
Abstract
Our study provides substantial evidence that the host genome affects the comprehensive function of the microbiome in the rumen of bovines. Of 1,107/225/1,141 rumen microbial genera/metagenome assembled uncultured genomes (RUGs)/genes identified from whole metagenomics sequencing, 194/14/337 had significant host genomic effects (heritabilities ranging from 0.13 to 0.61), revealing that substantial variation of the microbiome is under host genomic control. We found 29/22/115 microbial genera/RUGs/genes host-genomically correlated (|0.59| to |0.93|) with emissions of the potent greenhouse gas methane (CH4), highlighting the strength of a common host genomic control of specific microbial processes and CH4. Only one of these microbial genes was directly involved in methanogenesis (cofG), whereas others were involved in providing substrates for archaea (e.g. bcd and pccB), important microbial interspecies communication mechanisms (ABC.PE.P), host-microbiome interaction (TSTA3) and genetic information processes (RP-L35). In our population, selection based on abundances of the 30 most informative microbial genes provided a mitigation potential of 17% of mean CH4 emissions per generation, which is higher than for selection based on measured CH4 using respiration chambers (13%), indicating the high potential of microbiome-driven breeding to cumulatively reduce CH4 emissions and mitigate climate change.
Similar content being viewed by others
Introduction
Ruminants harbour a unique symbiotic gut microbial population that transforms indigestible fibrous feed into high-quality products such as meat and milk for human consumption. Moreover, ruminant livestock is vital to meet global food security and contribute to poverty1 reduction in an increasing world population2. Yet to be solved is the negative environmental impact, as dairy and beef cattle account for 9.5% of all anthropogenic greenhouse gas (GHG) emissions3. Of those, ruminal microbial fermentation represents 40–50%; in particular, due to the highly potent GHG methane (CH4)4. Additionally, CH4 emissions imply a considerable energy loss to the animal, ranging from 2 to 12% of gross energy intake5. Therefore, decreasing CH4 emissions is acknowledged to contribute to the mitigation of climate change and optimize the economic efficiency of cattle production6. Ruminal methanogenesis is a complex process dependent on the cooperation of taxonomic communities with different metabolic activities7,8,9,10. A diverse community of bacteria, ciliate protozoa, and anaerobic fungi11 convert complex diet carbohydrates, proteins, and lipids into volatile fatty acids, lactate, microbial proteins, and vitamins while releasing CO2, H2, and other compounds. Four orders of ruminal methanogenic archaea use electrons derived from H2, formate or methyl compounds to reduce carbon dioxide into CH4 to obtain energy for growth12. Dietary interventions designed to alter the microbiome for CH4 mitigation (e.g. protozoa defaunation13,14, seaweed15, and 3-NOP16 additives) have often failed in the long term due to microbiota adaptation to the new environment17 or are associated with increased production costs. In contrast, the genomic selection that targets the part of the host genome modulating microbiome composition related to low CH4-emitting cattle opens up the opportunity to provide a cost-effective permanent solution to reduce CH4 emissions from ruminants.
There is increasing evidence of a host genetic impact on the composition of the microbiota in the rumen of bovines7,18,19,20,21,22,23,24,25, monogastric livestock26,27, and humans28,29,30,31,32,33. These previous metagenomic studies were based on microbiota profiles mostly identified using sequence polymorphisms of the 16S rRNA gene and therefore did not consider the functional versatility of unique rumen microbial species, nor the ability of some microbial organisms to integrate foreign DNA from other microbial organisms into their DNA. Novel microbial species in the rumen have recently been identified using metagenome-assembled genomes generated from whole metagenomic sequence data of microbial DNA from rumen samples8,34, but how their abundances are shaped by host genomics is still unknown. There is also a lack of knowledge of the host genomic associations with the abundances of functional microbial KEGG genes which were found to predict methane emissions on the phenotypic level with a prediction ability of r2 = 0.817. Moreover, a microbiome-driven selection strategy based on this functional information of microbial genes in animal breeding and a study of its effect on the response to selection is to the best of our knowledge not available, since previous research emphasized host genetic effects on the taxonomical composition of the microbiome22,35,36. To identify the functionality of the rumen microbiome directly, we applied genome-resolved metagenomics to generate abundances of the microbial KEGG genes based on whole metagenomic sequencing. Based on these data, we carried out comprehensive research that elucidates how host genomics influences complex functions of ruminal microbes (determined by their microbial genes) genomically correlated to CH4 emissions. Furthermore, we developed a microbiome-driven selection strategy showing how this information can best be included in cattle breeding to directly change the rumen microbiome function and reduce these emissions.
We identified the host genomic (using single-nucleotide polymorphism (SNP) information) influence on an extensively characterized microbiome in relation to CH4, using whole metagenome sequencing of rumen microbial DNA samples from a bovine population designed for a powerful host genomic analysis37,38,39 with high standardization of diets and other husbandry effects. We characterized the core ruminal microbiome by identifying 1,108 cultured microbial genera by mapping our sequences to the Hungate1000 reference genome collection40 and RefSeq41 databases (Supplementary Data 1); 225 ruminal uncultured genomes (RUGs) by de novo metagenome-assembly of genomes34, 34 of them classified at strain level (Supplementary Data 2), and 1142 functional microbial genes (Supplementary Data 3); present in all (n = 359) and for RUGs in >200 of our animals. Our specific hypothesis is that the host genome influences the abundance of not only functional microbial genes involved in metabolism, but also in interspecies communication, host–microbiome interactions, and genetic information processing. These functions may play a key integrating role in achieving a ruminal balance where fermentation of feed into essential nutrients utilized by the host is optimized and substrates utilized by methanogenesis e.g. H2 excess are minimized. Our comprehensive description of ruminal microbiome functionalities includes the abundances of 34 microbial genes carried by methanogenic archaea directly implicated in CH4 metabolism; 511 involved in other metabolic pathways of bacteria, archaea, ciliate protozoa or fungi, indirectly influencing methanogenesis by minimizing required substrates through non-methanogenic routes that yield beneficial nutrients for ruminants42 (e.g. acetogenesis, propionogenesis13,43,44,45), or generating methanogen-inhibitor metabolites46,47,48; 207 in microbial communication processes and host-microbiome interaction (e.g. ABC transporters of different metabolites or fucose sensing) carried by fungi, bacteria and archaea49,50,51, of importance because the synthesis of CH4 in cooperation with other main metabolic routes in the rumen52,53,54 are syntrophic processes amongst microbial communities55; 330 involved in genetic information processes (e.g. ribosomal biosynthesis) related to microbial growth56; and 60 at present not functionally characterized. For each of these 2475 functional and taxonomic characteristics of the rumen microbiome, the host genomic determination and correlation with CH4 emissions were analysed. After stringent adjustment for multiple testing, we demonstrate that our hypothesis of a common host genomic control is valid by discovering heritabilities (h2) of microbial profiles and host-genomic correlations with CH4 emissions (rgCH4) significantly deviating from zero, which shows the effectiveness of this strategy. Our results are obtained in bovines, but also provide an indication of potential host genomic effects on functional microbial genes and their biological processes in other species.
Besides providing a better understanding of the complex host genomic effects on the rumen microbiome function, this research provides the basis for a cost-effective microbiome-driven breeding strategy to mitigate CH4 emissions from cattle without measuring it directly, which is necessary considering the cost-prohibitive limitations of measuring individual animal CH4 emissions.
Results
Bovine host genomics affected CH4 emissions produced by ruminal archaea
CH4 emissions57 were accurately measured from individual beef cattle (n = 285) using the gold-standard method of respiration chambers. Animals within the same breed or diet expressed high phenotypic variability in CH4 emissions with coefficients of variation from 16.3 to 28.5% (Supplementary Fig. 1a, b). Genomic h2 of CH4 emissions revealed that 33% (Bayes Factor for genomic effects (BF) = 5.91) of this phenotypic variation was explained by host genome variation, which is consistent with other studies58,59,60. The h2 obtained for CH4 emissions is at the level of other traits for which substantial gains due to breeding are achieved, such as growth rate61 and milk yield62. In addition, there was large genomic variation for CH4 emissions as deviation from the mean ranged from −2.67 to 3.51 g/kg of dry matter intake (DMI) with no difference between breeds (P > 0.16), which suggests that bovines have most likely not been indirectly selected for CH4 emissions as a result of a lack of genetic correlation to those traits under selection.
Host genomics affects the ruminal microbiome composition
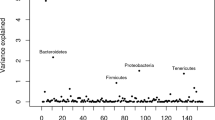
We next investigated the proportion of the ruminal microbiome variation at taxonomic and functional levels explained by the host genomic variation among individuals, by estimating h2 of the ruminal abundances of 1107 genera, 225 RUGs, and 1141 microbial genes. Our results demonstrate significant host genomic effects with h2 in a range between 0.13 and 0.61 for the abundances of 194 microbial genera, 14 RUGs, and 337 microbial genes representing cumulatively 58.4, 5.63, and 27.2%, respectively, of the total relative abundance (RA) (Fig. 1 and Supplementary Data 4–6). Among the 194 genera, 20 were highly heritable (h2 > 0.40), which belonged exclusively to bacteria (e.g. Firmicutes Acidaminococcus (RA = 0.3%), h2 = 0.54, BF = 8.82 × 10+5) and archaea (e.g. hydrogenotrophic methanogen Methanospirillum63 (RA = 0.0005%, h2 = 0.40, BF =; 1.04 × 10+2). Host genome also shaped the abundance of the hydrogenotrophic/methylotrophic55 methanogen Candidatus Methanoplasma (RA = 0.002%, h2 = 0.32, BF = 6.31 × 10+2), and to a lesser extent the abundance of ubiquitous Methanobrevibacter (RA = 5.02%, h2 = 0.24, BF = 9.10)—which is coherent with estimates from other studies18,19,20—Candidatus Methanomethylophilus (RA = 0.05%, h2 = 0.26, BF = 4.18), Methanothermus (RA = 0.002%, h2 = 0.25, BF = 1.26 × 10+1) and Methanoplanus (RA = 0.0008%, h2 = 0.24, BF = 9.56). Reinforcing the evidence of a host-genomic component in the abundance of methanogenic archaea, 5 RUGs annotated as uncultured Methanobrevibacter strains (RA > 0.27%) demonstrated moderate to high h2 estimates (0.39–0.48, BF from 3.5 to 4.65 × 10+1), indicating that more specific classification using RUGs provides the opportunity to find highly heritable strains. The most abundant complex carbohydrates degraders in the rumen—Eubacterium (RA = 1.02%), Prevotella (RA = 39.2%), Butyrivibrio (RA = 2.54%), Bacteroides (RA = 1.39%) and Pseudibutyrivibrio (RA = 0.54%)—were highly (h2 = 0.51 for Eubacterium, BF = 9.72 × 10+3) or moderately (h2 = 0.23–0.33 for the others, BF from 7.42 to 9.73 × 10+1) heritable; with 7 highly abundant RUGs (RA > 0.25%) classified as uncultured Prevotellaceae bacterium having h2 from 0.32 to 0.45 (BF from 7.48 to 1.67 × 10+2). These results support the concepts of a “core heritable microbiome”21,64 and stability over time of certain microbial genera abundance such as Prevotella65. None of the fungi and protist genera, which are considered to be non-essential for rumen function and highly variable within different host species66, were highly heritable.

