
Abstract
Chiral allene and N-heteroaryl motifs are present in an ever-growing list of biologically active natural products and synthetic drugs. Although significant progress has been made in asymmetric syntheses of chiral allenes, general and practical protocols for enantioselective syntheses of chiral N-heteroaryl-substituted allenes from readily available starting materials still remain rare. Here we report a highly enantioselective synthesis of quinolinyl-substituted chiral allenes through a copper-catalyzed asymmetric allenylation of quinoline N-oxides with readily available 1,3-enynes. A variety of 1,3-enynes react with quinoline N-oxides, affording the corresponding quinolinyl-substituted allenes in high yields (up to 95%) and high enantioselectivities (up to 99% ee). This transformation tolerates a variety of functional groups, such as chloro, bromo, trifluoromethyl ether, tertiary amine, siloxy, carboxylic ester, imide, pyridine, and thiophene moieties. DFT calculations suggest a pathway involving an intramolecular nucleophilic addition of an allenyl copper intermediate with a coordinated quinoline N-oxide through a five-membered, rather than seven-membered, transition state.
Similar content being viewed by others
Introduction
Chiral allene moieties are present in a variety of natural products, molecular materials, and a few marketed drugs due to their unique structural features, chemical reactivity, and biological properties1,2. In addition, chiral allenes also represent a class of useful starting materials for a variety of organic transformations3. Accordingly, substantial endeavors have been made to develop methodologies for the asymmetric synthesis of chiral allenes. Classic approaches typically involve (kinetic) resolution of racemic allenes4,5 or chirality transfer from optically pure propargylic precursors6,7,8,9,10. Recent years have witnessed a tremendous development on metal-catalyzed enantioselective synthesis of chiral allenes11,12,13,14,15,16, such as nucleophilic addition to 1,3-enynes17,18,19, enantioselective functionalization of racemic allenes20,21,22, β-hydride elimination from enol triflates23, rearrangement of propargylic compounds24,25,26,27, coupling of terminal alkynes with diazo compounds28,29,30,31, and enantioselective addition of terminal alkynes to aldehydes32. In addition, several asymmetric organocatalytic protocols have also been reported, such as nucleophilic addition to activated enynes33,34, isomerization of alkynes35,36, phase-transfer-catalyzed alleno-Mannich reaction37,38, alkynylogous Mukaiyama aldol reactions39, and chiral ion-pair catalysis involving a formal propargylic carbocation from racemic propargylic alcohols40. Notwithstanding these significant advances, asymmetric synthesis of chiral allenes, particularly N-heteroaryl-substituted chiral allenes36, from readily available starting materials is particularly important and still remains a highly challenging task.
Chiral functionalized quinoline motifs are found in numerous biologically and pharmaceutically active alkaloids41. In addition, chiral quinoline is also a privileged scaffold for cinchonidine-type organocatalysts42 and ligands for transition metal catalysis43. In view of the importance of functionalized quinolines and the importance of chiral allenes, we are interested in developing a general, enantioselective protocol for the asymmetric synthesis of quinoline-substituted chiral allenes. Furthermore, quinoline-substituted allenes, which are important but synthetically challenging, are unprecedented to the best of our knowledge.
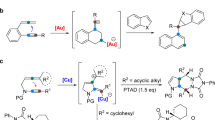
1,3-Enynes can be readily prepared via Sonogashira coupling reactions44, and are valuable potential precursors to access allenes. However, known synthetic methods to make chiral allenes from 1,3-enynes are rather limited to reactions of activated or electron-deficient 1,3-enynes17,18,19,33,34, and non-activated 1,3-enynes do not undergo these nucleophilic addition reactions due to their weak electrophilicity. Recent advances on Cu-catalyzed functionalization of alkenes indicate successful conversions of alkenes to nucleophilic alkylcopper species45,46,47,48,49,50,51,52,53,54. Furthermore, Cu-catalyzed functionalization of 1,3-enynes has also been studied (Fig. 1a)55,56. For example, Hoveyda reported a nucleophilic addition to aldehydes with nucleophiles generated from a borylcopper species and 1,3-enynes55. In 2016, Buchwald and Liu reported an asymmetric addition to ketones using nucleophiles derived from migratory insertion of 1,3-enynes to a chiral Cu-H intermediate56. Both reactions provide propargylic products selectively (Fig. 1a)55,56, although density functional theory (DFT)-calculation studies suggest a facile isomerization of initially generated propargylic copper species to allenyl copper intermediates during conversions of 1,3-enynes. Very recently, Hoveyda, Engle, and Ge groups have independently reported enantioselective copper-catalyzed hydroboration of 1,3-enynes through trapping of allenyl copper species with pinacolborane (Fig. 1b)57,58,59,60.

