho, K.-C. Single layer of nickel hydroxide nanoparticles covered on a porous Ni foam and its application for highly sensitive non-enzymatic glucose sensor. Sens. Actuators, B. 204, 159–166 (2014).
Hussain, M. et al. Synthesis of three dimensional nickel cobalt oxide nanoneedles on nickel foam, their characterization and glucose sensing application. Sensors 14, 5415–5425 (2014).
Guo, C., Wang, Y., Zhao, Y. & Xu, C. Non-enzymatic glucose sensor based on three dimensional nickel oxide for enhanced sensitivity. Anal. Methods 5, 1644–1647 (2013).
Akhtar, N., El-Safty, S. A., Khairy, M. & El-Said, W. A. Fabrication of a highly selective nonenzymatic amperometric sensor for hydrogen peroxide based on nickel foam/cytochrome c modified electrode. Sens. Actuators, B. 207, 158–166 (2015).
Yang, J., Cho, M. & Lee, Y. Synthesis of hierarchical Ni(OH)2 hollow nanorod via chemical bath deposition and its glucose sensing performance. Sens. Actuators, B. 222, 674–681 (2016).
Lu, P. et al. Nanosheets-assembled hierarchical microstructured Ni(OH)2 hollow spheres for highly sensitive enzyme-free glucose sensors. Electrochim. Acta 168, 148–156 (2015).
Lu, P., Lei, Y., Lu, S., Wang, Q. & Liu, Q. Three-dimensional rose like α-Ni(OH)2 assembled from nanosheet building blocks for non-enzymatic glucose detection. Anal. Chim. Acta 880, 42–51 (2015).
Liu, S., Hui, K. S. & Hui, K. N. Flower-like copper cobaltite nanosheets on graphite paper as high-performance supercapacitor electrodes and enzymeless glucose sensors. ACS Appl. Mater. Interfaces 8, 3258–3267 (2016).
Li, Z. et al. Electronic topological transition in Ag2Te at high-pressure. Sci. Rep. 5, 10617 (2015).
Li, G. et al. Controllable synthesis of 3D Ni(OH)2 and NiO nanowalls on various substrates for high-performance nanosensors. Small 11, 731–739 (2014).
Gao, A. et al. In situ synthesis of Ni(OH)2/TiO2 composite film on NiTi alloy for non-enzymatic glucose sensing. Sens. Actuators, B. 232, 150–157 (2016).
Andersen, M., Hornekær, L. & Hammer, B. Graphene on metal surfaces and its hydrogen adsorption: A meta-GGA functional study. Phys. Rev. B 86, 085405 (2012).
Xu, Z. & Buehler, M. J. Interface structure and mechanics between graphene and metal substrates: a first-principles study. J. Phys. Condens. Matter 22, 485301 (2010).
Vanin, M. et al. Graphene on metals: a van der Waals density functional study. Phys. Rev. B 81, 081408 (2010).
Kozlov, S. M., Viñes, F. & Görling, A. Bonding mechanisms of graphene on metal surfaces. J. Phys. Chem. C. 116, 7360–7366 (2012).
Adamska, L., Lin, Y., Ross, A. J., Batzill, M. & Oleynik, I. I. Atomic and electronic structure of simple metal/graphene and complex metal/graphene/metal interfaces. Phys. Rev. B 85, 198443 (2012).
Shi, Y. et al. Work function engineering of graphene electrode via chemical doping. ACS Nano 4, 2689–2694 (2010).
Yin, J. et al. Ni−C−N nanosheets as catalyst for hydrogen evolution reaction. J. Am. Chem. Soc. 138, 14546–14549 (2016).
Bittencourt, C. et al. Decorating carbon nanotubes with nickel nanoparticles. Chem. Phys. Lett. 436, 368–372 (2007).
Xia, H., Bai, S., Hartmann, J. R. & Wang, D. Synthesis of monodisperse quasi-spherical gold nanoparticles in water via silver(I)-assisted citrate reduction. Langmuir 26, 3585–3589 (2010).
Straney, P. J., Marbella, L. E., Andolina, C. M., Nuhfer, N. T. & Millstone, J. E. Decoupling mechanisms of platinum deposition on colloidal gold nanoparticle substrates. J. Am. Chem. Soc. 136, 7873–7876 (2014).
Kwon, K. C., Choi, K. S. & Kim, S. Y. Increased work function in few-layer graphene sheets via metal chloride doping. Adv. Funct. Mater. 22, 4724–4731 (2012).
Choi, H. C., Shim, M., Bangsaruntip, S. & Dai, H. Spontaneous reduction of metal ions on the sidewalls of carbon nanotubes. J. Am. Chem. Soc. 124, 9058–9059 (2002).
Hao, Y. et al. Probing layer number and stacking order of few-layer graphene by Raman spectroscopy. Small 6, 195–200 (2010).
Ni, Z., Wang, Y., Yu, T. & Shen, Z. Raman spectroscopy and imaging of graphene. Nano Res. 1, 273–291 (2008).
Safavi, A., Maleki, N. & Farjami, E. Fabrication of a glucose sensor based on a novel nanocomposite electrode. Biosens. Bioelectron. 24, 1655–1660 (2009).
Safavi, A. & Tohidi, M. Silver paste nanocomposite electrode as a new metallic electrode for amperometric determination of hydrazine. Anal. Methods 4, 2233–2241 (2012).
Nai, J., Wang, S., Bai, Y. & Guo, L. Amorphous Ni(OH)2 nanoboxes: fast fabrication and enhanced sensing for glucose. Small 9, 3147–3152 (2013).
Zhang, C. et al. Facile preparation of flower-like NiCo2O4/three dimensional graphene foam hybrid for high performance supercapacitor electrodes. Carbon N. Y. 89, 328–339 (2015).
Wang, J.-C. et al. Enhanced photoreduction CO2 activity over direct Z-scheme α-Fe2O3/Cu2O heterostructures under visible light irradiation. ACS Appl. Mater. Interfaces 7, 8631–8639 (2015).
Segall, M. D. et al. First-principles simulation: ideas, illustrations and the CASTEP code. J. Phys. Condens. Matter 14, 2717–2744 (2002).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Hamann, D. R., Schlüter, M. & Chiang, C. Norm-conserving pseudopotentials. Phys. Rev. Lett. 43, 1494–1497 (1979).
Voiry, D. et al. Enhanced catalytic activity in strained chemically exfoliated WS2 nanosheets for hydrogen evolution. Nat. Mater. 12, 850–855 (2013).
Acknowledgements
This work was generously supported by Global Frontier R&D Program on Center for Multiscale Energy System (NRF 2011-0031571), NRF grant (NRF-2016M1A2A2937137) funded by the Korea government, and Wong Magna Fund in Ningbo University, China. Authors also would like to thank the Korean Basic Science Institute at Jeonju (SEM and HRTEM analysis), Daejeon (TEM analysis), and CCRF in DGIST.
Author information
Authors and Affiliations
Contributions
C.Z. and J.-S.Y. conceived and designed this project. H.L. contributed to the characterization of work functions of related materials and mechanism analysis. B.-J.L. and J.S. contributed to the sensor application. B.-J.L. and T.-H.K. performed partial electrochemical analyses. C.Z. and J.-S.Y. wrote the manuscript. J.-S.Y. directed the research, and all the authors participated in manuscript corrections and revisions.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, C., Lee, BJ., Li, H. et al. Catalytic mechanism of graphene-nickel interface dipole layer for binder free electrochemical sensor applications. Commun Chem 1, 94 (2018). https://doi.org/10.1038/s42004-018-0088-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42004-018-0088-x
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