Bars show the h2 values of 194/14/337 rumen microbial genera/uncultured genomes (RUGs)/genes tested exhibiting non-zero h2 estimates and significant host genomic effects (based on Bayes Factor >3 and Deviance Information Criterion difference between models with or without host genomic effects ≤−20). a Cultured microbial genera and RUGs classified within the phylum. b Microbial genes grouped by microbial biological processes: microbial communication and host-microbiome interaction (Comm. & Host Interact.), genetic information processes (Genetic Inform.), metabolism other than methane (Other Metabol.), and methane metabolism (CH4 Metabol.). Source data is in Supplementary Data 4–6.
We elucidated that the specific functional capacity of the ruminal microbiome is heritable by estimating the h2 of a comprehensive set of microbial genes, of which 33 were highly (h2 > 0.4), and 304 were moderately (0.2 < h2 < 0.4) heritable. These microbial genes are involved in a wide variety of metabolic functions (Fig. 1b and Supplementary Data 6), e.g. synthesis of microbial proteins or volatile fatty acids, suggesting that the host genome influences the growth of microbes responsible for the release of nutrients during microbial fermentation67,68. Among 34 microbial genes involved in the CH4 metabolism pathway, 13 showed moderate h2 of 0.22–0.29 (BF from 3.96 to 7.82 × 10+1), e.g. mcrA, mcrB, mtrD, mtrE, and cofG. Ribosomal biosynthesis was revealed to be under strong host-genomic control with 56 heritable microbial genes, representing a cumulative RA of 6.57%, including 9 highly heritable genes (h2 = 0.40–0.53, BF = 1.62 × 10+2 to 1.18 × 10+8) synthesizing the large ribosomal subunit. Intracellular ribosomal biosynthesis reflects the growth rate of microbial organisms, given that ribosomes can account for up to 40% of their cellular dry mass56, and cell fitness and optimal growth are tightly coupled to efficient protein synthesis69. Demonstrating that differences among animals in complex microbiome functions are partly due to host genomic variation opens up opportunities to consider a new source of genetic variation not only in ruminants but also in humans, where the h2 of microbial gene abundances was estimated to be even larger (0.65–0.91)70.
Ruminal microbial mechanisms related to CH4 emissions are influenced by host genomics
The existence of a common host genomic influence on CH4 emissions and the rumen microbiome was evaluated by estimating host-genomic correlations between CH4 emissions and each microbial genus/RUG/gene abundance (rgCH4). Based on the probability of rgCH4 being different from 0 (P0) ≥ 0.95, our study revealed 29 microbial genera, 22 RUGs, and 115 functional microbial genes strongly host-genomically correlated with CH4 emissions (rgCH4 from |0.59| to |0.93|, Supplementary Data 7–9). Among the significant microbial communities, most were bacteria (22 genera/17 RUGs) belonging to Bacteroidetes (5/14), Firmicutes (6/2), and Proteobacteria (9/1) phyla. Most microbial genes with strong rgCH4 were not directly involved in CH4 metabolism pathways, but rather mechanisms indirectly affecting CH4 production most likely by limiting substrates for methanogenesis10,71, inhibiting methanogens, playing a role in coordinating actions among microbial communities and the host or leading microbial genetic processes. Only H2-oxidizing Methanoregula (RA = 0.003%) with unknown activity in rumen49 and the microbial gene cofG involved in F420 coenzyme biosynthesis72,73 resulted in significant negative rgCH4 (−0.82 and −0.71, P0 ≥ 0.95), suggesting that these are abundant under ruminal conditions unfavourable for high CH4 producing methanogens. Four uncultured Methanobrevibacter strains showed negative rgCH4 (<−0.72, P0 ≥ 0.95) and one was positive (0.91, P0 = 0.99), indicating that the relationship between the abundance of Methanobrevibacter and CH4 emissions is complex as different strains may have functional versatility. We hypothesize that some Methanobrevibacter sp. can produce CH4 even under a challenging ruminal environment (e.g. low pH value), however, at a substantially lower level than those adapted to more favourable conditions. To visualize which microbial genus/gene abundances in the rumen are influenced by a common host genomic background, we constructed a co-abundance network based on Pearson correlations among deregressed host genomic effects for each microbial genus/RUG/gene (Supplementary Fig. 2 and Supplementary Data 10). This approach revealed co-abundance clusters of bacterial and fungal genera74 with strong rgCH4 and methanogenic archaea, e.g. fungal Metschnikowia (rgCH4 = 0.77, P0 = 0.96) and archaeal Methanosarcina (cluster 9 in Supplementary Fig. 2); and of microbial genes not directly involved in CH4 metabolism but with strong rgCH4 (e.g. RP-L6, rgCH4 0.71, P0 = 0.96) and those involved directly in CH4 metabolism (e.g. fbaA, cluster 1 in Supplementary Fig. 2). The most important host-genomically affected ruminal microbial mechanisms associated with CH4 production (based on rgCH4) are as follows:
Microbial metabolism
An extensive group of microbial genes involved in amino acid metabolic and transport pathways displayed negative rgCH4. Part of this group of microbial genes was involved in the biosynthesis of arginine75 and branched-chain amino acids76,77 via oxocarboxylic acid metabolism (argF, argD and ilvA with rgCH4 = −0.84 to −0.88, P0 ≥ 0.96; and argJ, argC, alaA, ilvH, leuB, and leuD with rgCH4 = −0.55 to −0.77 at lower evidence P0 ≥ 0.85, Figs. 2 and 3). Aconitate hydratase (ACO) catalysing the isomerization of citrate to isocitrate in the early stage of the oxocarboxylic chain extension, and bcd and pccB degrading branched-chain amino acids into branched-chain volatile fatty acids, which have an inhibitory effect on methanogens46, also expressed negative rgCH4 = −0.76 to −0.90 (P0 ≥ 0.95). We also estimated negative rgCH4 for microbial genes coding ABC transporters of polar and branched-chain amino acids (ABC.PA.A, ABC.PA.S, livH, livG, and livK rgCH4 = −0.83 and −0.93, P0 ≥ 0.95). Another group of microbial genes was related to the metabolism of aromatic amino acids tryptophan, tyrosine, and phenylalanine (AROA2, trpA, trpD, trpE, tyrA2 and paaH with rgCH4 = −0.74 to −0.87, P0 ≥ 0.95 and aroC, aroA, aroF, trpG, and trpB, with rgCH4 = −0.68 to −0.74 at lower evidence P0 ≥ 0.85, Fig. 4). More specifically, trpE, trpD, and trpA take part in the metabolism of L-tryptophan (Fig. 4) whose catabolites (e.g. indole) are important signalling molecules in biofilm formation78, and activation of the host immune system79. Moreover, 2-oxocarboxylic acid and tyrosine catabolites are precursors for the biosynthesis of coenzyme B77,80 and methanofuran73 methanogenic cofactors, and their diversion into the synthesis of other substrates (e.g. arginine, branched-chain amino acids or tryptophan) could explain their negative rgCH4. Lastly, four microbial genes with negative rgCH4 (−0.61 to −0.87, P0 ≥ 0.95) were associated with methionine metabolism (metE, DNMT1) and transport (metQ and metN). Methionine is associated with minor methylotrophic methanogenesis pathway81 in the rumen82,83 and with an enhancement of microbial long-chain fatty acid production84, a highly H2 demanding process44. Our study highlights that the negative association between microbial amino acid metabolism and CH485,86 has a host genomic component. This could be partly due to host genomic effects19 on ruminal feed retention times, which have opposite effects on microbial protein synthesis efficiency67 and CH4 production87.

Small rectangles symbolize proteins encoded by the microbial genes. Microbial genes are highlighted in red when their rgCH4 estimates range between −0.74 and −0.93 and show a probability of being different from 0 (P0) ≥ 0.95, and in orange when they range between |0.55| and |0.77| and P0 ≥ 0.85. Source data is in Supplementary Data 9. Compounds are denoted by their short names. Full names of compounds and microbial genes are given in Supplementary Data 17.

Small rectangles symbolize proteins encoded by the microbial genes. Microbial genes are highlighted in red when their rgCH4 estimates range between −0.74 and −0.93 and show a probability of being different from 0 (P0) ≥ 0.95, and in orange when they range between |0.55| and |0.77| and P0 ≥ 0.85. Source data is in Supplementary Data 9. Compounds are denoted by their short names. Full names of compounds and microbial genes are given in Supplementary Data 17.

Small rectangles symbolize proteins encoded by the microbial genes. Microbial genes are highlighted in red when their rgCH4 estimates range between −0.74 and −0.93 and show a probability of being different from 0 (P0) ≥ 0.95, and in orange when they range between |0.55| and |0.77| and P0 ≥ 0.85. Compounds are denoted by their short names. Source data is in Supplementary Data 9. Full names of compounds and microbial genes are given in Supplementary Data 17.
We obtained negative rgCH4 (from −0.60 to −0.85, P0 ≥ 0.95) for the abundance of several microbial genes responsible for sucrose metabolism (sacA, maIZ, bgLB, SPP, and sucrose phosphorylase, Fig. 5), including the highly abundant sucrose fermenter88 Eubacterium (RA = 1.02%), transporters of multiple sugars across the membrane85 (ABC.MS.P1, ABC.MS.S, and ABC.MS.P), and the microbial gene PTS-EI that catalyses the phosphorylation of incoming sugar substrates concomitantly with their translocation across the cell membrane. Microorganisms capable of fast growth on soluble sugars are suggested to be favoured in hosts with low rumen size and high digesta turnover rate85,89, features also associated with low CH4 emissions87. Degradation of easily fermentable carbohydrates, such as sucrose or starch, causes a pH decline which has a strong CH4 reducing effect as a result of pH sensitivity of methanogens or H2-producing microbes90. Furthermore, previously mentioned microbial genes aroA and trpE are involved in the shikimate pathway91 linking sugar metabolism with the synthesis of microbial proteins (aromatic amino acids, tyrosine, phenylalanine, and tryptophan) which are an important source of amino acids for the host. Microbial protein yield from sucrose is suggested to be more persistent over time in comparison to other carbohydrates92, and partially stored by sucrose utilizers (e.g. Eubacterium) for the maintenance of the microbial population92.

Small rectangles symbolize proteins encoded by the microbial genes. Microbial genes are highlighted in red when their rgCH4 estimates range between −0.74 and −0.93 and show a probability of being different from 0 (P0) ≥ 0.95, and in orange when they range between |0.55| and |0.77| and P0 ≥ 0.85. Source data is in Supplementary Data 9. Compounds are denoted by their short names. Full names of compounds and microbial genes are given in Supplementary Data 17.