Copper-catalyzed enantioselective hydrofunctionalization of 1,3-enynes. a Trapping allenyl copper intermediates with carbonyls to form propargylic products. b Copper-catalyzed asymmetric hydroboration of 1,3-enynes. c This work: trapping allenyl copper intermediates with quinoline N-oxide
We recently reported an asymmetric alkylation of quinoline N-oxides with chiral alkylcopper species60. Inspired by the suitability of quinoline N-oxides as electrophiles for alkylcopper nucleophiles, we envisioned that quinoline N-oxides could serve as proper electrophiles to react with allenyl copper species to form chiral allene products through a five-membered, rather than seven-membered, transition state (Fig. 1c). Herein, we report our studies toward the development of an enantioselective synthesis of quinolinyl-containing chiral allenes.
Results
Evaluation of reaction conditions
We began our studies on this asymmetric allenylation by identifying an effective and selective chiral copper catalyst for the reaction between but-3-en-1-yn-1-ylbenzene (2a) and quinoline N-oxide (1a). We tested copper complexes generated in situ from CuOAc and various chiral bisphosphine ligands as catalysts. The results are summarized in Fig. 2. These reactions were conducted with (EtO)2MeSiH as hydride source in cyclohexane at 5 °C for 20 h, and the allene 3a was identified as a major product for these reactions. The reaction catalyzed by the combination of CuOAc and (R)-DTBM-segphos (L1) occurred smoothly and afforded 3a in good yield (74%), but with modest enantioselectivity (46%). Similar results were obtained for the reactions when catalysts generated from CuOAc and chiral ligands L2–L5 were employed. However, the reaction conducted with CuOAc and (S,S)-Ph-BPE (L6) produced 3a in high isolated yield (90%) with excellent enantioselectivity (95%). In addition, we also studied other reaction parameters, such as solvents, temperature, and hydride sources, and found that the reactions afforded the desired product 3a either in lower yields or with lower enantioselectivities under these tested conditions (for the details, see Table S1).

Evaluation of ligands for this copper-catalyzed chiral allene synthesis. Reaction conditions: quinoline N-oxide 1a (0.400 mmol), but-3-en-1-yn-1-ylbenzene 2a (0.200 mmol), (EtO)2MeSiH (0.400 mmol), CuOAc (8.0 μmol), ligand (12.0 μmol), cyclohexane (1 mL), 5 °C, 20 h, isolated yield, and ee was determined by chiral HPLC analysis
Scope of 1,3-enynes for Cu-catalyzed enantioselective allenylation of quinoline N-oxide
With an active chiral copper catalyst and reliable conditions identified, we studied the scope of alkyl-substituted 1,3-enynes for this Cu-catalyzed asymmetric allenylation, and the results are summarized in Fig. 3. In general, a variety of alkenyl- or alkyl-substituted 1,3-enynes (2b–2k) smoothly underwent this Cu-catalyzed enantioselective transformation to afford the corresponding allene products (3b–3k) in high isolated yields (61−89%) with good to excellent enantioselectivities (88–99%). Enynes bearing reactive groups, such as ester (3f and 3j), benzyl ether (3g), siloxy (3h), and imide (3k), are compatible with the reaction conditions. The absolute configuration of 3k was assigned as (S) by single-crystal X-ray diffraction (XRD) analysis.

Scope of aliphatic-substituted 1,3-enynes. Reaction conditions: quinoline N-oxide 1a (0.400 mmol), enyne (0.200 mmol), (EtO)2MeSiH (0.400 mmol), CuOAc (8.0 μmol), (S,S)-Ph-BPE (12.0 μmol), cyclohexane (1 mL), 5 °C, 20 h, isolated yields, and ee was determined by chiral HPLC analysis
The scope of aryl-substituted 1,3-enynes for this reaction is summarized in Fig. 4. In general, a wide range of aryl-substituted 1,3-enynes reacted under standard condition to produce the corresponding allene products (3l–3ag) in high yields (70−95%) and with high enantioselectivities (80–96%). The data in Fig. 4 indicate that the electronic properties of the aryl groups in aryl-substituted enynes have noticeable influence on the enantioselectivity of this reaction. For example, enynes containing electron-neutral and electron-rich aryl groups reacted to afford allenes 3a and 3l–3n with high enantioselectivities (90–94%), but the reactions of enynes containing electron-deficient aryl groups (2q–2s) took place with lower enantioselectivities (80–83% for 3q–3s). The steric hindrance at the para- or meta-positions of the aryl groups in aryl-substituted enynes has little effect on the enantioselectivity of this transformation (90–94% ee for 3l–3o). However, the reaction of an enyne containing ortho-substituted aryl group (2p) occurred with significantly lower enantioselectivity (80% ee for 3p).

