
Abstract
It is crucial to control the reactivity of surface silicon atoms for applications in miniaturized silicon-based nanodevices. Here we demonstrate that reactive silicon atoms are made unreactive by forming a Si16 cage that encapsulates a metal atom. Specifically, group 5 metal-encapsulating Si16 nanoclusters (M@Si16: M = V, Nb, and Ta) exhibit alkali-like superatomic behavior on n-type C60 substrates, where charge transfer between M@Si16 and C60 satisfies the 68-electron shell closure as M@Si16+. The oxidation properties of M@Si16+ are investigated by X-ray photoelectron spectroscopy, revealing that the chemical stability of the caged silicon surface towards oxygen is enhanced by a factor of 104 compared to a crystalline silicon surface, and that M@Si16 are oxidized stepwise from the outer Si16 cage to the central metal atom. While the nanoclusters share a common Si16 cage, their chemical robustness depends on a superatomic “periodicity” (Ta@Si16 > V@Si16 > Nb@Si16) which is explained by the electron density distributions of M@Si16 investigated by DFT calculations.
Similar content being viewed by others

Introduction
Silicon is the most important element in the development of modern technology. The demands of the latest functional devices (e.g. to be lightweight, flexible, energy saving, etc.) make it especially important that the functional units are constructed to be as small as possible, on the nanometer scale1,2. Since miniaturization of functional units with conventional patterning methods like photolithography has almost reached its limit3, it is urgent that we discover methods to construct Si-based low-dimensional functional nanomaterials (e.g. silicon nanodots, nanowires, nanosheets) that are fabricated with bottom-up technologies utilizing fine synthesis methods in the gas and liquid phases4,5,6,7,8,9, where each silicon nanomaterial is regarded as a building block of functionality. Furthermore, from a material science perspective, the chemical stability of the fabricated silicon nanomaterials is also very important because the surface oxidation reaction in the nanomaterials often changes their original functions. Therefore, strategies to make Si-based nanomaterials chemically robust are indispensable. In particular, chemical reactivity towards reactive oxygen gas is a common issue in the development of practical nanodevices and is also worth comparing to that of naked Si crystalline surfaces that exhibit an extremely reactive dangling-bond of Si atoms.
Silicene, a counter part of graphene, is a two-dimensional (2D) functional nanomaterial with Si atoms, achieving high in-plane conductivity combined with flexibility9,10,11,12,13. Although the Si atom, unlike the C atom, preferably forms sp3 hybridized chemical bonds, the 2D conformation of silicene forces it to make sp2-like Si−Si bonds, resulting in a nonflat 2D network (buckling structure)9,10,11,12. In spite of the structural deviation from the ideally flat sp2 conformation (cf. graphene), an enhancement of chemical robustness towards oxygen has been reported for silicene, which reaches a factor of 103 as compared to that of sp3 bonded crystalline Si11,12. To make a stable Si compound with “zero dimensional” nanostructure, by analogy with C60 fullerene, one can assume that a rounded silicene will have stable caged surface. From this viewpoint, much experimental and theoretical research on the simplest Si caged materials have been conducted, including Si6014,15,16,17. However, such a stable caged Si compound has not so far been clearly identified, although the Si compound is highly expected to work in harmony with well-established Si nanotechnology.
Making a nanoscale rounded Si surface, i.e. caged Si, with metal encapsulation is one promising approach to realize chemically stabilized Si-based functional compounds18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36. Here the central metal atom enhances the electronic stability of the Si cage by sharing their valence electrons to form a superatom30,33,34,35,36. In fact, transition metal-encapsulating Si16 nanocluster superatoms (M@Si16) can be synthesized in the gas phase, exhibiting a magic number behavior like a C60 fullerene23,27. Since their chemical properties can be tuned by choosing a different metal atom in the center of the geometrically close-packed M@Si16, the assembly of a series of M@Si16 could be a pathway to building Si-based nanomaterials.
In the literature, there are two electron counting rules for “superatoms”26,28,29,34,36 and “18-electrons”19,21,25,30 to explain the electronic properties of M@Sin. Including a charge state, the electronic stabilities of M@Si16 (M = Sc, Ti, and V) are governed by their superatomic nature and exhibit a shell-closure of 68 electron (16 × 4 e− (Si) + 4 e− (M) = 68 e−)26,28,29,34,36. In addition, the extended 18-electron rule can contribute to form M@Sin (n = 12, 14, and 16)19,21,25,30. In the DFT calculations for Ti@Si1634, its Kohn−Sham orbitals demonstrate that the 3s and 3p orbitals of the Si atoms are delocalized into superatomic orbitals, where the 68-electron shell closure, (1S)2(1P)6(1D)10(1F)14(2S)2(1G)18(2P)6(2D)10, governs their enhanced stability as found experimentally in gas phase27,28. In addition, a part of electronic states in the calculated Kohn−Sham orbitals can also be described by 18-electron filling to the 4s, 4p, and 3d orbitals of the central Ti atom. Since the two electron counting rules of “superatoms” of 68 electrons and “18-electrons” for a transition metal atom are not exclusive to each other, both may contribute to stabilizing the M@Si16 superatoms.
Since the group-5 transition metals V, Nb, and Ta possess five valence electrons, the M@Si16 superatom (M = V, Nb, and Ta) is stabilized as cationic species as M@Si16+ to satisfy the 68-electron shell23,27,34,36,37,38,39,40,41, and therefore the M@Si16 neutrals are classified as alkali-like superatoms. In addition, the Si cages that include a group-3 atom (M = Sc, Y, and Lu) or a group-4 atom (M = Ti, Zr, and Hf) exhibit halogen-like and rare gas-like behaviors in a superatom periodic table23,27,34,36.
We previously reported the chemical characterization of Ta@Si16 deposited on a graphite substrate as a demonstration of the use of X-ray photoelectron spectroscopy (XPS) to element-specifically clarify the local electronic structure of the caged Si atoms39, although the analysis of the chemical reactivity was qualitative and the species were limited to Ta@Si16 in an alkali-like superatomic family.
Here we provide chemical characterization of alkali-like superatoms, including group 5 central atoms, to demonstrate periodicity in a superatom periodic table together with quantitative kinetic analyses for oxidation reactions which suggest metal-dependent superatom chemistry. The degree of chemical robustness of these M@Si16 superatoms, which is about 104 higher than a naked crystalline Si surface, depends on the central metal atom: Nb@Si16 is the most reactive owing to the electron density spreading outside the Si16 cage, as suggested by DFT calculations.