We also found negative rgCH4 for the abundance of hydrogenotrophic acetogenic bacteria Blautia45, together with Eubacterium93 (rgCH4 = −0.60 and −0.73, P0 ≥ 0.95), and the fhs microbial gene involved in the reductive Wood–Ljungdahl acetyl-CoA pathway (rgCH4 = −0.79, P0 = 0.98). Acetogens produce volatile fatty acids (mainly acetate but also propionate and butyrate94), which serve as host nutrients to improve animal performance42 and simultaneously compete against methanogens for metabolic H29,43,45. Despite acetogenesis being thermodynamically less favourable than the reduction of CO2 into CH495 in the rumen, this may vary upon microbial interactions and host-genomically influenced ruminal environmental factors42,45,68. Propionogenesis via acrylate34,85,89,96 and lactaldehyde routes40 was another microbial mechanism under host genomic influence lowering CH4 emissions as indicated by negative rgCH4 (−0.76 to −0.90, P0 ≥ 0.95) for the abundances of microbial genes bcd and pccB involved in propanoyl-CoA metabolism and fucO catalysing the reduction of lactaldehyde into 1,2-propanediol, as well as the highly abundant (RA = 0.08%) lactate-producing bacteria Kandleria (rgCH4 = −0.87, P0 = 0.99). Propionate production from lactate not only reduces H2 availability for methanogenesis13,97 but also prevents rumen acidosis98 and results in a more efficient rumen fermentation99,100. The abundance of six microbial genes encoding [4Fe-4S] cluster containing proteins (bioB, cobL, cofG, nifU, ACO, and pflA) involved in electron transfer mechanisms in redox reactions presented rgCH4 from −0.71 to −0.87 (P0 ≥ 0.96). The first two proteins are involved in the synthesis of substrates required for methanogenic cofactors; i.e. bioB catalyses the conversion of dethiobiotin to biotin101, which competes with coenzyme B for the synthesis of its alkyl portion102,103; and cobL together with hemC (rgCH4 = −0.91, P0 = 1.00) take part in porphyrin metabolism, required for different processes including the synthesis of porphyrin-based cofactors vitamin B12 and F430104. Nitrogen fixation protein nifU carries out N2 reduction into ammonia105, which can act as an alternative H2-consuming sink competing with ruminal methanogenesis. Further negative rgCH4 were obtained for microbial genes in thiamine metabolism (iscS, thiD, thiH, and thiE with rgCH4 from −0.88 to −0.70, P0 ≥ 0.91)106; hydration of long-chain fatty acid oleate into anti-tumoral hydroxystearic acid107,108 (ohyA, −0.81, P0 = 0.95), or import of methanogen inhibitors long-chain fatty acids47 (ABCB-BAC109, rgCH4 = −0.9, P0 = 0.99). Moreover, highly abundant bacteria genera with ruminal fatty acid biohydrogenation activity110,111, Eubacterium and Butyrivibrio (RA = 2.54%, rgCH4 = −0.37, P0 = 0.80) were negatively correlated with CH4.
Microbial communication and host–microbiome interaction mechanisms
The majority of methanogens in the rumen are integrated into the biofilm on the surface of feed particles where H2 producing bacteria are active112,113,114. We found strong negative rgCH4 (−0.78 to −0.92, P0 ≥ 0.96) for abundances of microbial genes mediating microbial interactions, involved in ABC transport of cobalt/nickel (cbiO and cbiQ) and quorum sensing-related peptide/nickel ions (ABC.PE.P, ABC.PE.S, ABC.PE.A, ABC.PE.P1)—cobalt and nickel being detrimental for hydrogenotrophic and aceticlastic methanogenic activity48—protein export (secD and secF) and chemotaxis (cheA and mcp); and positive rgCH4 for transcription protein cbpA (0.85, P0 = 0.97) acting as a microbial response to maintain plasmids replication during amino acid starvation115. CH4 emissions were also host-genomically correlated with abundances of microbial genes mediating host-microbiome interaction; e.g. cbh and baiN116 (rgCH4 = −0.80, P0 ≥ 0.96) involved in bacterial biosynthesis of secondary bile acids which activate metabolic receptors within gut, host liver, and peripheral tissues116,117 and inhibit CH4 production in the rumen by transferring metabolic H2 into propionate production118. Another interesting finding is that TSTA3, involved in the metabolism of host-microbiome crosstalk mediator fucose119, displays a positive rgCH4 (0.85, P0 = 0.98). Fucose is a component of mucins present in saliva120, which is produced abundantly by ruminants and acts as a pH buffer during ruminal fermentation due to its phosphate and bicarbonate content121. Cellulolytic Fibrobacter, an indicator of high pH levels in rumen122, was positively host-genomically correlated to TSTA3 in our data (0.66, P0 = 0.94), while lactic acid producer Kandleria, generally associated with low pH levels and negative rgCH4, was host-genomically correlated to TSTA3 negatively (−0.70, P0 = 0.90). Thus TSTA3 could be involved in signalling enhanced saliva production, resulting in increased rumen pH that is known to stimulate the growth of methanogenic archaea and CH4 emissions123.
Genetic information processes
Ribosomal biogenesis represented by RP-S10, RP-S12, RP-S17, RP-L2, RP-L3, RP-L6, RP-L23, RP-L28, RP-L34, and RP-L35, was one of the few microbial mechanisms with positive rgCH4 from 0.71 to 0.84 (P0 ≥ 0.95). All of them are universal ribosomal proteins homologous in bacteria, archaea, and eukarya; except for RP-L28, RP-L34, and RP-L35 exclusively found in bacteria124,125. Given that protein synthesis is highly coupled with cellular growth69, these results suggest that the rumen environment provided by low CH4-emitter host genomes are related to lower growth or activities of specific microbes directly or indirectly involved in methanogenesis.
RUGs enriched with CH4-related microbial genes are strongly host-genomically correlated to CH4 emissions
The 20 highly prevalent (present in >200 animals) RUGs containing the highest number of unique proteins from the 115 microbial genes with strong rgCH4 were all bacterial RUGs carrying between 114 to 180 unique proteins classified into 60 to 84 microbial genes (Fig. 6 and Supplementary Data 11 and 12). Of these 20 highly enriched bacterial RUGs, 18 showed negative rgCH4 consistently with the majority of the microbial genes; 6 of them with rgCH4 < −0.65 (P0 ≥ 0.85) from which 5 RUGs were classified as uncultured Lachnospiraceae bacterium (RUG10082, RUG13438, RUG13308, RUG13002, RUG12132) and 1 as uncultured Clostridiales bacterium (RUG10940). The abundance of Blautia and Dorea microbial genera within Lachnsopiraceae family (identified by alignment to Hungate1000 collection and Refseq databases) also presented negative rgCH4 < −0.72 (P0 ≥ 0.95, Supplementary Data 7). We also investigated the enrichment of these 115 microbial genes in the 6 RUGs with rgCH4 (P0 ≥ 0.95) annotated at the genus level (Supplementary Data 8), and in those RUGs annotated in the same phylogeny level as any of the 29 microbial genera with rgCH4 (P0 ≥ 0.95, Supplementary Data 7), which had low occupancies in our cattle population (<200 animals) and therefore were not included in the 225 considered for breeding (see methods). Our findings show that part of the mechanisms identified in this study occurs in the 5 RUGs classified as uncultured Methanobrevibacter strains, each carrying at least 45 out of the 115 microbial genes (Supplementary Fig. 3). The uncultured Methanobrevibacter strain with positive rgCH4 (RUG12982) carried fewer unique proteins (67 vs. 75 to 93) and microbial genes (51 vs. 55 to 62) than the other 4 uncultured Methanobrevibacter sp. RUGs with negative rgCH4; lacking, for example, argD in arginine biosynthesis, tyrA2 in tyrosine and tryptophan metabolism, and DNMT1 in methionine metabolism, which reinforces the hypothesis of functional versatility amongst different Methanobrevibacter strains explaining their different effects and estimated rgCH4 on CH4 emissions. Low-occupancy RUGs annotated as Eubacterium ruminatum, Eubacterium pyruvativorans, Kandleria vitulina, and uncultured sp. of Blautia RUGs carried at least 49 out of the 115 microbial genes each (Supplementary Fig. 3). Interestingly, their counterparts identified at the genera level presented negative rgCH4 < −0.60 (P0 ≥ 0.95, Supplementary Data 7).

Microbiome-driven breeding of the bovine host for mitigation of CH4 emissions
The comprehensive findings of the host-genomic associations between microbial genus/RUG/gene abundances and CH4 emissions enabled us to predict its mitigation potential when applying genomic selection targeting each of them individually (Supplementary Data 13), indirectly informing about the impact of each microbial mechanism on methanogenesis. Considering 30% of our cattle population being selected based on the abundances of each microbial gene, maiZ in sucrose metabolism, ABC.PE.P in quorum sensing peptide/nickel transport, hemc in porphyrin or upp in pyrimidine metabolism are predicted to result in the highest CH4 mitigation potential (−5.2, −5.3, −5.8 and −6.54% of CH4 emissions mean respectively, P0 ≥ 0.99). Subsequently, our study aimed to find a group of heritable (BF > 3 and Deviance Information Criterion difference between models with or without host genomic effects ≤−20) ruminal microbial genera/RUGs/genes (RA > 0.01%) with strong rgCH4 (P0 ≥ 0.95) to be used collectively for selecting the host genomes associated with low CH4 emissions. We identified 2 microbial genera (Eubacterium and Blautia), 3 RUGs (two annotated as uncultured Methanobrevibacter sp. and one as uncultured Prevotellaceae bacterium) and 38 microbial genes meeting these requirements (Supplementary Data 14). We selected 30 out of the 38 microbial genes (Fig. 7a) covering several microbial mechanisms, e.g. sugar and nickel transport (ABC.PE.P, ABC.MS.P1 and ABC.MS.S), fucose sensing (TSTA3), chemotaxis (mcp), ribosomal biosynthesis (RP-L6, RP-L23, RP-L28, RP-L35, RP-S12 and RP-S17), reductive acetogenesis (fhs) and metabolism of amino acids (argD), sucrose (SPP), CH4 (cofG), biotin (bioB), propionate (pccB), porphyrin (hemC), thiamine (thiD) and pyrimidine (upp). A deep study of the host-genomic correlations among these 30 selected microbial genes showed a common host genomic background influencing the abundance of ABC.PE.P, ABC.MS.P1, fhs, cofG, argD, hemC, thiD, upp, tlyC, NTH, and copB with host-genomic correlations among each other ranging from 0.62 (P0 = 0.90) to 0.99 (P0 = 1.00) (Fig. 7b).