Scope of aromatic-substituted 1,3-enynes. Reaction conditions: quinoline N-oxide (0.400 mmol), enyne (0.200 mmol), (EtO)2MeSiH (0.400 mmol), CuOAc (8.0 μmol), (S,S)-Ph-BPE (12.0 μmol), cyclohexane (1 mL), 5 °C, 20 h, isolated yields, and ee was determined by chiral HPLC analysis. #CuOAc (16.0 μmol), (S,S)-Ph-BPE (24.0 μmol)
This Cu-catalyzed asymmetric transformation tolerates a range of functionalities, including fluoro (3v), chloro (3w), bromo (3x), thioether (3y), tertiary amine (3z), trifluoromethyl ether (3aa), silylether (3ab), and carboxylic ester (3s, 3ac, and 3ah) moieties. 1,3-Enynes bearing nitrogen- and sulfur-containing heteroaryl substituents also reacted to produce the corresponding allenes (3ad and 3ae) in high yields and high enantioselectivity. In addition, 1,3-enynes with substitution at the olefenic terminus of the enynes ((Z)-2af and (Z)-2ag) also reacted to afford the corresponding allenes (3af and 3ag) in high isolated yields and good enantioselectivities. (Z)-2-Alken-4-ynoate (2ah), an activated 1,3-enyne, also reacted to give the allene product 3ah in high yield, albeit with a modest enantioselectivity. However, (Z)-oct-2-en-4-yne, a 1,3-enyne containing two alkyl groups at 1,4-positions, does not undergo this Cu-catalyzed asymmetric reaction.
Scope of quinoline N-oxides
Fig. 5 summarizes the scope of quinoline N-oxides for this Cu-catalyzed asymmetric allenylation transformation. In general, quinoline N-oxides with substituents at various positions reacted smoothly with the enyne 2a to yield the corresponding allenes (3ai–3ar) in high yields and high enantioselectivities. However, 8-methyl and 8-methoxyquinoline N-oxides reacted with decreased enantioselectivities (3as and 3at), which might be due to the increased steric hindrance around the N–O bond. Various reactive groups, such as chloro (3am and 3an), bromo (3ao and 3ap), siloxy (3aq), and carboxylic ester (3ar), at different positions of quinoline N-oxides were tolerated, and the allene products were isolated in high yields (61–97%) with high to excellent enantioselectivities (84–97% ee). However, the allenylation reactions of N-oxides of pyridines and isoquinolines do not occur under identified conditions.

Scope of quinoline N-oxides. Reaction conditions: quinoline N-oxide 1a (0.400 mmol), enyne (0.200 mmol), (EtO)2MeSiH (0.400 mmol), CuOAc (8.0 μmol), (S,S)-Ph-BPE (12.0 μmol), cyclohexane (1 mL), 5 °C, 20 h, isolated yields, and ee was determined by chiral HPLC analysis
Synthetic potential
To show the utility of this asymmetric protocol, the reaction of quinoline N-oxide 1a with enyne 2a was conducted on gram scale with 2 mol% copper catalyst at 5 °C. This reaction proceeded to full conversion of 2a in 20 h and afforded allene 3a in 89% isolated yield and with 93% ee (Fig. 6a). The enantioselectivity of this gram-scale reaction is comparable to that of a 0.200 mmol scale reaction (Fig. 2). In addition, we also showed that the quinoline-substituted allene products 3a and 3d underwent NIS-initiated cyclization to generate the corresponding 2-iodo-pyrrolo[1,2-a]quinolones in good yields (Fig. 6b, NIS = N-iodosuccinimide)61.

Synthetic applications. a Gram-scale reaction. b NIS-promoted cyclization of quinoline-substituted allenes. c–e Allenylation reactions with chiral allenes derived from vitamin E and cholesterol. Yields are reported for isolated materials after column chromatography on silica gel
This copper-catalyzed asymmetric allenylation was utilized for the modification of complex molecules. For example, the allenylation of quinoline N-oxide 1a with enantiometrically pure 1,3-enynes 5 and 7 (5 derived from vitamin E and 7 derived from cholesterol, respectively) occurred smoothly in the presence of 4 mol% CuOAc/(S,S)-Ph-BPE, affording chiral allenes 6 and 8 in high isolated yields with excellent diastereoselectivity (dr > 20:1), respectively (Fig. 6c, d). The corresponding reaction with enyne 7 conducted with CuOAc/(R,R)-Ph-BPE produced the opposite diastereomers 8′ in comparable yield and with similarly high diastereoselectivity. The results of this experiment indicate a mechanism involving catalyst control rather than substrate control when enantiometrically pure 1,3-enynes are used. The absolute configuration of compounds 6, 8, and 8′ was assigned by analogy to that of compound 3k.
Mechanistic considerations
To gain some mechanistic insights into this Cu-catalyzed asymmetric allenylation of quinoline N-oxides, we conducted a series of deuterium-labeling experiments (Fig. 7). The reaction of quinoline-2-D N-oxide 1a-d1 with 1,3-enyne 2a under standard conditions afforded 3a in high isolated yield after purification with flash chromatography on silica gel, but no deuterium was incorporated into the product (Fig. 7a). When conducted with Ph2SiD2, the reaction of quinoline N-oxide 1a with 1,3-enyne 2a produced 3a-d1 in high isolated yield after purification with flash chromatography on silica gel, and the deuterium was located in the methyl group of 3a (Fig. 7b).

