
Abstract
Pyrrole is a privileged five-membered aromatic nitrogen heterocycle, which is ubiquitous in natural products, drug molecules, and functional materials. Therefore, numerous synthetic routes to substituted pyrroles have been extensively developed. Nevertheless, the efficient and short-step synthesis of highly substituted and/or fused pyrroles has remained a significant challenge in organic chemistry. Here we report a ruthenium-catalyzed nitrogen-transfer [2 + 2 + 1] cycloaddition of α,ω-diynes involving cyclic biscarbenoid intermediates. To achieve the key nitrogen transfer to carbenoid carbons, sulfoximines are employed as nitrene surrogates. Consequently, diverse fused pyrroles are successfully synthesized in good yields with wide functional group compatibility. Moreover, this method allows the synthesis of N-alkyl, N-aryl, and even N-H pyrroles, which are difficult to obtain using previous [2 + 2 + 1]-type reactions. Nitrogen transfer from sulfoximines to cyclic biscarbenoid intermediates is supported by density functional theory calculations and control experiments.
Similar content being viewed by others
Introduction
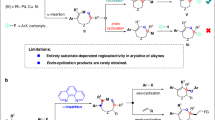
Transition metal (TM)-catalyzed nitrene-transfer reactions (NTRs) have been extensively investigated as they provide efficient access to valuable nitrogen compounds via C–H amination, aziridination, sulfimidation, etc1. Because nitrenes are extremely reactive, the combination of TM catalysts with nitrene surrogates is generally employed to control both the reactivity and selectivity of NTRs1,2,3,4,5. Thus, the development of methods involving a novel nitrene surrogate would significantly expand the synthetic potential of NTRs. A recent study by Tonks and coworkers demonstrated that Ti(IV) imido species, which were derived from N,N′-diaryl diazenes, participated in a [2 + 2 + 1] cycloaddition with alkynes to afford N-aryl pyrroles (Fig. 1a)6. Our group has also been engaged in the development of atom-transfer [2 + 2 + 1] cycloadditions of α,ω-diynes7,8,9. However, a similar nitrogen transfer has never been observed when conventional nitrene surrogates were employed.

[2 + 2 + 1] Approaches to substituted pyrroles. a Ti-catalyzed reaction of alkynes with diazenes. b Pd-catalyzed reaction of arylalkynes with amines. c Ru-catalyzed reaction of α,ω-diynes with sulfoximines. d Examples of highly substituted and fused pyrroles
Pyrroles are among the most important five-membered heteroaromatic compounds that are ubiquitously found in natural products, bioactive compounds, and functional molecules10,11,12,13,14,15. Although diverse synthetic routes to substituted pyrroles have been continuously devised beyond the conventional Paal–Knorr and related methods, there is still an ample need for mild, efficient, selective, and short-step synthesis of highly substituted pyrroles16,17,18,19,20,21. In this regard, [2 + 2 + 1]-type cyclocondensation reactions of alkynes with amines provide promising routes to fully substituted pyrroles in a single operation22,23,24,25,26. However, activated alkyne substrates are essential for these reactions, and stoichiometric promoters are often required. In contrast, Wang, Lu, and coworkers developed Pd-catalyzed methods to synthesize fully substituted pyrroles from arylalkynes and primary amines (Fig. 1b)27,28. Buchwald and coworkers also reported an indirect [2 + 2 + 1] synthesis of N-Boc pyrroles via a Cu-catalyzed coupling of BocNH2 with 1,4-diiodo-1,3-dienes, which were prepared beforehand from two alkyne molecules29. Recently, Tonks’ group reported a Ti-catalyzed [2 + 2 + 1] cycloaddition of alkynes using N,N’-diaryl diazenes as nitrene surrogates (Fig. 1a)6. Although these advances enable the catalytic synthesis of highly substituted pyrroles using simple amines or diazenes, most of the methods are limited to monocyclic and/or less functionalized pyrroles. Thus, an efficient protocol for the catalytic synthesis of functionalized polycyclic pyrroles remains undeveloped. Because fused pyrroles are found in bioactive molecules and functional materials (Fig. 1d), development of a catalytic nitrogen-transfer [2 + 2 + 1] synthesis of fused pyrroles would contribute significantly to pyrrole-based design of drugs and materials30,31. Nevertheless, the straightforward application of the state-of-the-art catalytic nitrogen-transfer systems to the targeted [2 + 2 + 1] reaction poses a major challenge: late-transition-metal nitrenoids generally undergo C–H insertion and/or [2 + 1] aziridination with unsaturated compounds1, and thus, the chemoselective transformation of functionalized α,ω-diynes into the target pyrroles requires a novel strategic approach: nitrogen transfer without involving metal nitrenoids.