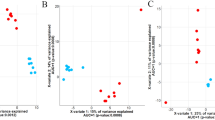
a Estimates of h2 and rgCH4 (error bars represent the highest posterior density interval enclosing 95% probability). Microbial genes grouped by microbial biological processes: methane metabolism (CH4), microbial communication and host–microbiome interaction (Comm. & host interact.), genetic information processes and metabolism other than CH4 (Metabolism). b Correlogram showing the median of the pairwise host-genomic correlations estimates among the additive log-ratio transformed microbial gene abundances selected for breeding purposes. Source data is in Supplementary Data 14. Full names of microbial genes selected for breeding purposes are given in Supplementary Data 19.
Finally, we evaluated the accuracies and response to selection in CH4 emission mitigation in our population based on the prediction of CH4 host genomic effects using three different sources of information: (1) CH4 emissions measured by the “gold-standard” technique of respiration chambers, (2) the 30 microbial gene abundances exhibiting strong rgCH4, and (3) combining both preceding criteria. A single (1) or multiple (2, 3) trait genomic estimation approach was applied in each case. In (2) and (3), CH4 host genomic effects were estimated based on observations of the 30 microbial gene abundances, the genomic relationship matrix amongst individuals, and the estimated host-genomic and residual (co)variance matrix comprising CH4 and the 30 microbial gene abundances; assuming unknown (2) or known (3) CH4 observations (see methods). Using microbiome-driven breeding based on the abundance of 30 specific microbial genes (2) resulted in the mean estimation accuracy of host genomic effects for CH4 emissions to be 34% higher than using measured CH4 emissions (1) (0.70 ± 0.18 vs. 0.52 ± 0.11) and confirmed that functional microbial genes are an extremely valuable source of information to perform host genomic evaluations for CH4 emissions. Using the combined selection criteria (3), the accuracy of estimation was 14% larger than using rumen microbial gene information alone (0.80 ± 0.20). Response to selection in CH4 emissions achieved by selecting animals with low CH4 emission host genomic effects predicted exclusively by microbial gene abundance information resulted in a reduction in emissions of −1.43 ± 0.14 to −3.32 ± 0.77 g CH4/kg DMI per generation, depending on selection intensity (from 1.16 to 2.67 in the analysed population, Fig. 8 and Supplementary Data 16). These results indicate that in our population, microbiome-driven breeding for CH4 emissions reduced its magnitude by 7–17% of its mean per generation, without the necessity for costly measures of CH4 emissions.

Intensities of selection 1.1590, 1.400, 1.755, 2.063, or 2.665 are equivalent to selecting 30, 20, 10, 5, or 1%, respectively, of our n = 285 animal population with CH4 and metagenomic data based on the above-described selection criteria. Dots display the medians and violin plots represent the estimated marginal posterior distributions of the response to selection for each intensity of selection and breeding strategy. Source data is in Supplementary Data 16.
Robustness of estimation of genomic parameters of CH4 emissions and microbiome traits from a cross-classified design of breeds and basal diets
The data, from the highly environmentally standardized experiments, comprised of animals from different breeds that were offered different diets, which could be challenging as different breeds might have different genomic backgrounds for the analysed traits, and different diets could have inflated their variances. To consider the difference in means of these effects we fitted a combination of experiment, breed, and diet effects so that an adjustment of each of these effects and their interactions was achieved. To analyse whether after this adjustment the genomic variances of CH4 emissions and the abundances of 30 microbial genes selected for microbiome-driven breeding are homogeneous across breeds and diets, we computed their posterior distributions separately (Supplementary Figs 4 and 5), following the partition of the variance suggested by Sorensen et al.126, and recently used by Lara et al.127. Using this methodology, we found that our genomic parameter estimates for CH4 emissions are based on similar genomic variances across breeds (with medians of 3.8, 3.7, 4.0, and 3.9 (g/kg DMI)2 for Aberdeen Angus, Limousin, Charolais crosses and pure breed Luing, respectively) and diets (with medians of 3.8 and 3.9 (g/kg DMI)2 for Forage and Concentrate based diets) with almost entirely overlapping distributions indicating their homogeneity (Supplementary Fig. 4). We also identified homogeneous genomic variances for the abundances of microbial genes with overlapping distributions across breeds (Supplementary Fig. 5a) and diets (Supplementary Fig. 5b); for example, we estimated genomic variances with medians between 0.024–0.028 (g/kg DMI)2 across breeds and 0.028–0.029 (g/kg DMI)2 across diets for RP-L35, or between 0.25–0.30 (g/kg DMI)2 across breeds and 0.292–0.296 (g/kg DMI)2 across diets for cofG (Supplementary Fig. 5). These results indicate that the data recorded under controlled experimental conditions with a cross-classified breed and diet two-way experimental design and progeny groups balanced over diets resulted in reliable and robust genomic parameters estimates.
Discussion
Previous metagenomic studies using 16S rRNA identified microbiota revealed host genomic effects on the rumen microbial community7,18,19,20,21,22,23,24,25, e.g., Wallace et al.21 found significant heritabilities for several members of Prevotella and Butyrivibrio genera. These species, together with other non-heritable OTUs from the core microbiota, explained up to 40% of the phenotypic variation of CH4 emissions21 that is expected to be the result of both genetic and environmental correlations, which we estimated in the present study separately for each taxa and microbial gene. Moreover, the challenge of previous studies is the lack of taxonomic resolution to sufficiently explain these associations21, which we resolved using metagenome-assembled RUGs. For example, we revealed that different Methanobrevibacter strains expressed divergent host-genomic correlations with CH4 emissions which correspond to the diverse microbial gene content of these strains.
To increase the predictability of CH4 emissions, Roehe et al.7, suggested genome-resolved metagenomics and identified 20 microbial gene abundances, mainly involved in the methanogenesis pathway, which explained 81% of the phenotypic variation in the emissions. In contrast, the present study uses a host genomic-microbiome analysis strategy and provides a robust and comprehensive insight into joint host-genomic correlations between rumen microbial genes known to affect complex functional mechanisms and CH4 emissions which further enabled us to predict the expected response of selection in the emissions based on each and a combination of the microbial genes. The findings of this research will be of major importance for the mitigation of the highly potent GHG CH4 in bovine through genomic selection on the functional microbiome associated with CH4 referred to as microbiome-driven breeding. The highlights of our research are that the host genome influences CH4 emissions by favouring the growth of reductive acetogenic microbes limiting the excess of metabolic H2 substrate (specifically, Blautia, Eubacterium genera and microbial gene fhs found in the genome of uncultured Lachnospiraceae bacterium, Eubacterium pyruvativorans, Eubacterium ruminatum, uncultured Eubacterium and Blautia sp. RUGs); and promoting the shift in the fermentation towards volatile fatty acids (Kandleria genera and microbial genes bcd, pccB, fucO, carried by uncultured Lachnospiraceae bacterium and Kandleria vitulina RUGs) and microbial proteins yield including arginine and branched-chain amino acids (argF, argD, ilvA, AROA2), tryptophan, tyrosine and phenylalanine (trpA, trpD, trpE, tyrA2, paaH) and methionine (metE or DNMT1), which are expected to lead to animals with improved efficiency of converting feed into nutrients42,128. Moreover, host genome contributes to lower CH4 emissions by enhancing the growth of microbes that consume H2 in alternative pathways (e.g. nitrogen fixation (nifU)); by promoting the pathways that divert specific substrates (e.g. tyrosine and 2-oxocarboxylic acid catabolites) required to produce methanogenic coenzymes or cofactors (coenzyme B and methanofuran) to other routes; and that inhibit methanogenic organisms (e.g. by the presence of branched-chain amino acids or cobalt/nickel (cbiO, cbiQ)) and maintain a lower optimum ruminal pH (sucrose metabolism (sacA, maIZ, bgLB, SPP, sucrose phosphorylase)) preventing gut disorders (e.g. thiamine metabolism (iscS, thiD, thiH, thiE)). The latter result supports our hypothesis that hosts who are genomically resilient to gut disorders produce less CH4, which is compatible with nutritional studies demonstrating that blocking methanogenesis has no undesirable effects on cattle health status or feed intake86. A further highlight of our study is that the host genome influenced the ABC transport of different metabolites (some of them in quorum sensing processes (ABC.PE.P, ABC.PE.S, ABC.PE.A, ABC.PE.P1)), interspecies electron transfers (bioB, cobL, cofG), sensitivity of environmental conditions (cheA, mcp), and host–microbiome interaction mechanisms (cbh, baiN, TSTA3), all host-genomically associated with CH4 emissions. These results shed light on the complex processes of methanogenesis regulated by different microbial mechanisms where communication between microbial communities and their interactions with the host plays an essential role. Genetic information processes in the microbiota (e.g. ribosomal biosynthesis (e.g. RP-S10)) also had a substantial host genomic effect on CH4 emissions, potentially reflecting different microbial community growth profiles.
Our findings on the functional microbial level are complementary to studies investigating the biological mechanisms underlying host genome influence on the colonization and maintenance of specific ruminal microbial groups, such as host genomic effects on rumen size87, muscle contraction associated with passage rate19, or ruminal pH20. Other studies in bovines have elucidated host candidate genes for CH4 emissions involved in similar mechanisms87,129,130, fitting into our demonstrated hypothesis that the host genome commonly influences rumen microbiome profile and CH4 emissions. From nutritional studies66, it is well known that the rumen pH has an overarching effect on the rumen microbial community and its metabolism. The rumen pH is intimately related to the production level of buffer-acting saliva98 that is rich in fucose120. We found that the abundance of TSTA3, encoding the sensor for host–microbiome crosstalk mediator fucose119, was host-genomically positively correlated to CH4 emissions and to Fibrobacter genera, an indicator for high pH122, making TSTA3 a highly valuable biomarker for rumen pH, CH4 metabolism and potentially, for host–microbiome mediation to enhance saliva production.
Our results provide comprehensive insight into which communities and functions of the rumen microbiome can be modified by genomic selection to obtain low CH4-emitter animals. We revealed that specific microbiome functionalities (i.e. microbial gene abundances) are more informative for breeding purposes than specific taxonomies, as indicated by a higher number of microbial genes than genera/RUGs being host-genomically correlated to CH4 emissions. This could be due to the closely defined function of those genes, e.g. being involved in producing specific substrates or mediating a specific pathway that interferes with CH4 metabolism; while each microbial genera expresses many microbial gene functions as indicated by functional versatility within different niche-specific species or clades classified in the same genus12,34,89,131,132 (as observed within different RUGs annotated as uncultured Methanobrevibacter strains); or as a result of horizontal transfer of genes among microbial species133,134. Thus, the knowledge generated in this study overcomes previous efforts exploring the breeding possibilities of rumen microbiome, where overall functional description was not considered and the number of taxonomic units associated with CH4 was limited21. Our previous research has shown that the abundance of microbial communities, in particular their genes and interactions, are excellent biomarkers for the phenotypic prediction of CH4 emissions7,10,37; however, the data sets were insufficient to estimate host genomic influence on these biomarkers7. The present study using a unique data set with highly standardized hosting and management conditions represents a large step further by discovering 38 heritable microbial gene abundances strongly host-genomically correlated with CH4 emissions and by designing a microbiome-based breeding strategy to evaluate their potential to mitigate CH4. Microbiome-driven (indirect) genomic selection for CH4 emissions collectively using 30 of these microbial gene abundances resulted in our population in substantial mitigation of CH4 (up to 17% of its mean per generation; approximately 8% per year using genomic selection), even larger than direct genomic selection based on the accurately measured CH4 emissions. This mitigation potential is permanent and can be cumulatively increased over generations. The selection strategy would at least partially avoid the high cost involved in measuring CH4 emissions, and the cost-effectiveness of indirect selection could be further improved by the development of a microarray to quantify the abundances of the most informative microbial genes135. Another advantage of the proposed selection strategy is that it is based on host-genomic correlations between microbial gene abundances and CH4 emissions which as we discussed have specific biological meanings.