Mechanistic studies. a–d Deuterium-labeling experiments. e Proposed catalytic pathway for this Cu-catalyzed asymmetric allenylation reaction
To detect the potential intermediates for this Cu-catalyzed asymmetric allenylation, we monitored by 1H nuclear magnetic resonance (NMR) analysis the reaction of quinoline N-oxide 1a and enyne 2a under standard conditions, and found that this reaction afforded I-3a with a modest diastereoselectivity (dr = 3.6:1) in high yield prior to the isolation of final product by flash choromatography on silica gel (Fig. 7c). Correspondingly, the reaction of quinoline-2-D N-oxide 1a-d1 with 1,3-enyne 2a produced the intermediate I-3a-d1 in 95% yield with a diastereoselectivity of 3.6:1 (Fig. 7d). Upon workup by column chromatography on silica gel, both intermediates I-3a and I-3a-d1 underwent re-aromatization to yield 2-allenyl quinoline 3a in high yields.
Based on the precedent of reactions of quinoline N-oxides with metal-alkyl reagents62,63, the Cu-catalyzed hydrofunctionalization of 1,3-enynes57,58,59, and the results of the above-mentioned mechanistic experiments, we proposed a plausible catalytic pathway for this Cu-catalyzed asymmetric allenylation of quinoline N-oxides (Fig. 7e). Activation of CuOAc with (EtO)2MeSiH in the presence of (S,S)-Ph-BPE (L*) produces a chiral copper hydride species (L*)Cu-H (A). Enantioselective migratory insertion of 1,3-enyne into (L*)Cu-H forms a propargylic copper intermediate B, which then undergoes a stereospecific isomerization to an allenyl copper species C. This allenyl copper intermediate reacts with quinoline N-oxide to form a copper species D, which reacts with (EtO)2MeSiH to afford a siloxane intermediate E and to regenerate the catalytically active Cu-H species A. The siloxane intermediate E then aromatizes to form the 2-allenyl quinoline product.
DFT calculations
With DFT calculations, we explored the mechanism and origins of chemoselectivity based on the reaction of quinoline N-oxide 1a and 1,3-enyne 2a catalyzed by the combination of CuOAc and (S,S)-Ph-BPE. The computed free energy diagram of the most favorable pathway is shown in Fig. 8a (see Supplementary Figure 18 for the detailed calculations on other pathways). From the Cu-H species int1, the hydrocupration of 2a occurs via TS2 and generates the propargyl copper intermediate int3 enantioselectively. The origins of this enantioselective hydrocupration step have been discussed, by Buchwald and Liu, for a Cu-catalyzed asymmetric addition of olefin-derived nucleophiles to ketones51. This hydrocupration step determines the enantioselectivity of this Cu-catalyzed allenylation reaction. From int3, the stereospecific 1,3-isomerization via TS4 to form int5 is facile. Thus, the propargyl copper species int3 and the corresponding allenyl copper species int5 are in a fast equilibrium. The final C–C bond formation proceeds through the five-membered ring transition state TS7 leading to the intermediate int8. Further exchange with (EtO)2MeSiH affords int9 and regenerates the Cu-H species int1 for the next catalytic cycle. Based on the free energy changes of the whole catalytic cycle, the allenyl copper intermediate int5 is the on-cycle resting state, and the rate-determining step is the regeneration of active catalyst int1 via TS9, which is consistent with previous related experimental mechanistic results64.

DFT studies. a DFT-computed free energy changes of the most favorable pathway for the Cu-catalyzed enantioselective allenylation of quinoline N-oxide 1a with 1,3-enyne 2a. b A competition experiment between sterically differentiated quinoline N-oxides
In addition, we also discovered an epimerization pathway from int5 to intS4 via TS-S5 (depicted in Fig. 8a), which could be responsible for the varied enantioselectivities obtained for the reactions with different quinoline N-oxides (Fig. 5). For example, 8-methyl quinoline N-oxide reacted with a significantly lower enantioselectivity (61% ee for 3at in Fig. 5) than quinoline N-oxide (95% ee for 3a in Fig. 2). We attributed this decreased enantioselectivity to a slower rate for the reaction of 8-methyl quinoline N-oxide, caused by the increased steric hindrance around the N–O bond. To further prove the difference in reactivity, we conducted a competition experiment of 1,3-enyne 2a between quinoline N-oxide and 8-methyl quinoline N-oxide in a single reaction vessel (Fig. 8b). Indeed, the allene product (3av) from the reaction of 8-methyl quinoline N-oxide was produced in a much smaller quantity than the product (3a) from the reaction of quinoline N-oxide.