Here we investigate the ruthenium-catalyzed nitrogen-transfer [2 + 2 + 1] cycloaddition of α,ω-diynes using sulfoximines as effective nitrene surrogates for ruthenacyclic intermediates bearing a cyclic biscarbenoid character (Fig. 1c). It should be noted that because sulfoximines are rather stable, their loss of nitrogen to give sulfoxides is very rare32,33. Sulfoximines can be synthesized via sulfimidation of the corresponding sulfoxides including ruthenium-catalyzed nitrene transfer34,35,36, and a reverse reaction, i.e., nitrogen transfer from sulfoximines, has not been previously disclosed, to the best of our knowledge. The newly developed process is operationally simple and compatible with a wide variety of diyne substrates bearing reactive functional groups. Moreover, the use of sulfoximines enables the introduction of reactive N-substituents on the pyrrole products and even an N-unprotected pyrrole can be directly obtained without a deprotection step. A plausible mechanism of nitrogen transfer from sulfoximines to cyclic biscarbenoid intermediates is proposed based on control experiments and density functional theory calculations.
Results
Reaction optimization
The key to the successful realization of the elusive catalytic nitrogen-transfer [2 + 2 + 1] synthesis of fused pyrroles is the identification of the optimal nitrene surrogates toward ruthenacycle intermediates as electrophilic biscarbenoids. Thus, we screened various alkyl-substituted and aryl-substituted N-methyl sulfoximines toward the reaction with ether-tethered diyne 1a in the presence of ruthenium catalyst [CpRu(MeCN)3]PF6 (Ru1, Cp = η5-cyclopentadienyl), as shown in Fig. 2a. When diphenyl sulfoximine 2a was used, the expected product 3a was obtained, albeit in a low yield (25%). Although dialkyl sulfoximine 2b was totally inefficient, methyl phenyl sulfoximine 2c was found to be more competent than 2a. Further electronic tuning was conducted based on 2c. The weakly electron-withdrawing p-chlorophenyl substituent in 2d had a negative impact on the product yield, while sulfoximine 2e bearing an electron-donating p-methoxyphenyl substituent increased the yield of 3a. Further improvement was observed when sulfoximine 2f with a weakly electron-donating p-fluorophenyl substituent was used, and thus, 2f was selected as the optimal nitrene surrogate. Other parameters were also examined. The more electron-rich catalyst [Cp*Ru(MeCN)3]PF6 (Ru2, Cp* = η5-pentamethylcyclopentadienyl) gave a much inferior result and common organic solvents such as AcOEt, THF, MeCN, and 1,2-dichloroethane (DCE) were less efficient than dimethylformamide (DMF). With increased loadings of Ru1 (2 mol%) and 2f (1.1 equiv), the reactions of 1a and sulfonamide-tethered diyne 1b proceeded to completion within 0.5 h in dry DMF at 80 °C, affording the desired pyrroles 3a and 3b in 92% and 98% yields, respectively (Fig. 2b).