Methods
Animals
Animal experiments were conducted at the Beef and Sheep Research Centre of Scotland’s Rural College (SRUC). The experiment was approved by the Animal Experiment Committee of SRUC and was conducted following the requirements of the UK Animals (Scientific Procedures) Act 1986. The data were obtained from 363 steers used in different experiments38,39,136,137,138 conducted over 5 years (2011, 2012, 2013, 2014, and 2017) in the same farm under the same hosting conditions. In these experiments, we tested different breeds (rotational cross from Aberdeen Angus and Limousin breeds, Charolais-crosses, and pure breed Luing) and two basal diets consisting of 480:520 and 80:920 forage: concentrate ratios (DM basis) and subsequently referred to as forage and concentrate diet. Supplementary Data 15 gives the distribution of the animals across experiments, breeds, and diets. Each experiment was balanced for the breed and diet effects, as well as the progenies of each sire was balanced over each diet, so that the experimental design by itself has high power to disentangle genomic effects from diet effects. Additionally, a power analysis indicated that for the given number of animals per experiment, a genetic design of sires with on average 8 progeny per sire showed the highest power to identify genetic differences between sires in methane yield with an achieved power of 0.93 using the sire estimates and root mean square error of 3.27 as obtained in experiment of our previous study7.
Methane emissions data
Methane emissions were individually measured in 285 of the 363 animals for 48 h within six indirect open-circuit respiration chambers39. One week before entering the respiration chambers, the animals were housed individually in training pens, identical in size and shape to the pens inside the chambers, to allow them to adapt to being housed individually. At the time of entering the chamber, the average age of the animals was 528 ± 38 days and the average live weight was 659 ± 54 kg. In each experiment, the animals were allocated to the respiration chambers in a randomized design within breed and diet. Animals were fed once daily, and the weight of the feed offered and refused was recorded. Methane emissions were expressed as g of CH4/kg of DMI, by dividing the average CH4 emissions (g/day) by the average DMI (kg/day) recorded both over 48 h.
Hosts genomic samples
For host DNA analysis, 6–10 ml of blood from the 363 steers were collected from the jugular or coccygeal vein in live animals or during slaughter in a commercial abattoir. In addition to the 363 samples, 7 blood and 23 semen samples from sires of the steers were available (n = 393 samples in total). Blood was stored in tubes containing 1.8 mg EDTA/ml blood and immediately frozen to −20 °C. Genomic DNA was isolated from blood samples using the Qiagen QIAamp toolkit and from semen samples using Qiagen QIAamp DNA Mini Kit, according to the manufacturer’s instructions. The DNA concentration and integrity were estimated with Nanodrop ND-1000 (NanoDrop Technologies). Genotyping was performed by Neogen Genomics (Ayr, Scotland, UK) using GeneSeek Genomic Profiler (GGP) BovineSNP50k Chip (GeneSeek, Lincoln, NE). Genotypes were filtered for quality control purposes using PLINK version 1.09b139. Single Nucleotide Polymorphisms were removed from further analysis if they met any of these criteria: no known chromosomal location according to Illumina’s maps140, non-autosomal locations, call rates less than 95% for SNPs, deviation from Hardy–Weinberg proportions (χ2 test P < 10−4), or minor allele frequency (MAF) <0.05. Seven animals, showing genotypes with a call rate <90%, were removed so that 386 animals and 36,780 autosomal SNPs remained for the analyses.
Hosts metagenomic samples
For microbial DNA analysis, post mortem digesta samples (approximately 50 ml) from 363 steers were taken at slaughter immediately after the rumen was opened to be emptied. Five ml of strained ruminal fluid was mixed with 10 ml of PBS containing glycerol (87%) and stored at −20 °C. DNA extraction from rumen samples was carried out following the protocol from Yu and Morrison141 based on repeated bead beating with column filtration and DNA concentrations and integrity was evaluated by the same procedure (Nanodrop ND-1000) as for blood samples. Four animals out of 363 did not yield rumen samples of sufficient quality for metagenomics analysis. DNA Illumina TruSeq libraries were prepared from genomic DNA and sequenced on Illumina HiSeq systems 2500 (samples from 4 animals from the experiment year 2011), HiSeq systems 4000 (samples from 284 animals from experiment years 2011, 2012, 2013 and 2014)8,34 or NovaSeq (samples from 76 animals from the experiment year 2017) by Edinburgh Genomics (Edinburgh, Scotland, UK). Paired-end reads (2 × 100 bp for Hiseq systems 2500 and 2 × 150 bp for Hiseq systems 400 and NovaSeq) were generated, resulting in between 7.8 and 47.8 GB per sample (between 26 and 159 million paired reads).
Bioinformatics
For phylogenetic annotation of rumen samples, the sequence reads of 359 samples were aligned to a database including cultured genomes from the Hungate 1000 collection40 and Refseq genomes41 using Kraken software142. From 1178 cultured microbial genera identified, we used only those present in all the samples and with a RA > 0.001% (1108 microbial genera) for downstream analysis, equivalent to 99.99% of the total number of counts. We used the 4941 RUGs generated by Stewart et al.34 with sequences of 282 rumen samples included in this study to identify and quantify the abundance of uncultured species. A detailed description of the metagenomics assembly and binning process and estimation of the depth of each RUG in each sample is described in Stewart et al.34. For breeding purposes, microbial taxa that are present in a large proportion of the animals are required; so we discarded those RUGs present in <200 animals (using a cut-off of 1× coverage) and kept 225 RUGs. RUGs coverages <1, which comprised 17.7% of the whole RUGs data set were imputed based on a Geometrical Bayesian-multiplicative method (GBM) of replacement by using cmultrepl function in zCompositions package143. This algorithm imputes zero values from a posterior estimate of the multinomial probability assuming a Dirichlet prior distribution with default parameters for GBM method144 and performs a multiplicative readjustment of non-zero components to respect original proportions in the composition. The 225 RUGs considered showed a mean relative abundance ≥0.15%. Bioinformatic analysis for the identification of rumen microbial genes was carried out as previously described by Wallace et al.145. Briefly, to measure the abundance of known functional microbial genes whole metagenome sequencing reads were aligned to the Kyoto Encyclopaedia of Genes and Genomes (KEGG) database (https://www.genome.jp/kegg/ko.html)146 using Novoalign (www.novocraft.com). Parameters were adjusted such that all hits were reported that were equal in quality to the best hit for each read and allowed up to a 10% mismatch across the fragment. The KEGG orthologous groups (KO) of all hits that were equal to the best hit were examined. If we were unable to resolve the read to a single KO, the read was ignored; otherwise, the read was assigned to the unique KO, the resulting KO grouping corresponding to a highly similar group of sequences. We identified 3,602 KO (also referred to as microbial genes), common in all animals. As for microbial genera, we used only core microbial genes present in all the samples and with a RA > 0.001% (1142 microbial genes) for downstream analysis, equivalent to 96.25% of the total number of counts. We combined information from KEGG, UniProt, and Clusters of Orthologous Groups of protein databases to classify 1141 microbial genes into classes depending on the biological processes they are involved in CH4 metabolism (34), metabolism other than CH4 pathway (511), genetic information processes (329), microbial communication and host-microbiome interaction (207) and other unknown or at present poorly characterized (61).
Statistics and reproducibility
Log-ratio transformation of metagenomic data
To describe the composition of the microbiome at the taxonomic level (cultured microbial genera and RUGs) and functional level (KO or microbial genes) we estimated their RA by dividing each microbial genus/gene (in counts) by the total sum of counts of microbial genera/genes identified in each sample (Supplementary Data 1–3). To compute host genomic analysis on the microbial cultured genera and gene abundances, we first applied a log-ratio transformation to attenuate the spurious correlations due to their compositional nature147. We used additive log-ratio transformation by using a reference microbial genera/gene because of the linear independence achieved between each variable and all the variables in the composition and because of the facility of its interpretation148,149. Assuming J denotes the number of variables in each microbial database (J = 1142 for microbial genes and 1108 for cultured microbial genera), and J−1 all of them excluding the reference microbial genera/gene, the RA of each microbial genus/gene within a sample was transformed as follows150:
where xj is the RA of each microbial genus/gene j and xref is the RA of a specific microbial genus/gene in the database selected as a reference. We selected the 16S rRNA gene and Oribacterium as reference microbial gene and microbial genus, respectively. These reference variables were selected based on the criteria recommended by Greenacre et al.151: (1) present in rumen samples of 359 animals; (2) highly abundant (mean RA 8.56% and 0.35%, respectively); (3) not mentioned to be associated with CH4 emissions in previous literature; (4) low log-ratio variance so the variation mainly proceeds to the numerator (0.09 and 0.24, both located in the first quartile when ordering the microbial variables by log-ratio variance in decreasing order) and (5) reproducing the geometry of the full set of log-ratios in the original data set shown by the estimate of the Procrustes correlation148,152 between the geometrical space defined by all log-ratios and the one defined by the selected additive log-ratios (Procrustes correlation is 0.95 and 0.92). Oribacterium is a strictly anaerobic and non-spore-forming bacterial genus from the order Clostridiales and family of Lachnospiraceae; commonly found in the rumen of cattle19,153 and also in the human oral cavity154,155. The abundance of RUGs was centred log-ratio transformed149 as additive log-ratio transformation was here hampered by the difficulty of selecting a reference RUG present in all animals. Assuming J denotes the total number of RUGs (J = 225):
where xj is the depth of each RUG j.