Results
Generation and deposition of M@Si16 +
Figure 1 shows the mass spectra of cationic M−Si binary nanoclusters (Fig. 1a, M = V, Fig. 1b, Nb, and Fig. 1c, Ta) generated with a magnetron sputtering (MSP) method34,36,42. In the mass region of 400−700 m/z, a series of M1Sin+ ions are predominantly observed. Although the series of Sin+ and M2Sin+ are observed as minor products especially for M = Nb (as marked in the figure) those species are almost negligible around the M@Si16+ region. Optimizing the production conditions of Ar and He flows and sputtering power, the M@Si16+ signals prominently appear as magic numbers; V@Si16+ = 500 m/z, Nb@Si16+ = 542 m/z, and Ta@Si16+ = 630 m/z. Although the MSP method usually generates cold nanoclusters with less magic number behavior compared to those made with a laser vaporization method23,27,34,36,42, the typical ion currents of generated M@Si16+ were larger than 500 pA with enhanced ion currents compared to their neighbors, as shown in Fig. 1. The flux of 3×109 clusters/s (at 500 pA) can deposit an amount of 1 monolayer (ML) on a substrate within 4 h, where the coverage is defined by the total ion current, and the amounts in the deposition area are consistent with those evaluated with step-by-step XPS measurements for C 1s and ultraviolet photoelectron spectroscopy (UPS) for C60-derived signals (Supplementary Figures 1 and 2). For XPS measurements, the peak profiles derived from the core levels of Si and M elements remain the same for coverages in the range of 0.3−1.4 ML, while the increase in their intensities show saturation behavior at >1 ML (Supplementary Figure 3), and M@Si16+ is typically deposited to 0.6 ML in all XPS analyses.

Mass spectra of MmSin binary nanocluster cations. The mass spectra for a VmSin, b NbmSin, and c TamSin cations show that the series of M1Sin+ ions (marked in the figures for n = 10−20) are mainly generated with a magic number behaviors of M@Si16+. The vertical scale bars represent ion currents of 500 pA, and all M@Si16+ are larger than 500 pA. While Sin+ and M2Sin+ are also resolved for M = Nb and Ta with minor products, those are negligible around the M@Si16+ region
Chemical environment of M@Si16 on C60
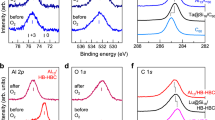
Figure 2 shows the XPS spectra for V@Si16, Nb@Si16, and Ta@Si16 deposited on the C60 substrate (photon energy (hν) = 1253.6 eV), where the binding energy regions encompass the core levels of Si 2p (Fig. 2a−c) and central metal atoms (Fig. 2d; V 2p, 2e; Nb 3d, and 2f; Ta 4f). Since all XPS signals derived from Si and M increase linearly with the deposition up to 1 ML (Supplementary Figure 3), all M@Si16 are nondestructively immobilized on the C60 substrate without forming three-dimensional aggregates, consistent with scanning tunneling microscopy (STM) observations37,38. Using an instrumental broadening (Voigt peak function; full width at half maximum (FWHM) = 1.09 eV with Lorentzian and Gaussian widths of 0.56 eV and 0.75 eV, respectively), the peaks of Si 2p core level are deconvoluted into two components which are due to spin-orbit splitting (2p2/1,3/2) with a known peak separation (0.608 eV) and branching ratio of 2J+1 (1:2)43. As seen in Fig. 2, the peak reproductions of Si 2p for all M@Si16 indicate that the chemical environment of Si atoms composing M@Si16 is uniform within the energy resolution of the XPS measurements; this implies that the surrounding Si atoms are almost equally distributed around the central metal atoms. Compared to the binding energy of Si 2p3/2 for bulk Si (99.5 eV)44, the Si 2p3/2 peaks for all M@Si16 at 99.6−99.7 eV indicates that the charge states of the surrounding Si atoms are close to that of bulk Si (Si0). Note that a small extra-component at a higher binding energy is seen for the Si 2p peak for Nb@Si16, which is presumably caused by partial oxidation due to lower chemical robustness of Nb@Si16 compared to V@Si16 and Ta@Si16, as discussed later on.

XPS spectra for M@Si16 superatom deposited on a C60 substrate. The binding energies are around core levels of Si 2p for a V@Si16, b Nb@Si16, and c Ta@Si16, and metal core levels of d V 2p for V@Si16, e Nb 3d for Nb@Si16, and f Ta 4 f for Ta@Si16. Reference binding energies of Si 2p3/2 for bulk Si (Si0), and M (V 2p2/3, Nb 3d5/2, and Ta 4f7/2) for M0 and M2+ are marked by vertical bars. Their binding energies show zerovalent Si and monovalent M+
For the central metal atoms of V, Nb, and Ta, a couple of XPS peaks are distinctly observed due to the spin-orbit splittings of core levels (V: 2p1/2,3/2, Nb: 3d3/2,5/2, and Ta: 4f5/2,7/2). Similarly to Si 2p, the peaks are reproduced by a single component using the spectroscopic parameters of energy separations and branching ratios of 2J+1; 7.5 eV, 2:1 for V 2p45, 2.8 eV, 3:2 for Nb 3d46, and 1.915 eV, 4:3 for Ta 4 f47. Strictly speaking, both V 2p and Nb 3d peaks show a slight asymmetry with tailing components at a higher binding energy, whereas the Ta 4f peaks can be accurately fitted with a symmetric peak function. The tail structures in XPS spectra are seemingly caused not by a partial oxidation of the sample but by a photoelectron energy loss with a subsequent electron-hole excitation48, which occurs more effectively at low photoelectron kinetic energy, Ek (higher binding energy). In fact, (1) an XPS spectrum for a standard sample of an Ag crystal, which has 3d derived core level at around 370 eV in binding energy (Ek ≈ 880 eV), shows asymmetric features (Supplementary Figure 4), and (2) the XPS spectra taken with the Mg Kα line for the clean surfaces of V, Nb, and Ta have similar spectral widths for V 2p (Ek ≈ 740 eV) and Nb 3d (Ek ≈ 1040 eV) core levels but not for Ta 4f (Ek ≈ 1230 eV)46,49,50. Therefore, the XPS spectra in Fig. 2 indicate that the chemical environments of both V and Nb in M@Si16 are also uniform, similarly to Ta@Si16, showing that all M@Si16 superatoms deposited preserve the original metal encapsulating Si16 cage structures on a C60 substrate. From a theoretical perspective, Sen et al. recently calculated the structure of neutral Ta@Si16 attaching to a graphite substrate, where all Ta−Si distances are similar (2.85−2.97 Å), thereby resulting in a single and uniform chemical component of Si 2p and Ta 4f core levels41. The calculation reasonably supports the above conclusion in the present C60 substrate.