Reaction optimization using diynes 1a and 1b. a Screening of sulfoximines as nitrene surrogates. b Reactions of diynes 1a and 1b with sulfoximine 2f under optimized conditions. Ru1: [CpRu(MeCN)3]BF4
Scope of [2 + 2 + 1] pyrrole synthesis
To further demonstrate the substrate scope, various diynes were subjected to the nitrogen-transfer [2 + 2 + 1] cycloaddition using 2f; the results obtained with optimal catalyst loadings and reaction times are shown in Fig. 3. Amide (3c), imide (3d), ketone (3e), nitrile (3f), ester (3g) and alcohol (3h) moieties on the tethers were well tolerated in the reaction protocol. Phenyl groups possessing electron-donating alkoxy (3k–m) or electron-withdrawing (3n, 3q, and 3s) groups, and thiophenes (3r) were compatible as alkyne terminal groups. Notably, reactive C–I and C–B bonds (3o and 3p) were also tolerated. Acenaphthene-fused and benzopyran-fused pyrroles (3i and 3j) as well as unsymmetrical pyrroles (3s and 3t) could be obtained in high yields. One of the terminal groups could be an alkyl group, as 3t was obtained in good yield. Although diyne 1u bearing methyl terminals did not afford the corresponding product 3u under similar conditions, the reaction of diyne 1v bearing cyclopropyl groups on both terminals afforded the corresponding product 3v in 68% yield with the cyclopropyl groups intact. Nevertheless, the use of 2d instead of 2f and a 10 mol% catalyst loading converted 1u into 3u in 52% yield. Silyl-substituted diynes also underwent nitrene-transfer [2 + 2 + 1] cycloaddition when DCE was used as the solvent instead of DMF under reflux. Unsymmetrical pyrrole 3w bearing a bulky SitBuMe2 group was obtained in 91% yield, albeit an increased catalyst loading (10 mol%) was required. The reaction of bis(trimethylsilyl)diyne 1x was performed similarly to afford 2,5-unsubstituted pyrrole 3x in 83% yield after exhaustive desilylation using nBu4NF [tetra-n-butylammonium fluoride (TBAF)].

Scope of [2 + 2 + 1] pyrrole synthesis in terms of diyne substrates 1. a2f (1.5 equiv). b2d was used instead of 2f. c1,2-Dichloroethane reflux. dDesilylated with TBAF
Because various sulfoximines are readily available34,35, the scope of N-substituents was investigated for the reaction of diyne 1a (Fig. 4a). Although increased loadings of sterically more demanding sulfoximines 2g–k (2 equivs) were required, the corresponding pyrroles 4–6, 8, and 9 were obtained in 69–82% yields. Although the reason was unclear, the reaction with N-prenyl sulfoximine 2i afforded N-H pyrrole 7 in 13% yield along with the expected product 6 (74%). N-aryl pyrrole 10 was also obtained in 67% yield. In this case, 1.5 equiv of sulfoximine 2l, bearing a more electron-donating p-methoxyphenyl substituent on the sulfur center, was required. In the syntheses of complex molecules including reactive pyrrole scaffolds, N-protecting groups play a critical role37. However, the removal of N-protecting groups often requires elaborate manipulations and/or affords low yields. Thus, the direct synthesis of N-unprotected highly substituted pyrroles has a significant advantage. Notably, the use of N-triethylsilyl sulfoximine 2m (1.5 equiv) as an N-H nitrene surrogate directly produced N-H pyrrole 7 in 70% yield, although a higher reaction temperature of 120 °C was required. The potency of sulfoximines as nitrogen donors is attributable to their sufficient nucleophilicity toward the electrophilic ruthenacycle intermediates as well as the leaving-group ability of the corresponding sulfoxides. In fact, much less nucleophilic N-Boc sulfoximine 2n (R = Boc, Y = F) proved to be totally inefficient. It should be noted that alkyne moieties, which can be used for further derivatizations, could be introduced on the N-substituents, because α,ω-diynes are selectively transformed into pyrroles in our nitrogen-transfer [2 + 2 + 1] cycloaddition. N-Propargyl pyrrole 8 underwent ruthenium-catalyzed Markovnikov hydrosilylation with triethylsilane and [3 + 2] cycloaddition with benzyl azide to regioselectively afford vinylsilane 11 and triazole 12 in 78% and 91% yields, respectively (Fig. 4b)38,39.