Influence of breed, diet, and experiment on CH4 emissions and microbial traits
In our genomic models, we applied an optimal adjustment of the fixed effects as a combination of experiment, breed, and diet so that adjustment of each of these effects and their interactions was achieved (see below section: Estimation of host genomic parameters of CH4 emissions and microbial traits). Sequencer effects were except for only 4 samples nested within the experiment and therefore were accounted for due to the inclusion of this effect. Since these are nuisance effects with the potential of influencing the estimation of genomic effects, we carried out further exploratory analyses and revealed that they were appropriately adjusted and do not interfere with the estimation of genomic parameters. In this exploratory analyses, we evaluated the effect of breed, diet, and experiment on microbial genes, genera, and RUGs using a PERMANOVA analysis with 999 permutations computed with the R package vegan156. Diet and experiment showed the largest effects on the composition of the rumen microbiome. Diet accounted for 18.9, 12.0, and 13.19% of the total phenotypic variance in the microbial genes/microbial genera/RUGs databases, while experiment explained 7.5, 11.17, and 6.09%, respectively (P-value < 0.001 in all cases). Breed effects on microbial genes/microbial genera/RUGs databases were depreciable (0.49, 0.65, and 0.78%) and non-significant (P-values = 0.668, 0.385, and 0.141) indicating that the breeds considered in this study did not show substantial differences in their functional or taxonomical microbiome composition. As expected, all effects became negligible (explaining 0% of the phenotypic variance) after their adjustment as a combined fixed effect. We also evaluated the effects of breed and diet on CH4 emissions mean and variance (Supplementary Tables 1 and 2). Before adjustment, diet had a significant (P-value = 2.2 × 10−16) large effect on CH4 (explained 41.2% of the phenotypic variance), while the effect of breed (2.92%, P-value = 0.0007) was smaller but still significant, however, both turned to 0% after the fixed-effects adjustment. Importantly, for the reliability of estimation of genetic parameters, animals from different breeds, or fed different diets presented homogenous CH4 phenotypic variances (P-values of Levene test = 0.19 and 0.48).
Estimation of host genomic parameters of CH4 emissions and microbial traits
Genomic heritabilities (h2) of CH4 emissions, log-transformed microbial genera (n = 1107), RUGs (n = 225) and microbial genes (n = 1141) abundances were estimated by fitting 2474 GBLUP univariate animal models described as:
Data were assumed to be conditionally distributed as:
where y is the n = 359 or n = 285 observations of the microbiome or CH4 emissions trait, b is the vector of fixed effects including a combination of breed, diet, and experiment effect, g is the random host genomic effect, e is the residual of the model, and X and Z are known incidence matrices for fixed and random effects. Host genomic effects were normally distributed as:
Residuals were independently normally distributed as:
in which \({\sigma }_{g}^{2}\) and \({\sigma }_{e}^{2}\) are the host-genomic and residual variances, I is an identity matrix of the same order as the number of data, and GRM is the host-genomic relationship matrix between the individuals defined as157:
where W contains genotypes adjusted for allele frequency, and pn is the allele frequency for marker n in the whole genotyped population. Host genomic and residual effects were assumed to be uncorrelated between them. Host genomic (rgCH4) and residual correlations among CH4 emissions and log-transformed abundances of microbial genera, RUGs and microbial genes were estimated by fitting 2473 GBLUP bivariate animal models including the same effects as Eq. (3). Host genomic effects were distributed as:
and residuals as:
where G0 and R0 are the 2 × 2 host genomic and residual (co)variance matrices between CH4 emissions and each microbial genus, RUG, or microbial gene, I is an identity matrix of the same order as the number of individuals with data. Bayesian statistics were used158, assuming priors for all unknowns as implemented in THRGIBBSF90 program159. Results were based on Markov chain Monte Carlo chains consisting of 1,000,000 iterations, with a burn-in period of 200,000, and to reduce autocorrelations only 1 of every 100 samples was saved for inferences. In all analyses, convergence was tested using the POSTGIBBSF90159 program by calculating the Z criterion of Geweke (varying between −0.05 and 0.05 in univariate and −0.09 and 0.1 in bivariate models). Monte Carlo sampling errors were computed using time-series procedures and checked to be at least 10 times lower than the standard deviation of the marginal posterior distribution. As h2 estimates, we used the median of its marginal posterior distribution of CH4, each microbial genus, RUG, or microbial gene, and the highest posterior density interval at 95% probability (HPD95%). We considered microbial abundances with h2 estimates <0.20 being lowly heritable, 0.20 < h2 < 0.40 being moderately heritable and h2 estimates >0.40 being highly heritable. To test the significance of host genomic effects we analysed the fitness of the full univariate genomic model vs. the univariate model without host genomic effect by comparing the deviance information criterion (DIC)160 between models and computed the BF using an approximation of the marginal likelihood probability161. BF was corrected for multi-testing by assuming prior odds ratios equal to 1/number of hypothesis tests performed (n = 2473). We assumed evidence of a host genomic effect on the microbial trait when the DIC of the full model was at least 20 points lower than the DIC in the reduced model, and corrected BF was >3162. To study the homogeneity of genomic variances for CH4 emissions and microbial traits within each breed and within each diet, we computed the marginal posterior distribution of the genomic variance for the most relevant microbial traits in each of the 4 different breeds and in each of the 2 different diets, following the same steps as described for the partition of the variance in Sorensen et al.126,127. As an estimate for the host genomic correlations, we used the median of its marginal posterior distribution and the HPD95%. To investigate the confidence level of rgCH4, we estimated the posterior probability of rgCH4 being >0 when the median of the correlation was positive or <0 when the median was negative (P0). We only considered significant those rgCH4 estimates with (P0) ≥ 0.95. Additionally, univariate analyses were run using the frequentist approach using AIREMLF90159 and similar results were obtained.
To predict the impact of indirect selection for reduced CH4 emissions using microbial genera/genes significantly (P0 ≥ 0.95) host-genomically correlated with CH4 emissions, we estimated the marginal posterior distribution of the correlated response in CH4 emissions after host genomic selection for each of these microbial genera/genes, considering only the own performance of each individual163:
where RCH4j presents the selection response in CH4 emissions after selection for the abundance of each microbial genus/gene j, i is the intensity of selection considered to be 1.159 (equivalent to 30% of our cattle population being selected based on the selection criterion), hj is the marginal posterior distribution of the square root of the h2 estimate of the microbial genus/gene from univariate analyses, rgCH4j is the marginal posterior distribution of the host-genomic correlation between CH4 emissions and microbial genus/gene j from bivariate models, and σgCH4 is the squared root of the genomic variance of CH4 emissions. The median, standard deviation, and the probability (P0) of the correlated response to selection to be higher (lower) than 0 when the correlated response was positive (negative) were computed.
Co-abundance network analysis of host genomic effects on the rumen microbiome
To study the correlation structure among host genomic effects of the log-transformed abundances of 1107 microbial genera, 225 RUGs, and 1141 microbial genes, we built a co-abundance network analysis using deregressed host genomic effects (dGEBVs) for all microbial traits in the 359 samples. Deregressed host genomic effects were calculated from previously described univariate GBLUP models by using ACCF90 and DEPROOF90 programs159. Co-abundance network (Graphia software164) connected or edged microbial traits (nodes) based on a Pearson correlation >0.70 among their dGEBVs. The complexity of the graph was reduced by discarding nodes with a minimum number of incident edges (referred to as node degree) of 2, i.e. only those microbial traits Pearson-correlated (>0.70) with at least other 2 microbial traits were kept. The total number of microbial genera, RUGs, and microbial genes included in the network was 2129 out of the 2473 tested. The number of edges of each node was reduced by ranking the edges based on k-nearest neighbour algorithm and retaining only 80% of them. The software applies Markov Clustering algorithm by a flow simulation model165 to find discrete groups of nodes (clusters) based on their position within the overall topology of the graph. The granularity of the clusters, i.e. the minimum number of nodes that a cluster has to contain, was set to 2 nodes. The network showed 106 clusters, but only those 12 clusters including ≥3 methanogenic archaea genera, RUGs and microbial genes involved in CH4 metabolism pathway according to KEGG146 database or microbial genera/RUGs/genes host-genomically correlated with CH4 emissions (P0 ≥ 0.95) were studied in depth.
Enrichment analysis of microbial gene abundances in RUGs
To identify which of the 225 RUGs were carrying the microbial genes (KO) demonstrating a rgCH4 with a confidence level P0 ≥ 0.95, an enrichment analysis was performed by counting the number of unique proteins clustered in the 115 microbial genes mapped in each of the 225 RUGs.
Identification of most informative microbial traits to predict CH4 emission host genomic effects and maximize response to selection
Only microbial variables present in the 359 animals, showing a RA ≥ 0.01%, with significant h2 (P ≤ 2.02 × 10−5), and host-genomically correlated with CH4 emissions (P0 ≥ 0.95) were considered for breeding purposes. Four microbial genera and 36 microbial genes met these conditions. Due to computation reasons, only 30 microbial gene abundances were carried forward for downstream analysis. To use microbial gene information to select hosts emitting less CH4, the estimation between their host genomic and residual (co)variance matrices was required. Host-genomic and residual (co)variances among the 30 selected microbial gene abundances were estimated using 435 bivariate analyses. Bivariate analyses fitted the same model as previously described for estimation of rgCH4 with the same assumptions (Eqs. (8) and (9)). Results were based on Markov chain Monte Carlo chains consisting of 1,000,000 iterations, with a burn-in period of 200,000, and only 1 of every 100 samples was saved for inferences. Convergence was tested with POSTGIBBSF90 program by checking Z criterion of Geweke to be between −0.12 and 0.15. Monte Carlo sampling errors were computed using time-series procedures and checked to be at least 10 times lower than the standard deviation of the posterior marginal distribution158. The 31 × 31 host-genomic and residual variance–covariance matrices, including CH4 emissions and the 30 microbial genes were built based on medians of the estimated variance components from the bivariate analyses and mean across all previous bivariate models for host genomic and residual variances of CH4 emissions. Both matrices needed bending to be positive definite (tolerance for minimum eigenvalues = 0.001). The difference between original and bent matrices was never higher than the posterior standard error of the corresponding parameters.
Estimation of the selection response of CH4 emissions based on different sources of information
We analysed three different scenarios to predict host-genomic effects of CH4 emissions: (1) by using measured CH4 emissions only, (2) by using the 30 microbial gene abundances only, and (3) by using a combination of both, measured CH4 emissions and the 30 microbial gene abundances. The three scenarios were computed with data from 285 animals with CH4 emissions and metagenomics information. All scenarios were calculated by GBLUP analysis assuming as fixed variance components the previously estimated 31 × 31 host genomic and residual variance-covariance matrices of the traits after bending. Scenario (1) was performed using a univariate GBLUP analysis including only measured CH4 emissions; scenario (2) was computed by fitting a multivariate GBLUP model including the 30 microbial gene abundances host-genomically correlated to CH4 emissions (using measured CH4 emissions as missing value166); and scenario (3) considered besides the abundance of the 30 microbial genes, the measured CH4 emission values. In all cases, models included the same effects as in Eq. (3). Host genomic values estimates for CH4 emissions were based on Markov chain Monte Carlo chains consisting of 100,000 iterations, with a burn-in period of 20,000, and to reduce autocorrelation only 1 of every 100 samples was saved for inferences. Response to selection was estimated as the marginal posterior distributions of the difference between the mean of CH4 emissions host genomic values of all animals with data and the mean of selected animals when alternatively, 1, 5, 10, 20, 30, 40, and 50% of our population were selected. The mean accuracy of the CH4 emissions genomic values in each scenario was estimated as the average of the individual accuracies:
where sdi is the standard deviation of the posterior marginal distribution of the host genomic value for animal i and gRMii is the GRM diagonal element for animal i.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Metagenomic sequence reads for all rumen samples are available under European Nucleotide Archive (ENA) under accession projects PRJEB31266, PRJEB21624, and PRJEB10338. The genotypes of the host animals are readily available from the authors.