The C–C bond formation step determines the chemoselectivity between allene and alkyne products. We carefully examined the possible transition states for the C–C bond formation, and the key competing ones are shown in Fig. 9. The propargyl copper intermediate int3 is 4.0 kcal/mol unfavorable as compared to the allenyl copper intermediate int5. This thermodynamic difference also reflects in the corresponding transition states, and TS12 and TS13 (from int3) are less favorable than TS7 and TS11 (from int5). Comparing TS7 and TS11, the calculations suggest a strong selectivity toward the formation of allene product via TS7, which is consistent with the experimental selectivity (Fig. 2). TS7 is a five-membered ring transition state, while the competing TS11 requires a seven-membered metallacycle, which is unfavorable due to the ring strain. The important role of metallacycle in determining the chemoselectivity provides insights for the reaction design with allenyl and propargyl copper species.

Optimized structures and relative free energies of competing transition states for C–C bond formation
Discussion
In summary, we report an efficient and enantioselective protocol for the asymmetric synthesis of trisubstituted allenes through a Cu-catalyzed allenylation of quinoline N-oxides with 1,3-enynes. A wide range of aryl- and alkyl-substituted 1,3-enynes reacted with quinoline N-oxides to afford the corresponding allenes in high isolated yields with good to excellent enantioselectivity. This asymmetric transformation could be readily conducted on a gram scale with a reduced catalyst loading (2 mol%), and thus this protocol presents a practical route to prepare enantiomerically enriched quinoline-containing trisubstituted allenes from readily accessible quinoline N-oxides and 1,3-enynes. DFT calculations suggest that this Cu-catalyzed allenylation occurs through a nucleophilic attack of quinoline N-oxide with an allenyl copper intermediate via a five-membered transition state, thereby providing a rationale to explain the obtained chemoselectivity toward an allene rather than a propargylic product. These findings will enable future development of new reactions with an allenyl or a propargyl copper intermediate, and these studies will be the subject of future work.
Methods
General procedure for the synthesis of chiral allenes
CuOAc (4 mol%, 0.9 mg) and (S,S)-Ph-BPE (6 mol%, 6.1 mg) were dissolved in cyclohexane (1 mL) in a screw-capped vial under argon atmosphere. The resulting mixture was stirred for 5 min, to which was added (EtO)SiHMe (0.400 mmol, 65 μL), 1,3-enynes (0.200 mmol), and quinoline N-oxide (0.400 mmol) successively. The vial was stirred at 5 °C for 20 h. The mixture was concentrated under vacuum and the crude product was purified by flash chromatography on silica with a mixture of ethyl acetate/hexane (1:100 to 1:50). The ee values were determined by high-performance liquid chromatography.
(Note: The allene products are unstable and prone to racemization in acidic solvent such as CHCl3. The ee values and optical rotation should be determined immediately after isolation.)
Mechanistic studies
Spectra pertaining to deuterium labeling, competition, and other mechanistic studies are available in Supplementary Figures 1-16.
Synthesis and characterization
Full synthetic procedures are available in the Supplementary Methods. The XRD structure of 3k is available in Supplementary Figure 17 and Supplementary Data 1-2. NMR spectra of purified organic compounds are available in Supplementary Figures 19-128.
Screening conditions
Further details of reaction conditions screened are available in Supplementary Table 1.
Computational study
Full procedures and results are available in the Supplementary Methods, Supplementary Figure 18, and Supplementary Table 2. Cartesian coordinates of calculated intermediates and transition states are available in Supplementary Table 3.
Data availability
The authors declare that all data supporting the findings of this study are available within the article and Supplementary Information files, and also are available from the corresponding author upon reasonable request. The X-ray crystallographic coordinates for the structure reported in this article has been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition number CCDC 1823673. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre at www.ccdc.cam.ac.uk/data_request/cif.
References
Hoffmann-Röder, A. & Krause, N. Synthesis and properties of allenic natural products and pharmaceuticals. Angew. Chem. Int. Ed. 43, 1196–1216 (2004).
Rivera-Fuentes, P. & Diederich, F. Allenes in molecular materials. Angew. Chem. Int. Ed. 51, 2818–2828 (2012).
Krause, N. & Hashmi, A. S. K. Modern Allene Chemistry (Wiley, 2004).
Trost, B. M., Fandrick, D. R. & Dinh, D. C. Dynamic kinetic asymmetric allylic alkylations of allenes. J. Am. Chem. Soc. 127, 14186–14187 (2005).
Manzuna Sapu, C., Bäckvall, J.-E. & Deska, J. Enantioselective enzymatic desymmetrization of prochiral allenic diols. Angew. Chem. Int. Ed. 50, 9731–9734 (2011).
Sherry, B. D. & Toste, F. D. Gold(I)-catalyzed propargyl claisen rearrangement. J. Am. Chem. Soc. 126, 15978–15979 (2004).