To compare the uniformity of chemical environments with a “nonencapsulating” metal-silicon nanocluster, we chose TaSi8+; eight Si atoms insufficient to encapsulate a Ta atom51. Accordingly, TaSi8+ cations were deposited onto a C60 substrate (Supplementary Figure 5), using MSP conditions that slightly enhance TaSi8+ cation formation (Supplementary Figure 6). For the TaSi8 deposited film, indeed, the XPS peaks for Si 2p and Ta 4f are fitted by broader peak functions (1.66 eV for Si 2p and 1.36 eV for Ta 4f in FWHM, see Supplementary Figure 5), which is 1.52−1.25 times broader than those of Ta@Si16. Together with the shift of the peak position towards a higher binding energy, the peak broadenings show inhomogeneous chemical environments of Si and Ta atoms due to the nonencapsulating structure of TaSi8. In other words, the uniform chemical environment in M@Si16 on a C60 substrate reveals the presence of the symmetric metal-encapsulating feature of the Si16 cage.
The charge state of the Si and central metal atoms are worth discussing to emphasize the characteristics of the alkali-like M@Si16. As shown in the right hand of Fig. 2, since the peaks of metal core levels appear between metallic (M0) and divalent (M2+) charge states45,46,47,52,53, the charge state of each central metal atom is assignable to monovalent (M+), although such monovalent compounds of group-5 transition metals are rarely seen in the nature. Since the surrounding Si atoms are in a neutral charge state (Si0) (Fig. 2a−c), the overall charge state of M@Si16 is characterized to be +1 (namely M@Si16+), which favorably satisfies the 68 e− shell closure for M@Si16 with group 5 central metals. On the other hand, the C 1s peak shifts towards lower binding energy with depositing the M@Si16 (Supplementary Figure 7). The results show that C60 is negatively charged due to a high electron affinity (2.68 eV) of C60 (n-type organic semiconductor)54, forming a charge transfer (CT) complex of M@Si16+C60−37,38,40. Furthermore, the stoichiometric ratio between M@Si16 and C60 is 1:1 (M@Si16+C60−) because the coverage dependence of the XPS/UPS spectra exhibits peak shift behaviors that saturated at 1 ML (Supplementary Figures 1, 2).
Oxidative reaction of M@Si16
As shown in Fig. 2, the extra chemical component is observed in the Si 2p XPS signal for Nb@Si16, implying that the chemical robustness of M@Si16 is dependent on the central metal atom. Indeed, only for Nb@Si16 is the Si 2p profile sensitive to change during the XPS measurements which irradiate trace amount of atomic oxygen from the X-ray source (Supplementary Figure 8). Chemical robustness is an important issue in the characterization of the alkali-like superatom family for Si-based nanomaterials. Here, we evaluate the chemical robustness of M@Si16 as their oxidative reactivities towards a reactive gas of O2, where both XPS profiles for Si 2p and the central metal core levels are obtained after step-by-step O2 exposures at room temperature. The amount of oxygen is defined by Langmuir (1 L = an exposure of 1.33×10−4 Pa during 1 s). For every 1 L exposure, an O2 molecule hits the M@Si16 on the substrate approximately two times. As shown in Fig. 3, at 1×104 L exposure, the original peak components for Si 2p and the central metal M are mostly retained, showing the enhanced chemical robustness of M@Si16 on C60. This compares with the behavior of clean crystalline Si surfaces as well as those of the central metal elements which are easily oxidized by less than 10 L oxygen exposure55,56,57,58,59,60,61. In particular, the enhanced chemical robustness of “Si surface” at M@Si16 is worth comparing quantitatively with that at naked crystalline Si, whose oxidative reactivity towards oxygen has been extensively studied with many surface science techniques55,56,57,58. Through oxygen exposure, the multiple oxidation stages of Si atoms (e.g. Si1+−Si4+) are formed in both M@Si16 (see next section) and bulk Si surfaces. Here the chemical robustness towards oxygen for M@Si16 is considered as an XPS intensity ratio between unreacted Si (Si0) and reacted Si (Si1+−Si4+, denoted Si1~4+ in Fig. 4) after oxygen exposures. This simple assumption is considered valid because all Si atoms in M@Si16 are equally exposed to oxygen in the present dispersive immobilization on the C60 substrate. From an XPS study of the oxidation of Si single crystal surfaces, it has been reported that 80% of the surface Si atoms react with oxygen by 5 L O2 exposure, thereby showing oxidized Si (Si1+−Si4+)-derived peak components55. As analyzed in the next section, for M@Si16, it shows only 20% (Ta@Si16)−50% (Nb@Si16) of the oxidized Si atoms after 1×104 L O2 exposures (Si1~4+ in Fig. 4). Compared to the case of a bulk Si surface, approximately one-fourth of the oxidative reaction occurs at the Si atoms of Ta@Si16 despite 2×103 times higher oxygen dosages. By metal-encapsulation of Si16 cage, therefore, the chemical robustness of the Si surface towards oxygen is enhanced by a factor of 104.