Reactions with other sulfoximines and transformations of N-propargyl pyrroles. a Reactions of diyne 1a with sulfoximines 2g–m. bTransformations of N-propargyl pyrrole 8. Ru1: [CpRu(MeCN)3]BF4, Ru2: [Cp*Ru(MeCN)3]BF4, Ru3: Cp*RuCl(cod), cod = 1,5-cyclooctadiene
Mechanistic considerations
To gain insights into the mechanism, several control experiments were carried out. The reaction of diyne 1a with enantiopure sulfoximine (R)-2c (99% ee, 1.1 equiv) was performed using acetone as the solvent to ensure the isolation of the resultant sulfoxide. After reaction for 6 h, methyl phenyl sulfoxide (R)-13 was obtained in 68% yield with 98% ee along with pyrrole 3a (Fig. 5a). Thus, it was confirmed that the nitrogen transfer from sulfoximines occurs with retention of configuration at the sulfur center. The influence of the sulfoxide side products on the reaction efficiency was also examined by comparing rates for the reactions of diyne 1a and sulfoximine 2c with or without adding sulfoxide 13 (Supplementary Methods). As a result, the reaction rate was slightly decreased by initially adding 1 equiv of 13, suggesting that sulfoxides slow the catalyst turnover by their coordination to the ruthenium center.

Control experiments and proposed mechanism. a Reaction of enantiopure sulfoximine 2c. b Reaction of preformed ruthenacycle 14 with sulfoximine 2f in the presence of AgPF6. c Plausible catalytic cycle. Ru1: [CpRu(MeCN)3]BF4
Moreover, the involvement of cationic ruthenacycle intermediates was experimentally corroborated by the formation of 3a from the reaction of isolated ruthenacycle 1440 with sulfoximine 2f (1.1 equiv) in the presence of AgPF6 (1.1 equiv) in DMF at 80 °C (Fig. 5b). In this case, the cationic ruthenacycle derived from 14 was less electrophilic than that derived from Ru1 because the Cp* ligand in 14 was more electron-donating than the Cp ligand in Ru1. Nevertheless, 3a was obtained in a moderate yield of 58%.
Based on these observations and the results obtained in our previous studies7,8,9, a plausible mechanism for the nitrogen-transfer [2 + 2 + 1] cycloaddition is proposed, as shown in Fig. 5c. The reaction of Ru1 with a diyne substrate produces the ruthenacycle key intermediate I. Because I behaves as a cyclic biscarbenoid40,41,42,43, nitrogen transfer from sulfoximines 2 to one of the carbenoid carbons in I occurs via TS I–II to produce azadienylcarbene complex II. Subsequent cycloisomerization of II affords η5-pyrrole complex III. Finally, the pyrrole ligand of III is replaced by diyne substrate 1 to afford free pyrrole 3, and subsequent oxidative cyclization regenerates I. This mechanism is quite distinct from that of Tonks’ titanium-catalyzed reaction, which involves the formation of Ti(IV) imido species and its sequential reactions with alkynes6. Our reaction is the first example of a [2 + 2 + 1] cycloaddition via a catalytic nitrogen transfer to a five-membered metallacycle intermediate. However, stoichiometric reactions of cobaltacyclopentadienes or titanacyclopentadienes with nitrene surrogates leading to pyrroles has been reported44,45,46.