Code availability
Metagenomic data processing was carried out using Kraken (https://ccb.jhu.edu/software/kraken/) for taxonomic annotation and Novoalign (http://www.novocraft.com/support/download/ available under license) for functional annotation. SNP data filtering was performed PLINK (https://www.cog-genomics.org/plink2). Host genomic analysis were carried out using the RENUMF90, THRGIBBSF90, POSTGIBBSF90, ACCF90, and DEPROOF90 software, which have free access in http://nce.ads.uga.edu/wiki/doku.php?id=application_programs, except for ACCF90 and DEPROOF90 available only under research agreement. Network analysis was carried out by free access to Graphia software whose code source can be found at https://graphia.app/download.html.
References
OECD/FAO. OECD-FAO Agricultural Outlook 2020-2029 (OECD Publishing/Food and Agriculture Organization of the United Nations, 2020).
Vollset, S. E. et al. Fertility, mortality, migration, and population scenarios for 195 countries and territories from 2017 to 2100: a forecasting analysis for the Global Burden of Disease Study. Lancet 396, 1285–1306 (2020).
Gerber, P. J. et al. Tackling Climate Change Through Livestock – A Global Assessment of Emissions and Mitigation Opportunities (Food and Agriculture Organization of the United Nations (FAO), 2013).
Myhre, G. et al. Anthropogenic and Natural Radiative Forcing: Supplementary Material. Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change (IPCC, 2013).
Johnson, K. A. & Johnson, D. E. Methane emissions from cattle. J. Anim. Sci. 73, 2483–2492 (1995).
Manafiazar, G. et al. Methane and carbon dioxide emissions and grazed forage intake from pregnant beef heifers previously classified for residual feed intake under drylot conditions. Can. J. Anim. Sci. 101, 71–84 (2021).
Roehe, R. et al. Bovine host genetic variation influences rumen microbial methane production with best selection criterion for low methane emitting and efficiently feed converting hosts based on metagenomic gene abundance. PLoS Genet. 12, 1–20 (2016).
Stewart, R. D. et al. Assembly of 913 microbial genomes from metagenomic sequencing of the cow rumen. Nat. Commun. 9, 1–11 (2018).
Wallace, R. J. et al. Archaeal abundance in post-mortem ruminal digesta may help predict methane emissions from beef cattle. Sci. Rep. 4, 5892 (2015).
Martínez-Álvaro, M. et al. Identification of complex rumen microbiome interaction within diverse functional niches as mechanisms affecting the variation of methane emissions in bovine. Front. Microbiol. 11, 1–13 (2020).
Barrett, K., Jensen, K., Meyer, A. S., Frisvad, J. C. & Lange, L. Fungal secretome profile categorization of CAZymes by function and family corresponds to fungal phylogeny and taxonomy: example Aspergillus and Penicillium. Sci. Rep. 10, 1–12 (2020).
Tapio, I., Snelling, T. J., Strozzi, F. & Wallace, R. J. The ruminal microbiome associated with methane emissions from ruminant livestock. J. Anim. Sci. Biotechnol. 8, 7 (2017).
Cottle, D. J., Nolan, J. V. & Wiedemann, S. G. Ruminant enteric methane mitigation: a review. Anim. Prod. Sci. 51, 491–514 (2011).
Martin, C., Morgavi, D. P. & Doreau, M. Methane mitigation in ruminants: from microbe to the farm scale. Animal 4, 351–365 (2010).
Roque, B. M. et al. Red seaweed (Asparagopsis taxiformis) supplementation reduces enteric methane by over 80 percent in beef steers. PLoS ONE 16, e0247820 (2021).
Dijkstra, J., Bannink, A., France, J., Kebreab, E. & van Gastelen, S. Short communication: Antimethanogenic effects of 3-nitrooxypropanol depend on supplementation dose, dietary fiber content, and cattle type. J. Dairy Sci. 101, 9041–9047 (2018).
Hristov, A. N. et al. Special topics-Mitigation of methane and nitrous oxide emissions from animal operations: I. A review of enteric methane mitigation options. J. Anim. Sci. 91, 5045–5069 (2013).
Difford, G. F. et al. Host genetics and the rumen microbiome jointly associate with methane emissions in dairy cows. PLoS Genet. 14, 1–22 (2018).
Zhang, Q. et al. Bayesian modeling reveals host genetics associated with rumen microbiota jointly influence methane emission in dairy cows. ISME J. 14, 2019–2033 (2020).
Li, F. et al. Host genetics influence the rumen microbiota and heritable rumen microbial features associate with feed efficiency in cattle. Microbiome 7, 1–17 (2019).
Wallace, J. R. et al. A heritable subset of the core rumen microbiome dictates dairy cow productivity and emissions. Sci. Adv. 5, eaav8391 (2019).
Saborío‐Montero, A. et al. Structural equation models to disentangle the biological relationship between microbiota and complex traits: Methane production in dairy cattle as a case of study. J. Anim. Breed. Genet. 137, 36–48 (2020).
Sasson, G. et al. Heritable bovine rumen bacteria are phylogenetically related and correlated with the cow’s capacity to harvest energy from its feed. MBio 8, 1–12 (2017).
Weimer, P. J., Stevenson, D. M., Mantovani, H. C. & Man, S. L. C. Host specificity of the ruminal bacterial community in the dairy cow following near-total exchange of ruminal contents. J. Dairy Sci. 93, 5902–5912 (2010).
Abbas, W. et al. Influence of host genetics in shaping the rumen bacterial community in beef cattle. Sci. Rep. 10, 15101 (2020).
Bergamaschi, M. et al. Heritability and genome-wide association of swine gut microbiome features with growth and fatness parameters. Sci. Rep. 10, 1–12 (2020).
Chen, C. et al. Contribution of host genetics to the variation of microbial composition of cecum lumen and feces in pigs. Front. Microbiol. 9, 1–13 (2018).
Poole, A. C. et al. Human salivary amylase gene copy number impacts oral and gut microbiomes. Cell Host Microbe 25, 553–564.e7 (2019).
Kurilshikov, A. et al. Large-scale association analyses identify host factors influencing human gut microbiome composition. Nat. Genet. 53, 156–165 (2021).
Turpin, W. et al. Association of host genome with intestinal microbial composition in a large healthy cohort. Nat. Genet. 48, 1413–1417 (2016).
Qin, Y. et al. Combined effects of host genetics and diet on human gut microbiota and incident disease in a single population cohort. Nat. Genet. 54, 134–142 (2022).
Hughes, D. A. et al. Genome-wide associations of human gut microbiome variation and implications for causal inference analyses. Nat. Microbiol. 5, 1079–1087 (2020).
Goodrich, J. K. et al. Human genetics shape the gut microbiome. Cell 159, 789–799 (2014).
Stewart, R. D. et al. Compendium of 4,941 rumen metagenome-assembled genomes for rumen microbiome biology and enzyme discovery. Nat. Biotechnol. 37, 953–961 (2019).
Perez-Enciso, M. Opportunities and limits of combining microbiome and genome data for complex trait prediction. Genet. Sel. Evol. 53, 65 (2021).
Weishaar, R., Wellmann, R., Camarinha-Silva, A., Rodehutscord, M. & Bennewitz, J. Selecting the hologenome to breed for an improved feed efficiency in pigs—a novel selection index. J. Anim. Breed. Genet. 137, 14–22 (2020).
Auffret, M. D. et al. Identification, comparison, and validation of robust rumen microbial biomarkers for methane emissions using diverse Bos Taurus breeds and basal diets. Front. Microbiol. 8, 1–15 (2018).
Duthie, C. A. et al. The impact of divergent breed types and diets on methane emissions, rumen characteristics and performance of finishing beef cattle. Animal 11, 1762–1771 (2017).
Rooke, J. A. et al. Hydrogen and methane emissions from beef cattle and their rumen microbial community vary with diet, time after feeding and genotype. Br. J. Nutr. 112, 398–407 (2014).
Seshadri, R. et al. Cultivation and sequencing of rumen microbiome members from the Hungate1000 Collection. Nat. Biotechnol. 36, 359–367 (2018).
Pruitt, K. D., Tatusova, T. & Maglott, D. R. NCBI Reference Sequence (RefSeq): a cuarted non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 33, D501–D504 (2005).
Joblin, K. N. Ruminal acetogens and their potential to lower ruminant methane emissions. Aust. J. Agric. Res. 50, 629–650 (1999).
McAllister, T. A. & Newbold, C. J. Redirecting rumen fermentation to reduce methanogenesis. Aust. J. Exp. Agric. 48, 7–13 (2008).
Hegarty, R. S. Mechanisms for competitively reducing ruminal methanogenesis. Aust. J. Agric. Res. 50, 629–650 (1999).
Greening, C. et al. Diverse hydrogen production and consumption pathways influence methane production in ruminants. ISME J. 13, 2617–2632 (2019).
Hajarnis, S. R. & Ranade, D. R. Inhibition of methanogens by n- and iso-volatile fatty acids. World J. Microbiol. Biotechnol. 10, 350–351 (1994).
Henderson, C. The effects of fatty acids on pure cultures of rumen bacteria. J. Agric. Sci. 81, 107–112 (1973).
Paulo, L. M., Ramiro-Garcia, J., van Mourik, S., Stams, A. J. M. & Sousa, D. Z. Effect of nickel and cobalt on methanogenic enrichment cultures and role of biogenic sulfide in metal toxicity attenuation. Front. Microbiol. 8, 1–12 (2017).
Zhou, M., Chen, Y. & Guan, L. L. in Rumen Microbiology: From Evolution to Revolution (eds. Puniya, A. K., Singh, R. & Kamra, D. N.) 79–95 (Springer, 2015).
van Wolferen, M., Orell, A. & Albers, S. V. Archaeal biofilm formation. Nat. Rev. Microbiol. 16, 699–713 (2018).
Chen, H. & Fink, G. R. Feedback control of morphogenesis in fungi by aromatic alcohols. Genes Dev. 20, 1150–1161 (2006).
Thauer, R. K. Anaerobic oxidation of methane with sulfate: On the reversibility of the reactions that are catalyzed by enzymes also involved in methanogenesis from CO2. Curr. Opin. Microbiol. 14, 292–299 (2011).