XPS spectra for M@Si16 deposited on a C60 substrate measured after different O2 exposures. The XPS spectra obtained before and after O2 exposures for V@Si16 (a Si 2p and b V 2p), Nb@Si16 (c Si 2p and d Nb 3d), and Ta@Si16 (e Si 2p and f Ta 4f), where the amounts of O2 exposures are defined as Langmuir (L). All spectra shift towards higher binding energy with oxygen exposures, indicating the M@Si16 are oxidized and finally proceed to SiO2 and M2O5 species. Reference binding energies of oxidized Si (Si3+and Si4+) and M (M5+) are marked by vertical bars. The spectra of Si 2p are deconvoluted into three components of initial (Si0), intermediate (Si1,2+), and final (Si3,4+) ones. For the metals, the spectra are dominated by M+ and M5+ components, although for Nb 3d, the spectra are deconvoluted with Nb2+ as well as Nb+ and Nb5+. For Ta 4f, an O 2s contribution is also considered in the deconvolution. More details of the fitting procedures are seen in Supplementary Note 1

Area ratio of spectral components of Si 2p and core levels of M. The initial (Si0), intermediate (Si1,2+), and final (Si3,4+) charge states for Si 2p for a V@Si16, b Nb@Si16, and c Ta@Si16, and initial (M1+) and final (M5+) charge states for core levels of d V@Si16 (V 2p), e Nb@Si16 (Nb 3d), and f Ta@Si16 (Ta 4f) are obtained from the analysis of XPS spectra in Fig. 3. The summation of Si1,2+ and Si3,4+ (solid pink circles) is also shown. With increasing oxygen exposure, for Si 2p, the intermediate states of Si1,2+ (solid green triangles) increase at first, and then Si3,4+ (solid orange triangles) dominates at higher O2 exposures. The intensity evolutions of M5+ (including M2+ for Nb 4d) are similar to ones for Si3,4+, corresponding to Si cage collapse
At 5×1010 L (=1 atm × 60 s), some changes in the Si 2p signal are evident. However, the spectra of central metal M mostly retain their original profiles, showing that the metal atom is still protected against oxidation by the partially oxidized Si16 cage. The enhanced stability by the metal encapsulation suggests that air exposure within a few minutes lead to insignificant degradation of M@Si16. In fact, a “nonencapsulating” TaSi8 nanocluster obviously shows substantial reactivity towards the oxygen (Supplementary Figure 9); most of Si and Ta atoms react with oxygen at 5×1010 L.
By an extreme exposure of M@Si16 to 1×1014 L (=1 atm × 40 h) oxygen, the main Si 2p and metal core peaks completely shift towards higher binding energies, whose charge states are Si3+ or Si4+, and M5+, respectively, as marked in Fig. 344,45,53,61. The results indicate that the deposited M@Si16 is finally oxidized into SiO2 and M2O5 in the O2-rich atmosphere, where the oxidation of the central metals is associated with the coreless structure as a final product. The detailed mechanism of the oxidative reaction, and the degree of reactivity of the central atoms are discussed as follows.
Mechanism of oxidative reaction on M@Si16
As described previously, the O2 exposure dependences are different between outer Si and inner central metal atoms. The surrounding 16 Si atoms exhibit initial oxidation showing an intermediate charge state of Si1+−Si2+ (noted as Si1,2+), and finally reach an oxidation state of Si3+−Si4+ states (noted as Si3,4+). On the other hand, the initial oxidation state of the metal atom (M1+) is promptly changed into the final oxidation state of M5+. In order to discuss the mechanism of oxidative reaction on M@Si16, the XPS spectra of Fig. 3 are deconvoluted using three components of Si0, Si1,2+, and Si3,4+ for Si 2p and two components of M1+ and M5+ for core levels of M. As shown in Fig. 3, the XPS spectra can be deconvoluted well, although for Nb@Si16, the minor intermediate component of Nb2+ has to be included (see Supplementary Note 1 for details).
Figure 4 shows the ratios of the Si0, Si1,2+, and Si3,4+ states, and that of the M1+ and M5+ states against oxygen dosages. For Si 2p of all M@Si16, it is clear that the oxidation proceeds as a sequential two-step reaction via the intermediate oxidation states of Si1,2+, and then the Si3,4+ component appears at higher oxygen exposures. For the oxidation of the metal atom, the XPS peaks exhibit a behavior indicating a primary reaction where the rise of M5+ is followed by the decay of M1+. Since the intensity evolutions of M5+ closely correspond to those for the Si3,4+ component, the intermediate states of Si1,2+ are ascribed to Si16 cage oxidation, keeping the metal atom protected, such as with 2O adducts via dissociative O2 adsorption products (Supplementary Figure 10), and those of Si3,4+ are resulted from the Si16 cage collapse along with metal oxidation. Note that, for the Nb@Si16, the minor intermediate oxidized component of Nb 4d5/2 at 203.7 eV, corresponding to the binding energy of NbO (Nb2+)60, is extracted in the fitting procedure owing to the higher reactivity of Nb@Si16, where Nb oxidation seemingly progresses as the Si cage collapses.
From the above intensity analysis, it is fair to conclude that the surrounding Si atoms are oxidized initially preserving the caged structure, and then the caged structure collapses, resulting in the highest oxidation states like SiO2 and M2O5. The maximum intensity ratio of the Si1,2+ component is about 30%, implying that the number of oxygen atoms attached is 4−5 per M@Si16 before the collapse of the Si16 cage. The experimental observation is in fair agreement with the theoretical work by Sen et al., who suggest from their calculation that: (1) in the oxidation reaction of Ta@Si16 cage, the central Ta atom is protected by the surrounding 16 Si atoms from the oxygen, and (2) six Si atoms (38% of 16 Si atoms) in Ta@Si16 are affected by four oxygen molecules in a possible final structure after exposure41.
In the oxidation of crystalline Si surfaces it is well-known that O2 molecules dissociatively chemisorb on the top Si atoms at room temperature as atomic O, which forms the oxidized surface in a “Si-O-Si” bridging configuration44,56,57,58,62,63,64. Further oxidation (formation of a SiO2 film) is followed by the oxygen penetrating deep into the Si surface, accompanying the thermal evaporation of molecular SiO to relax lattice mismatches65,66. The M@Si16 superatoms are similarly oxidized by atomic oxygen dissociatively adsorbed on the Si16 cage, because during initial oxidation (1×104 L) the XPS peak of the O 1s core level shows almost the same binding energy (532.3 eV for Ta@Si16 on C60) with that observed in the first oxidative product of SiO at crystalline Si surfaces with O2 exposure (see Supplementary Figure 11)62. On the crystalline Si surface, further oxidation followed by oxygen penetration deeper into the surface results in a slight energy shift of the O 1s core level towards a higher binding energy by 0.55 eV (in pure SiO2)62. In fact, the O 1s core level for Ta@Si16 on C60 (Supplementary Figure 11) consistently shifts by 0.35 eV towards a higher binding energy at an O2 exposure of 1×1014 L compared with that at 1×104 L. The results show that the final product of the oxidized Si16 cage is mainly SiO2 compounds without any bonding to the central metal atom.