To support the proposed mechanism, the reaction of diyne 1a with sulfoximine 2f was analyzed by density functional theory calculations at the PCM (DMF) rM06L/6-311 G + + (d,p)–SDD//rB3LYP/6-31 G(d)–LanL2DZ level of theory (for details, see Supplementary Methods). This method has been reliably used for the computational analysis of TM-catalyzed reactions47. The calculated energy surface is shown in Fig. 6. The initial oxidative cyclization of cationic diyne complex A with one DMF ligand proceeds via TS AB with an activation energy of ΔG‡ = + 12.1 kcal/mol. After passing through TS AB , ruthenacyclopentadiene B is located at a slightly lower energy level than A. Subsequently, B undergoes facile isomerization to planar delocalized ruthenacycle C through a small activation barrier of ΔG‡ = + 5.1 kcal/mol. The geometry of C is comparable to that of previously reported ruthenacyclopentatrienes37: the Cα–Cβ bonds (1.413 and 1.415 Å) are only slightly longer than the Cβ–Cβ’ bond (1.389 Å). In addition, the Ru–Cα bonds (1.994 and 1.995 Å) are shorter than a normal Ru–C single bond (ca. 2.0 Å), indicating that C has a biscarbenoid character. The formation of C from A is estimated to be 18.2 kcal/mol exergonic.

Calculated energy surface for the model reaction of diyne 1a with sulfoximine 2f. Relative Gibbs free energies (298 K, 1 atm) are shown. Computed models: hydrogen, white; carbon, gray; nitrogen, blue; oxygen, red; fluorine, yellow green; sulfur, yellow; and Ru, green
After oxidative cyclization, ligand exchange of DMF-bound ruthenacycle C with sulfoximine 2f generates N-bound sulfoximine complex D, which undergoes the key nitrogen transfer with an activation energy of ΔG‡ = + 27.7 kcal/mol to generate azadienyl carbene complex E with the concomitant extrusion of the sulfoxide residue. The shortened Ru–N/Cα–N and elongated Ru–Cα/N–S distances in TS DE suggest that the nitrogen transfer is a concerted process. The formation of E from D is thermodynamically favorable as it is exergonic (–50.4 kcal/mol). The subsequent cycloisomerization of azadienyl carbene complex F is very facile and thermodynamically favorable as it proceeds via TS FG with a small activation energy of ΔG‡ = + 7.2 kcal/mol, and the formation of η5-pyrrole complex G from F is exergonic (–21.9 kcal/mol). The rate-determining step is the nitrogen transfer, and the largest energetic span between C and TS DE is estimated to be 28.4 kcal/mol, which is reasonably small under the experimental conditions. Moreover, the high exergonicity (–96.6 kcal/mol) of the formation of G from A suggests that the overall process is thermodynamically feasible.
Discussion
In conclusion, a catalytic [2 + 2 + 1] cycloaddition route to fused pyrroles, using sulfoximines as the nitrene surrogates, has been successfully developed. Screening of several sulfoximines led to the identification of methyl p-fluorophenyl sulfoximines as the optimal surrogates. The optimized protocol enabled the short-step synthesis of diverse fused pyrroles with good functional group compatibility. A plausible mechanism, involving the nitrogen transfer from sulfoximines to cationic ruthenacycle intermediates, was proposed based on several control experiments. The kinetic and thermodynamic feasibility of the proposed mechanism was also inspected by DFT calculations on a model system.
Methods
Synthesis and characterization
Full synthetic procedures are available in the Supplementary Methods. HPLC charts for rac-2c and (R)-2c, and rac-13 and (R)-13 are available in Supplementary Figures 1 and 2. NMR spectra of purified compounds are available in Supplementary Figures 6-49.
Kinetic study
Full details are available in the Supplementary Methods, Supplementary Table 1, and Supplementary Figure 3.
Computational study
Full details are available in the Supplementary Methods, Supplementary Table 2, and Supplementary Figures 4 and 5. Cartesian coordinates of calculated molecules are available in Supplementary Table 3.
Data availability
The authors declare that the data supporting the findings of this study are available within the article and Supplementary Information file, or from the corresponding author upon reasonable request.