Pu, X. & Ready, J. M. Direct and stereospecific synthesis of allenes via reduction of propargylic alcohols with Cp2Zr(H)Cl. J. Am. Chem. Soc. 130, 10874–10875 (2008).
Uehling, M. R., Marionni, S. T. & Lalic, G. Asymmetric synthesis of trisubstituted allenes: copper-catalyzed alkylation and arylation of propargylic phosphates. Org. Lett. 14, 362–365 (2012).
Wu, S., Huang, X. & Wu, W. A C-H bond activation-based catalytic approach to tetrasubstituted chiral allenes. Nat. Commun. 6, 7946 (2015).
Ruchti, J. & Carreira, E. M. Rh-catalyzed stereospecific synthesis of allenes from propargylic benzoates and arylboronic acids. Org. Lett. 18, 2174–2176 (2016).
Ogasawara, M. Catalytic enantioselective synthesis of axially chiral allenes. Tetrahedron.: Asymmetry 20, 259–271 (2009).
Yu, S. & Ma, S. How easy are the syntheses of allenes? Chem. Commun. 47, 5384–5418 (2011).
Neff, R. K. & Frantz, D. E. Recent advances in the catalytic syntheses of allenes: a critical assessment. ACS Catal. 4, 519–528 (2014).
Ye, J. & Ma, S. Conquering three-carbon axial chirality of allenes. Org. Chem. Front. 1, 1210–1224 (2014).
Shirakawa, S., Liu, S. & Kaneko, S. Organocatalyzed asymmetric synthesis of axially, planar, and helical chiral compounds. Chem. Asian J. 11, 330–341 (2016).
Chu, W.-D., Zhang, Y. & Wang, J. Recent advances in catalytic asymmetric synthesis of allenes. Catal. Sci. Technol. 7, 4570–4579 (2017).
Nishimura, T., Makino, H. & Nagaosa, M. Rhodium-catalyzed enantioselective 1,6-addition of arylboronic acids to enynamides: asymmetric synthesis of axially chiral allenylsilanes. J. Am. Chem. Soc. 132, 12865–12867 (2010).
Wang, M., Liu, Z.-L. & Zhang, X. Synthesis of highly substituted racemic and enantioenriched allenylsilanes via copper-catalyzed hydrosilylation of (Z)-2-alken-4-ynoates with silylboronate. J. Am. Chem. Soc. 137, 14830–14833 (2015).
Yao, Q., Liao, Y. & Lin, L. Efficient synthesis of chiral trisubstituted 1,2‐allenyl ketones by catalytic asymmetric conjugate addition of malonic esters to enynes. Angew. Chem. Int. Ed. 55, 1859–1863 (2016).
Imada, Y., Nishida, M. & Kutsuwa, K. Palladium-catalyzed asymmetric amination and imidation of 2,3-allenyl phosphates. Org. Lett. 7, 5837–5839 (2005).
Wan, B. & Ma, S. Enantioselective decarboxylative amination: synthesis of axially chiral allenyl amines. Angew. Chem. Int. Ed. 52, 441–445 (2013).
Wang, G., Liu, X. & Chen, Y. Diastereoselective and enantioselective alleno-aldol reaction of allenoates with isatins to synthesis of carbinol allenoates catalyzed by gold. ACS Catal. 6, 2482–2486 (2016).
Crouch, I. T., Neff, R. K. & Frantz, D. E. Pd-catalyzed asymmetric β-hydride elimination en route to chiral allenes. J. Am. Chem. Soc. 135, 4970–4973 (2013).
Li, Z., Boyarskikh, V. & Hansen, J. H. Scope and mechanistic analysis of the enantioselective synthesis of allenes by Rhodium-catalyzed tandem ylide formation/[2,3]-sigmatropic rearrangement between donor/acceptor carbenoids and propargylic alcohols. J. Am. Chem. Soc. 134, 15497–15504 (2012).
Wang, Y., Zhang, W. & Ma, S. A room-temperature catalytic asymmetric synthesis of allenes with ECNU-Phos. J. Am. Chem. Soc. 135, 11517–11520 (2013).
Liu, Y. et al. Nickel(II)-Catalyzed asymmetric propargyl and allyl Claisen rearrangements to allenyl- and allyl-substituted β-ketoesters. Angew. Chem. Int. Ed. 53, 11579–11582 (2014).
Liu, Y. et al. Synergistic kinetic resolution and asymmetric propargyl claisen rearrangement for the synthesis of chiral allenes. Angew. Chem. Int. Ed. 55, 4054–4058 (2016).
Xiao, Q., Xia, Y. & Li, H. Coupling of N-tosylhydrazones with terminal alkynes catalyzed by copper(I): synthesis of trisubstituted allenes. Angew. Chem. Int. Ed. 50, 1114–1117 (2011).