The structural dynamics in the M@Si16 oxidation are discussed from the total XPS intensity analysis of Si 2p with O2 exposure. Figure 5 displays the integrated intensities of core levels at each O2 exposure, in which the XPS signal intensity is carefully normalized (Supplementary Figure 12). The Si 2p intensities (Fig. 5a) for all M@Si16 increase with O2 exposure, and the Si 2p signals are enhanced to 125−135%. At a coverage of M@Si16 of 0.6 ML, the enhancement is ascribed to a cage collapse of M@Si16 via an oxidative reaction, which is caused by the extension of the mean Si surface area with relief of the attenuation of photoemission from the back half of the Si16 cage. In fact, as mentioned above, the dosage dependences of Si3,4+ and M5+ in Fig. 4 are similar to each other, and moreover their behaviors are similar also to the dosage dependence of the total Si 2p intensities in Fig. 5b. The results consistently show Si cage collapse at the higher O2 dosages. Concurrently, the intensity enhancement of Si 2p indicates that the oxidation of M@Si16 occurs without SiO desorption, although the oxidation of bulk Si surface is accompanied by desorption of SiO65,66.

Total XPS intensities at each oxygen dosage. The XPS intensities of a Si 2p and b metals core levels for V@Si16, Nb@Si16, and Ta@Si16. The intensity fluctuations of the X-ray source are normalized by the XPS signal at a binding energy of 110 eV, where background signals are obtained that only vary with the light intensity. The detailed procedures and the XPS data are shown in Supplementary Figure 12. Error bars: the uncertainty in the intensity analysis is estimated to be ±10%, except for V 2p (±20%). The Si 2p signals for all M@Si16 increase with oxygen exposures, and are 1.25−1.35 times stronger at >1×1014 L than that of as-deposited ones, implying Si cage collapse of M@Si16. The changes of the core levels of the central metals are less significant compared to the Si 2p because M atoms are located beneath a layer of Si
Although the XPS analyses of the central metals contain some uncertainties due to satellites (in V 2p and Nb 4d) and O 2s contributions (in Ta 4f) (see Supplementary Figure 12), the XPS intensities of the metals increase slightly against the O2 dosage by 120% at most (Fig. 5b). The result suggests that the metal atoms are unsealed from the Si cage, as described in detailed discussion in Supplementary Note 2.
Periodicity of chemical stability for M@Si16
Although the oxidation robustness of M@Si16 compounds are distinct compared to naked Si surfaces, the degree of robustness depends on the central metal atom; Ta@Si16 > V@Si16 > Nb@Si16, as seen in Figs. 3, 4. Here we evaluate the rate constant of M@Si16 oxidation, kM (M = V, Nb, and Ta), assuming that (1) the oxidation proceeds with step-by-step attachment of oxygen molecules, and (2) the kM@Si16 of each oxidation step is identical at an early stage of oxygen exposure (1×104 L). These assumptions are known as the Eley-Rideal mechanism67, where gas phase molecules directly react with adsorbed species. Importantly, the kinetic analyses are valid when the M@Si16 superatoms are immobilized monodispersively on a surface. In fact, the C60 decoration makes the M@Si16 superatoms individually immobilized due to local chemical interactions of a CT nature between M@Si16 and C60, which has been examined by STM37,38.
By exposing oxygen molecules to deposited M@Si16, the Si16 cage is oxidized sequentially to M@Si16O2n, where dissociative O2 adsorption plausibly produces 2O adducts as shown in Supplementary Figure 10, similarly to bulk Si surface oxidation63,64. Along the stepwise oxidation of M@Si16 → M@Si16O2 → M@Si16O4 → …, some of the Si 2p XPS signal shifts from Si0 to Si1,2+, and the drop of the Si0 component in Fig. 4 indicates the progress of oxidation reaction in units of L. Since the exposure amount can be converted to the corresponding reaction time under elementary reactions, the reaction rates, kM@Si16, are qualitatively evaluated by the conversion of 1 L → 1 s. At 1×104 L, the ratio of nonoxidized Si atoms (Si0), RM@Si16, are RV@Si16 = 0.73, RNb@Si16 = 0.54, and RTa@Si16 = 0.81 (Fig. 4a−c), and thus these ratios provide kV@Si16 = 1.4×10−4/s, kNb@Si16 = 2.5×10−4/s, and kTa@Si16 = 1.0×10−4/s (see Supplementary Note 3 and Supplementary Figure 13 for details).
The difference in chemical reactivities, depending on the central metal atoms, can be discussed in terms of electronic properties of free M@Si16 superatoms obtained from DFT calculations, in which an electron transfer from M@Si16 superatom to O2 molecules is described as a key process in the dissociative O2 adsorption58,63,64. When the second ionization energies are calculated for M@Si16 corresponding to the ionization energy (Ei) of cationic M@Si16+, they are calculated as: Ei(V@Si16+); 10.93 > Ei(Nb@Si16+); 10.90 > Ei(Ta@Si16+); 10.87 in eV. This energetic parameter is important for reaction of O2 as postulated for metal nanoclusters35. Although the delicate difference in the second Eis implies that Ta@Si16 is most reactive for Ta@Si162+O2− formation, the second ionization energy appears to be an implausible argument to explain the reactivity difference. In terms of reaction energetics, the binding energies for the molecular adduct of 3[M@S16 – (O2)] and the dissociated adduct of 1[M@S16(O)2] gradually decrease with decreasing period number of the central metal (Supplementary Figure 10), by which the nonperiodic trend for Nb@Si16+ could not be explained. Since the oxidation reaction proceeds through spin-flip from the initially formed 3[M@Si16(O2)]+ to the dissociated product of 1[M@Si16(Odis)2]+ (Supplementary Figure 10), the HOMO-LUMO gaps of the nanoclusters correspond well with spin excitation energy from the initial state is also important35. However, the calculated HOMO-LUMO gaps for M@Si16+ (3.99 eV for M = Ta, 3.99 eV for M = Nb, and 3.88 eV for M = V) never correlate with the observed reactivity trend. Since the initial step of the molecular O2 adsorption on M@Si16+ seems sensitive to the degree of electron density spreading to the outside of the Si16 cage, the electron density was also calculated. For alkali metal atoms of K, Rb, and Cs, indeed, the ns electrons spread more in higher quantum number orbitals (K (4s) < Rb (5s) < Cs (6s)) with a lowering of Eis, and their oxidation reactivity becomes greater with heavier alkali atoms.