References
Dequirez, G., Pons, V. & Dauban, P. Nitrene chemistry in organic synthesis: still in its infancy? Angew. Chem. Int. Ed. 51, 7384–7395 (2012).
Müller, P. & Fruit, C. Enantioselective catalytic aziridinations and asymmetric nitrene insertions into CH bonds. Chem. Rev. 103, 2905–2919 (2003).
Katsuki, T. Azide compounds: nitrogen sources for atom-efficient and ecologically benign nitrogen-atom-transfer reactions. Chem. Lett. 34, 1304–1309 (2005).
Minakata, S. Utilization of N–X bonds in the synthesis of N-heterocycles. Acc. Chem. Res. 42, 1172–1182 (2009).
Darses, B., Rodrigues, R., Neuville, L., Mazurais, M. & Dauban, P. Transition metal-catalyzed iodine(III)-mediated nitrene transfer reactions: efficient tools for challenging syntheses. Chem. Commun. 53, 493–508 (2017).
Gilbert, Z. W., Hue, R. J. & Tonks, I. A. Catalytic formal [2+2+1] synthesis of pyrroles from alkynes and diazenes via TiII/TiIV redox catalysis. Nat. Chem. 8, 63–68 (2016).
Yamashita, K., Yamamoto, Y. & Nishiyama, H. Ruthenium-catalyzed transfer oxygenative cyclization of α,ω-diynes: Unprecedented [2+2+1] route to bicyclic furans via ruthenacyclopentatriene. J. Am. Chem. Soc. 134, 7660–7663 (2012).
Matsui, K., Shibuya, M. & Yamamoto, Y. Ruthenium-catalyzed transfer oxygenative [2+2+1] cycloaddition of silyldiynes using nitrones as adjustable oxygen atom donors. Synthesis of bicyclic 2-silylfurans. ACS Catal. 5, 6468–6472 (2015).
Matsui, K., Shibuya, M. & Yamamoto, Y. Catalytic [2+2+1] synthesis of fused thiophenes using thiocarbonyls as sulfur donors. Angew. Chem. Int. Ed. 55, 15397–15400 (2016).
Walsh, C. T., Garneau-Tsodikova, S. & Howard-Jones, A. R. Biological formation of pyrroles: nature’s logic and enzymatic machinery. Nat. Prod. Rep. 23, 517–531 (2006).
Loudet, A. & Burgess, K. BODIPY dyes and their derivatives: syntheses and spectroscopic properties. Chem. Rev. 107, 4891–4932 (2007).
Maeda, H. Supramolecular chemistry of acyclic oligopyrroles. Eur. J. Org. Chem. 2007, 5313–5325 (2007).
Fan, H., Peng, J., Hamann, M. T. & Hu, J.-F. Lamellarins and related pyrrole-derived alkaloids from marine organisms. Chem. Rev. 108, 264–287 (2008).
Domagala, A., Jarosz, T. & Lapkowski, M. Living on pyrrolic foundations—advances in natural and artificial bioactive pyrrole derivatives. Eur. J. Med. Chem. 100, 176–187 (2015).
Saha, I., Lee, J. T. & Lee, C.-H. Recent advancements in calix[4]pyrrole-based anion-receptor chemistry. Eur. J. Org. Chem. 2015, 3859–3885 (2015).
Gulevich, A. V., Dudnik, A. S., Chernyak, N. & Gevorgyan, V. Transition metal-mediated synthesis of monocyclic aromatic heterocycles. Chem. Rev. 113, 3084–3213 (2013).
Estévez, V., Villacampa, M. & Monéndez, J. C. Recent advances in the synthesis of pyrroles by multicomponent reactions. Chem. Soc. Rev. 43, 4633–4657 (2014).
Zhou, N.-N., Zhu, H.-T., Yang, D.-S. & Guan, Z.-H. Recent developments in the group-1B-metal-catalyzed synthesis of pyrroles. Org. Biomol. Chem. 14, 7136–7149 (2016).