Tang, Y., Chen, Q. & Liu, X. Direct synthesis of chiral allenoates from the asymmetric C-H insertion of α-diazoesters into terminal alkynes. Angew. Chem. Int. Ed. 54, 9512–9517 (2015).
Chu, W.-D., Zhang, L. & Zhang, Z. Enantioselective synthesis of trisubstituted allenes via Cu(I)-catalyzed coupling of diazoalkanes with terminal alkynes. J. Am. Chem. Soc. 138, 14558–14561 (2016).
Poh, J.-S., Makai, S. & Keutz, T. Rapid asymmetric synthesis of disubstituted allenes by coupling of flow-generated diazo compounds and propargylated amines. Angew. Chem. Int. Ed. 56, 1864–1868 (2017).
Tang, X. et al. CuBr2-catalyzed enantioselective routes to highly functionalized and naturally occurring allenes. Org. Chem. Front. 2, 688–691 (2015).
Zhang, W., Zheng, S. & Liu, N. Enantioselective bromolac-tonization of conjugated (Z)-enynes. J. Am. Chem. Soc. 132, 3664–3665 (2010).
Qian, H., Yu, X. & Zhang, J. Organocatalytic enantioselective synthesis of 2,3-allenoates by intermolecular addition of nitroalkanes to activated enynes. J. Am. Chem. Soc. 135, 18020–18023 (2013).
Liu, H., Leow, D. & Huang, K.-W. Enantioselective synthesis of chiral allenoates by guanidine-catalyzed isomerization of 3-alkynoates. J. Am. Chem. Soc. 131, 7212–7213 (2009).
Jiang, Y., Diagne, A. B. & Thomson, R. J. Enantioselective synthesis of allenes by catalytic traceless petasis reactions. J. Am. Chem. Soc. 139, 1998–2005 (2017).
Hashimoto, T., Sakata, K. & Tamakuni, F. Phase-transfer-catalysed asymmetric synthesis of tetrasubstituted allenes. Nat. Chem. 5, 240–244 (2013).
Mbofana, C. T. & Miller, S. J. Diastereo- and enantioselective addition of anilide-functionalized allenoates to N-acylimines catalyzed by a pyridylalanine-based peptide. J. Am. Chem. Soc. 136, 3285–3292 (2014).
Tap, A., Blond, A. & Wakchaure, V. N. Chiral allenes via alkynylogous mukaiyama aldol reaction. Angew. Chem. Int. Ed. 55, 8962–8965 (2016).
Qian, D., Wu, L. & Sun, J. Organocatalytic synthesis of chiral tetrasubstituted allenes from racemic propargylic alcohols. Nat. Commun. 8, 567 (2017).
Kumar, S., Bawa, S. & Gupta, H. Biological activities of quinoline derivatives. Mini Rev. Med Chem. 14, 1648–1654 (2009).
Tian, S., Chen, Y. & Huang, J. Asymmetric organic catalysis with modified cinchona alkaloids. Acc. Chem. Res. 37, 621–631 (2004).
Chen, G., Zhuang, G. & Li, G. Ligand-enabled β-C-H arylation of α-amino acids without installing exogenous directing groups. Angew. Chem. Int. Ed. 56, 1506–1509 (2017).
Cornelissen, L., Lefrancq, M. & Riant, O. Copper-catalyzed cross-coupling of vinylsiloxanes with bromoalkynes: synthesis of enynes. Org. Lett. 16, 3024–3027 (2014).
Miki, Y., Hirano, K. & Miura, M. Copper-catalyzed intermolecular regioselective hydroamination of styrenes with polymethylhydrosiloxane and hydroxylamines. Angew. Chem. Int. Ed. 52, 10830–10834 (2013).
Yang, Y., Shi, S.-L. & Niu, D. Catalytic asymmetric hydroamination of unactivated internal olefins to aliphatic amines. Science 349, 62–66 (2015).
Niu, D. & Buchwald, S. L. Design of modified amine transfer reagents allows the synthesis of α-chiral secondary amines via CuH-catalyzed hydroamination. J. Am. Chem. Soc. 137, 9716–9721 (2015).
Xi, Y., Butcher, T. W., Zhang, J. & Hartwig, J. F. Regioselective, asymmetric formal hydroamination of unactivated internal alkenes. Angew. Chem. Int. Ed. 55, 776–780 (2016).
Han, J. T., Jang, W. J., Kim, N. & Yun, J. Asymmetric synthesis of borylalkanes via copper-catalyzed enantioselective hydroallylation. J. Am. Chem. Soc. 138, 15146–15149 (2016).
Zhou, Y., Bandar, J. S. & Buchwald, S. L. Enantioselective CuH-catalyzed hydroacylation employing unsaturated carboxylic acids as aldehyde surrogates. J. Am. Chem. Soc. 139, 8126–8129 (2017).