Figure 6 shows the calculated difference of total electron density (Δρ) between monocationic M@Si161+ and dicationic M@Si162+ for M= (a) V, (b) Nb, and (c) Ta. The blue and red regions show the increase (Δρ > +0.0008 e−/a.u.3) and decrease (Δρ < −0.0008 e−/a.u.3) of electron density, respectively, by changing the charge state from M@Si161+ to M@Si162+, where the geometric frameworks are fixed to the optimized monocationic species. Since the charge distributions approximately correspond to an electron transfer channel to form an O2− adduct, the distribution seems relevant to the oxidation reactivity. Although the spatial shapes of Δρ distribution are similar to each other, it is clear that the electrons in Nb@Si16 spread further outside the Si frame (marked by a dotted line) compared to V@Si16 and Ta@Si16. This is presumably because the central metal’s d-orbitals contributing to the superatomic molecular orbitals of M@Si16 are distinctly delocalized over the Si16 cage, seemingly resulting in the higher chemical reactivity. The results show that the chemical properties of M@Si16 superatoms are governed not only by the electron counting rule (68 e−) but also by the species of the central metal hybridized with the Si16 cage.

Isosurfaces for the difference electron density between M@Si16+ and M@Si162+. Those for a M = V, b Nb, and c Ta are calculated with the def-TZVP basis set, where the blue (red) region corresponds to the decrease (increase) of electron density difference (Δρ) at Δρ < –0.0008 e–/a.u.3 (Δρ > + 0.0008 e–/a.u.3). In the calculations, the geometric structure is fixed to that optimized one for M@Si16+. The decrease of Δρ outside the Si cage is significant in Nb@Si16, as shown by circles with dotted lines
Discussion
Our results demonstrate that the oxidation reactivities of alkali-like M@Si16 nanocluster superatoms deposited on a C60 surface are characterized for M = V, Nb, and Ta using XPS spectra. The nanostructured metal−atom encapsulation makes Si atoms in a gas or a surface compound much less reactive with oxygen molecules by a factor of 104, particularly when M@Si16 forms a charge transfer complex with a C60 molecule as M@Si16+C60−, where M@Si16+ satisfies an electronically closed superatom of 68 e−. The oxidation proceeds stepwise via the oxidation of the Si16 cage and finally results in fully oxidized SiO2 and M2O5 with the collapse of the cage. The chemical reactivities of M@Si16 depends on the central metal atom; Nb@Si16 > V@Si16 > Ta@Si16, which is explained by the degree of electron density spreading outside the M@Si16 obtained by the DFT calculation. Since the nature revealed as a periodic dependence in a superatom periodic table is closely related to a transfer integral in their aggregates, this periodic variation in chemical and physical properties may inspire scientists to design assembled functional nanomaterials consisting of naked M@Si16 superatoms.
Methods
Nanocluster generation and deposition
M@Si16 nanoclusters were generated by an MSP technique, whose detailed experimental configuration has been described elsewhere37,38,39,40. Briefly, M−Si alloy targets were sputtered and clusterized in an aggregation cell with a cooled He flow. The cationic nanocluster beam involving M@Si16+ was introduced to a quadrupole mass filter through ion optics. The M@Si16 superatoms were deposited on to the substrate of 2 ML C60 film formed on highly oriented pyrolytic graphite. The collision energy of M@Si16+ to the substrate was set to as low as possible by applying the voltages to the substrate (soft landing condition), which was typically less than 2 eV per cluster. The amount of deposition was monitored with a pico-ammeter and was controlled to be 2.9×1013 nanoclusters except for the coverage dependence experiment, which is equal to 0.6 ML coverage, assuming a deposition area of 6 mm in diameter and the size of M@Si16 (0.8–0.9 nm diameter)37. The deposited samples were transferred to the photoelectron spectroscopy system connecting with the MSP nanocluster deposition system while keeping ultrahigh vacuum conditions. The morphology of the M@Si16+ deposited surface was confirmed by STM, where the Ta@Si16+ nanoclusters are independently immobilized on the C60 surface37,38.
Photoelectron spectroscopy
XPS measurements were performed using an Mg Kα (hν = 1253.6 eV) X-ray source. Photoelectrons emitted from the sample were collected with a commercial hemispherical electron analyzer (VG SCIENTA, R-3000), where the detection angle was 45° from the surface normal. In the XPS analysis, the peak fittings were performed using an instrumental broadening as a Voigt function (FWHMs amounted to 1.09 eV, for which Gaussian and Lorentzian components equaled 0.75 and 0.56 eV, respectively), after a subtraction of the Shirley background. The sample temperature during all XPS measurements was kept to room temperatures.
Calculations
All geometries of M@Si16+ (M = V, Nb, and Ta) nanocluster cations and their oxygen adducts of [M@Si16O]+ and [M@Si16O2]+ were optimized by the density functional incorporated in the Gaussian09 program68 until no imaginary frequency was found. The hybrid exchange-correlation functional of PBE069 and triple-zeta valence basis function of def-TZVP70 were utilized in the calculations. Difference electron density isosurfaces were visualized with GaussView5 based on the difference between the total electron density for M@Si16+ (optimized structure) and M@Si162+ (same structure with monovalent cation).
Data availability
All experimental and calculated data generated during the current study are available from the corresponding author on reasonable request.
References
Peercy, P. S. The drive to miniaturization. Nature 406, 1023–1026 (2000).
Flood, A. H., Stoddart, J. F., Steuerman, D. W. & Heath, J. R. Whence molecular electronics? Science 306, 2055–2056 (2004).
Ito, T. & Okazaki, S. Pushing the limit of lithography. Nature 406, 1027–1031 (2000).
Schmidt, V., Wittemann, J. V., Gösele, U., Bley, R. A. & Kauzlarich., S. M. A low-temperature solution phase route for the synthesis of silicon nanoclusters. J. Am. Chem. Soc. 118, 12461–12462 (1996).
Morales, A. M. & Lieber, C. M. A laser ablation method for the synthesis of crystalline semiconductor nanowires. Science 279, 208–211 (1998).
De Padova, P. et al. Burning match oxidation process of silicon nanowires screened at the atomic scale. Nano Lett. 8, 2299–2304 (2008).
Sugiyama, Y. et al. Synthesis and optical properties of monolayer organosilicon nanosheets. J. Am. Chem. Soc. 132, 5946–5947 (2010).
Okamoto, H., Sugiyama, Y. & Nakano, H. Synthesis and modification of silicon nanosheets and other silicon nanomaterials. Chem. Eur. J 17, 9864–9887 (2011).
Yaokawa, R. et al. Monolayer-to-bilayer transformation of silicenes and their structural analysis. Nat. Commun. 7, 10657 (2016).
Kara, A. et al. A review on silicene-new candidate for electronics. Surf. Sci. Rep. 67, 1–18 (2012).