Vessally, E. A new avenue to the synthesis of highly substituted pyrroles: synthesis from N-propargylamines. RSC Adv. 6, 18619–18631 (2016).
Chelucci, G. Metal-catalyzed dehydrogenative synthesis of pyrroles and indoles from alcohols. Coord. Chem. Rev. 331, 37–53 (2017).
Kornienko, A. & La Clair, J. J. Covalent modification of biological targets with natural products through Paal–Knorr pyrrole formation. Nat. Prod. Rep. 34, 1051–1060 (2017).
Liu, W., Jiang, H. & Huang, L. One-pot silver-catalyzed and PIDA-mediated sequential reactions: synthesis of polysubstituted pyrroles directly from alkynoates and amines. Org. Lett. 12, 312–315 (2010).
Sayyed-Alangi, S. Z., Hossaini, Z. & Rostami-Charati, F. Synthesis of highly functionalized pyrroles from primary amines and activated acetylenes in water. Chin. Chem. Lett. 23, 1119–1121 (2012).
Madabhushi, S. et al. Ceric (IV) ammonium nitrate (CAN) mediated synthesis of pyrrole-2,3,4,5-tetracarboxylates by reaction of dimethyl acetylenedicarboxylate with an amine. Tetrahedron Lett. 54, 6737–6739 (2013).
Zhang, L., Wang, X., Li, S. & Wu, J. Synthesis of pyrrole-2,3,4,5-tetracarboxylates via a copper-catalyzed reaction of amine with but-2-ynedioate. Tetrahedron 69, 3805–3809 (2013).
Chong, Q., Xin, X., Wang, C., Wu, F. & Wan, B. Synthesis of polysubstituted pyrroles via Ag(I)-mediated conjugate addition and cyclization reaction of terminal alkynes with amines. Tetrahedron 70, 490–494 (2014).
Chen, X., Jin, J., Wang, Y. & Lu, P. Palladium-catalyzed synthesis of 7,9-diaryl-8H-acenaphtho[1,2-c]pyrroles and their application in explosives detection. Chem. Eur. J. 17, 9920–9923 (2011).
Chen, X. et al. Palladium-catalyzed reaction of arylamine and diarylacetylene: solvent-controlled construction of 2,3-diarylindoles and pentaarylpyrroles. Eur. J. Org. Chem. 2012, 4380–4386 (2012).
Martin, R., Larsen, C. H., Cuenca, A. & Buchwald, S. L. Cu-catalyzed tandem C–N bond formation for the synthesis of pyrroles and heteroarylpyrroles. Org. Lett. 9, 3379–3382 (2007).
Bhardwaj, V., Gumber, D., Abbot, V., Dhiman, S. & Sharma, P. Pyrrole: a resourceful small molecule in key medicinal hetero-aromatics. RSC Adv. 5, 15233–15266 (2015).
Gholap, S. S. Pyrrole: an emerging scaffold for construction of valuable therapeutic agents. Eur. J. Med. Chem. 110, 13–31 (2016).
Cram, D. J. et al. Stereochemistry of sulfur compounds. I. Stereochemical reaction cycles involving an open chain sulfoxide, sulfimide, and sulfoximide. J. Am. Chem. Soc. 92, 7369–7384 (1970).
Li, Zhen, Yu, H. & Bolm, C. Dibenzothiophene sulfoximine as an NH3 surrogate in the synthesis of primary amines by copper-catalyzed C–X and C–H bond amination. Angew. Chem. Int. Ed. 56, 9532–9535 (2017).
Okamura, H. & Bolm, C. Sulfoximines: Synthesis and catalytic applications. Chem. Lett. 33, 482–487 (2004).
Bizet, V., Hendriks, C. M. M. & Bolm, C. Sulfur imidations: access to sulfimides and sulfoximines. Chem. Soc. Rev. 44, 3378–3390 (2015).