Xi, Y. & Hartwig, J. F. Mechanistic studies of copper-catalyzed asymmetric hydroboration of alkenes. J. Am. Chem. Soc. 139, 12758–12772 (2017).
Rendler, S. & Oestreich, M. Polishing a diamond in the rough: “Cu-H” catalysis with silanes. Angew. Chem. Int. Ed. 46, 498–504 (2007).
Deutsch, C., Krause, N. & Lipshutz, B. H. CuH-catalyzed reactions. Chem. Rev. 108, 2916–2927 (2008).
Pirnot, M. T., Wang, Y.-M. & Buchwald, S. L. Copper hydride catalyzed hydroamination of alkenes and alkynes. Angew. Chem. Int. Ed. 55, 48–57 (2016).
Meng, F., Haeffner, F. & Hoveyda, A. H. Diastereo- and enantioselective reactions of bis(pinacolato)diboron, 1,3-enynes, and aldehydes catalyzed by an easily accessible bisphosphine-Cu complex. J. Am. Chem. Soc. 136, 11304–11307 (2014).
Yang, Y., Perry, I. B. & Buchwald, S. L. Copper-catalyzed asymmetric addition of olefin-derived nucleophiles to ketones. Science 353, 144–150 (2016).
Huang, Y., del Pozo, J. & Hoveyda, A. H. Enantioselective synthesis of trisubstituted allenyl-B(pin) compounds by phosphine-Cu-catalyzed 1,3-enyne hydroboration. insights regarding stereo-chemical integrity of Cu-allenyl intermediates. J. Am. Chem. Soc. 140, 2643–2655 (2018).
Sang, H. L., Yu, S. & Ge, S. Copper-catalyzed asymmetric hydroboration of 1,3-enynes with pinacolborane to access chiral allenylboronates. Org. Chem. Front. 5, 1284–1287 (2018).
Gao, D.-W., Xiao, Y. & Engle, K. M. Catalytic, enantioselective synthesis of allenyl boronates. ACS Catal. 8, 3650–3654 (2018).
Yu, S., Sang, H. L. & Ge, S. Enantioselective copper-catalyzed alkylation of quinoline N-oxides with vinylarenes. Angew. Chem. Int. Ed. 56, 15896–15900 (2017).
Grandclaudon, C., Michelet, V. & Toullec, P. Y. Synthesis of polysubstituted 2-iodoindenes via iodonium-induced cyclization of arylallenes. Org. Lett. 18, 676–679 (2016).
Andersson, H., Gustafsson, M. & Bostrom, D. The regio- and stereoselective synthesis of trans-2,3-dihydropyridine N-oxides and piperidines. Angew. Chem. Int. Ed. 48, 3288–3291 (2009).
Larionov, O. V., Stephens, D. & Mfuh, A. Direct, catalytic, and regioselective synthesis of 2-alkyl-, aryl-, and alkenyl-substituted N-heterocycles from N-oxides. Org. Lett. 16, 864–867 (2014).
Bandar, J. S., Pirnot, M. T. & Buchwald, S. L. Mechanistic studies lead to dramatically improved reaction conditions for the Cu-catalyzed asymmetric hydroamination of olefins. J. Am. Chem. Soc. 137, 14812–14818 (2015).
Acknowledgements
This work was supported by the grant from NUS Young Investigator Award (R-143-000-630-133) and the Ministry of Education of Singapore (R-143-000-A07-112). Computational study was supported by the National Natural Science Foundation of China (21702128) and calculations were performed on high-performance computing system at Department of Chemistry, Zhejiang University.
Author information
Authors and Affiliations
Contributions
S.Y. planned and conducted most of the experiments; S.Y. and H.L.S. prepared substrates for the reaction scope evaluation; S.-Q.Z. and X.H. conducted DFT calculation studies. S.G. directed the projects and S.G., S.Y., and X.H. co-wrote the manuscript. All authors contributed to the discussion.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yu, S., Sang, H.L., Zhang, SQ. et al. Catalytic asymmetric synthesis of chiral trisubstituted heteroaromatic allenes from 1,3-enynes. Commun Chem 1, 64 (2018). https://doi.org/10.1038/s42004-018-0065-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42004-018-0065-4
This article is cited by
-
Copper(I)-catalyzed asymmetric 1,3-dipolar cycloaddition of 1,3-enynes and azomethine ylides
Nature Communications (2023)
-
Photoredox cobalt-catalyzed regio-, diastereo- and enantioselective propargylation of aldehydes via propargyl radicals
Nature Communications (2023)
-
Enantioselective synthesis of tetrasubstituted allenes via addition/arylation tandem reaction of 2-activated 1,3-enynes
Science China Chemistry (2023)
-
Photo and copper dual catalysis for allene syntheses from propargylic derivatives via one-electron process
Nature Communications (2022)
-
Copper(I)-catalyzed diastereo- and enantio-selective construction of optically pure exocyclic allenes
Nature Communications (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