De Padova, P., Quaresima, C., Olivieri, B., Perfetti, P. & Le Lay, G. Strong resistance of silicene nanoribbons towards oxidation. J. Phys. D. Appl. Phys. 44, 312001 (2011).
Molle, A. et al. Hindering the oxidation of silicene with non-reactive encapsulation. Adv. Funct. Mater. 23, 4340–4344 (2013).
Cerdá, J. I. et al. Unveiling the pentagonal nature of perfectly aligned single-and double-strand Si nano-ribbons on Ag(110). Nat. Commun. 7, 13076 (2016).
Zybill, C. Si60, an analogue of C60? Angew. Chem. Int. Ed. 31, 173–175 (1992).
Bower, J. E. & Jarrold, M. F. Properties of deposited size-selected clusters: reactivity of deposited silicon clusters. J. Chem. Phys. 97, 8312–8321 (1993).
Li, B.-X. & Cao, P. L. Distorted cage structures of Sin (n=20, 24, 26, 28, 30, 32) clusters. J. Phys. Condens. Matter 13, 10865–10872 (2001).
Yoo, S., Zhao, J., Wang, J. & Zeng, X. C. Endohedral silicon fullerenes SiN (27 ≤ N ≤ 39). J. Am. Chem. Soc. 126, 13845–13849 (2004).
Kumar, V. & Kawazoe, Y. Metal-encapsulated fullerenelike and cubic caged clusters of silicon. Phys. Rev. Lett. 87, 045503 (2001).
Khanna, S. N., Rao, B. K. & Jena, P. Magic numbers in metallo-inorganic clusters: chromium encapsulated in silicon cages. Phys. Rev. Lett. 89, 016803 (2002).
Kawamura, H., Kumar, V. & Kawazoe, Y. Growth behavior of metal-doped silicon clusters Sin M (M=Ti, Zr, Hf; n=8–16). Phys. Rev. B 71, 075423 (2005).
Reveles, J. U. & Khanna, S. N. Nearly-free-electron gas in a silicon cage. Phys. Rev. B 72, 165413 (2005).
Zheng, W., Nilles, J. M., Radisic, D. & Bowen, K. H. Jr. Photoelectron spectroscopy of chromium-doped silicon cluster anions. J. Chem. Phys. 122, 071101 (2005).
Koyasu, K., Akutsu, M., Mitsui, M., & Nakajima, A. Selective formation of MSi16 (M=Sc, Ti, and V). J. Am. Chem. Soc. 127, 4998–4999 (2005).
Jaeger, J. B., Jaeger, T. D. & Duncan, M. A. Photodissociation of metal-silicon clusters: encapsulated versus surface-bound metal. J. Phys. Chem. A 110, 9310–9314 (2006).
Reveles, J. U. & Khanna, S. N. Electronic counting rules for the stability of metal-silicon clusters. Phys. Rev. B 74, 035435 (2006).
Torres, M. B., Fernández, E. M. & Balbás, L. C. Theoretical study of isoelectronic Sin M clusters (M=Sc, Ti, V+, n=14-18). Phys. Rev. B 75, 205425 (2007).
Koyasu, K., Atobe, J., Akutsu, M., Mitsui, M. & Nakajima, A. Electronic and geometric stabilities of clusters with transition metal encapsulated by silicon. J. Phys. Chem. A 111, 42–49 (2007).
Lau, J. T. et al. X-ray spectroscopy reveals high symmetry and electronic shell structure of transition-metal-doped silicon clusters. Phys. Rev. A 79, 054301 (2009).
Cantera-López, H., Balbás, L. C. & Borstel, G. First-principles calculations of structural and electronic properties of Ta-doped Si clusters, wires, and bulk systems. Phys. Rev. B 83, 075434 (2011).
Jena, P. Beyond the periodic table of elements: the role of superatoms. J. Phys. Chem. Lett. 4, 1432–1442 (2013).
Abreu, M. B., Reber, A. C. & Khanna, S. N. Does the 18-electron rule apply to CrSi12? J. Phys. Chem. Lett. 5, 3492–3496 (2014).
Chauhan, V., Abreu, M. B., Reber, A. C. & Khanna, S. N. Geometry controls the stability of FeSi14. Phys. Chem. Chem. Phys. 17, 15718–15724 (2015).
Tomalia, D. A. & Khanna, S. N. A systematic framework and nanoperiodic concept for unifying nanoscience: hard/soft nanoelements, superatoms, meta-atoms, new emerging properties, periodic property patterns, and predictive Mendeleev-like nanoperiodic tables. Chem. Rev. 116, 2705–2774 (2016).
Tsunoyama, H. et al. Development of integrated dry−wet synthesis method for metal encapsulating silicon cage superatoms of M@Si16 (M=Ti and Ta). J. Phys. Chem. C 121, 20507–20516 (2017).
Reber, A. C. & Khanna, S. N. Superatoms: electronic and geometric effects on reactivity. Acc. Chem. Res. 50, 255–263 (2017).
Tsunoyama, H., Shibuta, M., Nakaya, M., Eguchi, T. & Nakajima, A. Synthesis and characterization of metal-encapsulating Si16 cage superatoms. Acc. Chem. Res. https://doi.org/10.1021/acs.accounts.8b00085 (2018).
Nakaya, M., Iwasa, T., Tsunoyama, H., Eguchi, T. & Nakajima, A. Formation of a superatom monolayer using gas-phase-synthesized Ta@Si16 nanocluster ions. Nanoscale 6, 14702–14707 (2014).
Nakaya, M., Iwasa, T., Tsunoyama, H., Eguchi, T. & Nakajima, A. Heterodimerization via the covalent bonding of Ta@Si16 nanoclusters and C60 molecules. J. Phys. Chem. C 119, 10962–10968 (2015).
Shibuta, M. et al. Chemical characterization of an alkali-like superatom consisting of a Ta-encapsulating Si16 cage. J. Am. Chem. Soc. 137, 14015–14018 (2015).
Ohta, T., Shibuta, M., Tsunoyama, H., Eguchi, T. & Nakajima, A. Charge transfer complexation of Ta-encapsulating Ta@Si16 superatom with C60. J. Phys. Chem. C 120, 15265–15271 (2016).
Sen, A. & Sen, P. Properties of highly oriented pyrolytic graphite supported TaSi16 clusters: a density functional investigation. J. Phys. Chem. C 121, 28490–28497 (2017).
Zhang, C. et al. Advanced nanocluster ion source based on high-power impulse magnetron sputtering and time-resolved measurements of nanocluster formation. J. Phys. Chem. A 117, 10211–10217 (2013).