Bizet, V., Buglioni, L. & Bolm, C. Light-induced ruthenium-catalyzed nitrene transfer reactions: a photochemical approach towards N-acyl sulfimides and sulfoximines. Angew. Chem. Int. Ed. 53, 5639–5642 (2014).
Jolicoeur, B., Chapman, E. E., Thompson, A. & Lubell, W. D. Pyrrole protection. Tetrahedron 62, 11531–11563 (2006).
Trost, B. M. & Ball, Z. T. Markovnikov alkyne hydrosilylation catalyzed by ruthenium complexes. J. Am. Chem. Soc. 123, 12726–12727 (2001).
Zhang, L. et al. Ruthenium-catalyzed cycloaddition of alkynes and organic azides. J. Am. Chem. Soc. 127, 15998–15999 (2005).
Yamamoto, Y., Arakawa, T., Ogawa, R. & Itoh, K. Ruthenium(II)-catalyzed selective intramolecular [2 + 2 + 2] alkyne cyclotrimerizations. J. Am. Chem. Soc. 125, 12143–12160 (2003).
Yamamoto, Y. et al. Ru(II)-catalyzed cycloadditions of 1,6-heptadiynes with alkenes: new synthetic potential of ruthenacyclopentatrienes as biscarbenoids in tandem cyclopropanation of bicycloalkenes and heteroatom-assisted cyclocotrimerization of 1,6-heptadiynes with heterocyclic alkenes. J. Am. Chem. Soc. 122, 4310–4319 (2000).
Le Paih, J. et al. Biscarbene-ruthenium complexes in catalysis: novel stereoselective synthesis of (1E,3E)-1,4-disubstituted-1,3-dienes via head-to-head coupling of terminal alkynes and addition of carboxylic acids. J. Am. Chem. Soc. 125, 11964–11975 (2003).
Schmid, R. & Kirchner, K. Ruthenium-mediated C–C coupling reactions of alkynes—the key role of ruthenacyclopentatriene complexes. Eur. J. Inorg. Chem. 2004, 2609–2626 (2004).
Wakatsuki, Y., Kuramitsu, T. & Yamazaki, H. Cobaltacyclopentadiene complexes as starting materials in the synthesis of substituted benzenes, cyclohexadienes, thiophenes, selenophenes and pyrroles. Tetrahedron Lett. 1974, 4549–4552 (1974).
Hong, P. & Yamazaki, H. Reaction of cobaltacyclopentadiene complexes with organic azides directed toward synthesis of highly substituted pyrroles. J. Organomet. Chem. 373, 133–142 (1989).
Hill, J. E., Fanwick, P. E. & Rothwell, I. P. Formation of a terminal aryl–imido compound of titanium by cleavage of the N = N double bond in benzo[c]cinnoline. Inorg. Chem. 30, 1143–1144 (1991).
Sperger, T., Sanhueza, I. A., Kalvet, I. & Schoenebeck, F. Computational studies of synthetically relevant homogeneous organometallic catalysis involving Ni, Pd, Ir, and Rh: an overview of commonly employed DFT methods and mechanistic insights. Chem. Rev. 115, 9532–9586 (2015).
Acknowledgements
This research is partially supported by the Platform Project for Supporting in Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research; BINDS) from the Japan Agency for Medical Research and Development, and JSPS KAKENHI (Grand Number JP 16KT0051).
Author information
Authors and Affiliations
Contributions
K.M. conceived the project, carried out the experimental works, and analyzed the experimental results. Y.Y. performed computational studies and wrote the manuscript. M.S. discussed the results.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Matsui, K., Shibuya, M. & Yamamoto, Y. Synthesis of pyrroles via ruthenium-catalyzed nitrogen-transfer [2 + 2 + 1] cycloaddition of α,ω-diynes using sulfoximines as nitrene surrogates. Commun Chem 1, 21 (2018). https://doi.org/10.1038/s42004-018-0022-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42004-018-0022-2
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