Rangelov, G. & Fauster, Th Thermally-induced evolution of codeposited Co-Si layers on Si(100)surfaces. Surf. Sci. 365, 403–410 (1996).
Himpsel, F. J., McFeely, F. R., Taleb-Ibrahimi, A., Yarmoff, J. A. & Hollinger, G. Microscopic structure of the SiO2/Si interface. Phys. Rev. B 38, 6084–6096 (1988).
Silversmit, G., Depla, D., Poelman, H., Marin, G. B. & Gryse, R. D. Determination of the V2p XPS binding energies for different vanadium oxidation states (V5+ to V0+). J. Electron Spectrosc. Relat. Phenom. 135, 167–175 (2004).
Latta, E.-E. & Ronay, M. Catalytic oxidation of niobium by rare earths. J. Vac. Sci. Technol. A 4, 1626–1630 (1986).
Riffe, D. M. & Wertheim, G. K. Ta(110) surface and subsurface core-level shifts and 4f 7/2 line shapes. Phys. Rev. B 47, 6672–6679 (1993).
Doniach, S. & Šunjić, M. Many-electron singularity in x-ray photoemission and x-ray line spectra from metals. J. Phys. C: Solid St. Phys. 3, 285–291 (1970).
Kasperkiewicz, J., Kovacich, J. A. & Lichtman, D. XPS studies of vanadium and vanadium oxides. J. Electron Spcectrosc. Relat. Phenom. 32, 123–132 (1983).
Atanassova, E. & Spassov, D. X-ray photoelectron spectroscopy of thermal thin Ta2O5 films on Si. Appl. Surf. Sci. 135, 71–82 (1998).
Guo, P. et al. Relativistic computational investigation: the geometries and electronic properties of TaSin + (n=1−13, 16) clusters. J. Phys. Chem. A 110, 7453–7460 (2006).
Horvathj, B., Strutzj, J., Geyer-Lippman, J. & Horva, E. G. Preparation, properties, and ESCA characterization of vanadium surface compounds on silicagel. I. Z. Anorg. Allg. Chem. 483, 181–192 (1981).
Ho, S. F., Contarini, S. & Rabalais, J. W. Ion-beam-induced chemical changes in the oxyanions (MOy n-) and oxides (MOx) where M=Cr, Mo, W, V, Nb, and Ta. J. Phys. Chem. 91, 4779–4788 (1987).
Huang, D. L., Dau, P. D., Liu, H. T. & Wang, L. S. High-resolution photoelectron imaging of cold C60 − anions and accurate determination of the electron affinity of C60. J. Chem. Phys. 140, 224315 (2014).
Hollinger, G. & Himpsel, F. J. Multiple-bonding configurations for oxygen on silicon surfaces. Phys. Rev. B 28, 3651–3653 (1983).
Höfer, U., Morgen, P., Wurth, W. & Unbach, E. Metastable molecular precursor for the dissociative adsorption of oxygen on Si(111). Phys. Rev. Lett. 55, 2979–2982 (1985).
Morgen, P., Höfer, U., Wurth, W. & Unbach, E. Initial stages of oxygen adsorption on Si(111):the stable state. Phys. Rev. B 39, 3720–3734 (1989).
Höfer, U., Morgen, P., Wurth, W. & Unbach, E. Initial stages of oxygen adsorption on Si(111). II. the molecular precursor. Phys. Rev. B 40, 1130–1145 (1989).
Shen, M., Ma, Q., Lee, I. & Zaera, F. Oxygen adsorption and oxide formation on V(100) surfaces. J. Phys. Chem. C 111, 6033–6040 (2007).
Hu, Z. P., Li, Y. P., Ji, M. R. & Wu, J. X. The interaction of oxygen with niobium studied by XPS and UPS. Solid State Commun. 71, 849–852 (1989).
van der Veen, J. F., Himpsel, F. J. & Eastman, D. E. Chemisorption-induced 4f-core-electron binding-energy shifts for surface atoms of W(111), W(100), and Ta(111). Phys. Rev. B 25, 7388–7397 (1982).
Hollinger, G., Jugnet, Y., Pertosa, P. & Duc., T. M. X-ray photoelectron spectroscopy of thermally grown silicon dioxide film on silicon. Chem. Phys. Lett. 36, 441–445 (1975).
Lee, S.-H. & Kang, M.-H. Identification of the initial-stage oxidation products on Si(111)-(7 × 7). Phys. Rev. Lett. 82, 968–971 (1999).
Lee, S.-H. & Kang, M.-H. Origin of O 1s core-level shifts on oxygen adsorbed Si(111)-(7 × 7). Phys. Rev. Lett. 84, 1724–1727 (2000).
Engel, Th The interaction of molecular and atomic oxygen with Si(100) and Si (111). Surf. Sci. Rep. 18, 93–144 (1993).
Raschke, M. B., Bratu, P. & Höfer, U. Optical second-harmonic investigations of the isothermal desorption of SiO from the Si(100) and Si(111)surfaces. Surf. Sci. 410, 351–361 (1998).
Weinberg, W. H. Eley-Rideal surface chemistry: direct reactivity of gas phase atomic hydrogen with adsorbed species. Acc. Chem. Res. 29, 479–487 (1996).
Frisch, M. J. et al. Gaussian 09, Revision E.01 (Gaussian, Inc., Wallingford CT, 2013).
Adamo, C. & Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 Model. J. Chem. Phys. 110, 6158–6170 (1999).
Schäfer, A., Horn, H. & Ahlrichs, R. Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr. J. Chem. Phys. 100, 5829–5835 (1994).
Acknowledgements
This work is partly supported by JSPS KAKENHI of Grant-in-Aid for Scientific Research (A) Grant Number 15H02002 and of Challenging Research (Pioneering) Grant Number 17H06226.
Author information
Authors and Affiliations
Contributions
M.S., T.K., T.O., H. T., and A.N. contributed to the experimental process. M.S., H.T., and A.N. carried out the simulations and theoretical interpretations. All authors have given approval to the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shibuta, M., Kamoshida, T., Ohta, T. et al. Oxidative reactivity of alkali-like superatoms of group 5 metal-encapsulating Si16 cage nanoclusters. Commun Chem 1, 50 (2018). https://doi.org/10.1038/s42004-018-0052-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42004-018-0052-9
This article is cited by
-
Solar Driven CO2 Hydrogenation on Ti-Doped Silicon Nanocages
Journal of Cluster Science (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