McInerney, M. J., Sieber, J. R. & Gunsalus, R. P. Syntrophy in anaerobic global carbon cycles. Curr. Opin. Biotechnol. 20, 623–632 (2009).
McInerney, M. J. et al. Physiology, ecology, phylogeny, and genomics of microorganisms capable of syntrophic metabolism. Ann. NY Acad. Sci. 1125, 58–72 (2008).
Evans, P. N. et al. An evolving view of methane metabolism in the Archaea. Nat. Rev. Microbiol. 17, 219–232 (2019).
Nomura, M., Gourse, R. & Baughman, G. Regulation of the synthesis of ribosomes and ribosomal components. Ann. Rev. Biochem. 53, 75–117 (1984).
Garnsworthy, P. C. et al. Comparison of methods to measure methane for use in genetic evaluation of dairy cattle. Animals 9, 1–12 (2019).
Manzanilla-Pech, C. I. V. et al. Genomewide association study of methane emissions in angus beef cattle with validation in dairy cattle. J. Anim. Sci. 94, 4151–4166 (2016).
Hayes, B. J. et al. Genomic heritabilities and genomic estimated breeding values for methane traits in Angus cattle. J. Anim. Sci. 94, 902–908 (2016).
Donoghue, K. A., Bird-Gardiner, T., Arthur, P. F., Herd, R. M. & Hegarty, R. F. Genetic and phenotypic variance and covariance components for methane emission and postweaning traits in Angus cattle. J. Anim. Sci. 94, 1438–1445 (2016).
Cesarani, A. et al. Beef trait genetic parameters based on old and recent data and its implications for genomic predictions in Italian Simmental cattle. J. Anim. Sci. 98, 1–8 (2020).
Gengler, N., Wiggans, G. R. & Gillon, A. Adjustment for heterogeneous covariance due to herd milk yield by transformation of test-day random regressions. J. Dairy Sci. 88, 2981–2990 (2005).
Gunsalus, R. P. et al. Complete genome sequence of Methanospirillum hungatei type strain JF1. Stand. Genom. Sci. 11, 1–JF10 (2016).
Henderson, G. et al. Rumen microbial community composition varies with diet and host, but a core microbiome is found across a wide geographical range. Sci. Rep. 5, 14567 (2015).
Snelling, T. J. et al. Temporal stability of the rumen microbiota in beef cattle, and response to diet and supplements. Anim. Microbiome 1, 1–14 (2019).
Morgavi, D. P., Forano, E., Martin, C. & Newbold, C. J. Microbial ecosystem and methanogenesis in ruminants. Animal 4, 1024–1036 (2010).
Uddin, M. J. K. Dynamics of microbial protein synthesis in the rumen - A Review. Ann. Vet. Anim. Sci. 2, 116–131 (2015).
Lovendahll, P. et al. Review: Selecting for improved feed efficiency and reduced methane emissions in dairy cattle. Animal 12, S336–S349 (2018).
Tobin, C. Removal and Replacement of Ribosomal Proteins. PhD thesis, Uppsala Univ. (2011).
Liu, X. et al. A genome-wide association study for the gut microbiome in Chinese adults illuminates on complex diseases. Cell Discov. 7, 9 (2019).
Vanwonterghem, I. et al. Methylotrophic methanogenesis discovered in the archaeal phylum Verstraetearchaeota. Nat. Microbiol. 1, 16170 (2016).
Graham, D. E., Xu, H. & White, R. H. Identification of the 7,8-didemethyl-8-hydroxy-5-deazariboflavin synthase required for coenzyme F420 biosynthesis. Arch. Microbiol. 180, 455–464 (2003).
Grochowski, L. L. & White, R. H. Biosynthesis of the methanogenic coenzymes. Compr. Nat. Prod. II Chem. Biol. 7, 711–748 (2010).
Peng, X. et al. Genomic and functional analyses of fungal and bacterial consortia that enable lignocellulose breakdown in goat gut microbiomes. Nat. Microbiol. https://doi.org/10.1038/s41564-020-00861-0 (2021).
Miyazaki, J., Kobashi, N., Nishiyama, M. & Yamane, H. Functional and evolutionary relationship between arginine biosynthesis and prokaryotic lysine biosynthesis through α-aminoadipate. J. Bacteriol. 183, 5067–5073 (2001).
Andries, J. I., Buysse, F. X., De Brabander, D. L. & Cottyn, B. G. Isoacids in ruminant nutrition: Their role in ruminal and intermediary metabolism and possible influences on performances - a review. Anim. Feed Sci. Technol. 18, 169–180 (1987).
Drevland, R. M., Waheed, A. & Graham, D. E. Enzymology and evolution of the pyruvate pathway to 2-oxobutyrate in Methanocaldococcus jannaschii. J. Bacteriol. 189, 4391–4400 (2007).
Lee, J. H. & Lee, J. Indole as an intercellular signal in microbial communities. FEMS Microbiol. Rev. 34, 426–444 (2010).
Roager, H. M. & Licht, T. R. Microbial tryptophan catabolites in health and disease. Nat. Commun. 9, 1–10 (2018).
Drevland, R. M., Jia, Y., Palmer, D. R. J. & Graham, D. E. Methanogen homoaconitase catalyzes both hydrolyase reactions in coenzyme B biosynthesis. J. Biol. Chem. 283, 28888–28896 (2008).
Neill, A. R., Grime, D. W. & Dawson, R. M. Conversion of choline methyl groups through trimethylamine into methane in the rumen. Biochem. J. 170, 529–535 (1978).
Janssen, P. H. & Kirs, M. Structure of the archaeal community of the rumen. Appl. Environ. Microbiol. 74, 3619–3625 (2008).
Liu, Y. & Whitman, W. B. Metabolic, phylogenetic, and ecological diversity of the methanogenic archaea. Ann. NY Acad. Sci. 1125, 171–189 (2008).
Chamberlain, D. G. & Thomas, P. C. The effect of supplemental methionine and inorganic sulphate on the ruminal digestion of grass silage in sheep. J. Sci. Food Agric. 34, 440–446 (1983).
Kamke, J. et al. Rumen metagenome and metatranscriptome analyses of low methane yield sheep reveals a Sharpea-enriched microbiome characterised by lactic acid formation and utilisation. Microbiome 4, 1–16 (2016).
Yanibada, B. et al. Inhibition of enteric methanogenesis in dairy cows induces changes in plasma metabolome highlighting metabolic shifts and potential markers of emission. Sci. Rep. 10, 1–14 (2020).
Goopy, J. P. et al. Low-methane yield sheep have smaller rumens and shorter rumen retention time. Br. J. Nutr. 111, 578–585 (2014).
Stewart, C. S., Flint, H. J. & Bryant, M. P. in The Rumen Microbial Ecosystem (eds. Hobson, P. N. & Stewart, C. S.) 10–72 (Blackie Academic and Professional, 1997).
Kittelmann, S. et al. Two different bacterial community types are linked with the low-methane emission trait in sheep. PLoS ONE 9, 1–9 (2014).
Strobel, H. J. & Russell, J. B. Effect of pH and energy spilling on bacterial protein synthesis by carbohydrate-limited cultures of mixed rumen bacteria. J. Dairy Sci. 69, 2941–2947 (1986).
Herrmann, K. M. & Weaver, L. M. The shikimate pathway. Annu. Rev. Plant Biol. 50, 473–503 (1999).
Hall, M. B. & Herejk, C. Differences in yields of microbial crude protein from in vitro fermentation of carbohydrates. J. Dairy Sci. 84, 2486–2493 (2001).
Nollet, L. & Verstraete, W. Gastro-enteric methane versus sulphate and volatile fatty acid production. Environ. Monit. Assess. 42, 113–131 (1996).
Demeyer, D., De Graave, K., Durand, M. & Stevani, J. Acetate: a hydrogen sink in hindgut fermentation as opposed to rumen fermentation. Acta Vet. Scabd Suppl. 86, 68–75 (1989).
Lopez, S., Mcintosh, F. M., Wallace, R. J. & Newbold, C. J. Effect of adding acetogenic bacteria on methane production by mixed rumen microorganisms. Anim. Feed Sci. Technol. 78, 1–9 (1999).
Baldwin, R. L., Wood, W. A. & Emery, R. S. Conversion of lactate-c’4 to propionate by the rumen microflora. J. Bacteriol. 83, 907–913 (1961).
Janssen, P. H. Influence of hydrogen on rumen methane formation and fermentation balances through microbial growth kinetics and fermentation thermodynamics. Anim. Feed Sci. Technol. 160, 1–22 (2010).
Owens, F. N., Secrist, D. S., Hill, W. J. & Gill, D. R. Acidosis in cattle: a review. J. Anim. Sci. 76, 275–286 (1998).
Doyle, N. et al. Use of lactic acid bacteria to reduce methane production in ruminants, a critical review. Front. Microbiol. 10, 2207 (2019).
Kruger Ben Shabat, S. et al. Specific microbiome-dependent mechanisms underlie the energy harvest efficiency of ruminants. ISME J. 10, 2958–2972 (2016).
Ugulava, N. B., Sacanell, C. J. & Jarrett, J. T. Spectroscopic changes during a single turnover of biotin synthase: destruction of a [2Fe-2S] cluster accompanies sulfur insertion. Biochemistry 40, 8352–8358 (2001).
Howell, D. M., Harich, K., Xu, H. & White, R. H. α-Keto acid chain elongation reactions involved in the biosynthesis of coenzyme B (7-mercaptoheptanoyl threonine phosphate) in methanogenic archaea. Biochemistry 37, 10108–10117 (1998).
Widdel, F. Growth of methanogenic bacteria in pure culture with 2-propanol and other alcohols as hydrogen donors. Appl. Environ. Microbiol. 51, 1056–1062 (1986).
Moore, S. J. et al. Elucidation of the biosynthesis of the methane catalyst coenzyme F430. Nature 543, 78–82 (2017).
Bulen, W. A. & LeComte, J. R. The nitrogenase system from Azotobacter: two-enzyme requirement for N2 reduction, ATP-dependent H2 evolution, and ATP hydrolysis. Proc. Natl Acad. Sci. USA 56, 979–986 (1966).
Wang, M., Wang, H., Zheng, H., Dewhurst, R. J. & Roehe, R. A heat diffusion multilayer network approach for the identification of functional biomarkers in rumen methane emissions. Methods https://doi.org/10.1016/j.ymeth.2020.09.014 (2020).
Jenkins, T. C., Abughazaleh, A. A., Freeman, S. & Thies, E. J. The production of 10-hydroxystearic and 10-ketostearic acids is an alternative route of oleic acid transformation by the ruminal microbiota in cattle. J. Nutr. 136, 926–931 (2006).
Abe, A. & Sugiyama, K. Growth inhibition and apoptosis induction of human melanoma cells by omega-hydroxy fatty acids. Anticancer. Drugs 16, 543–549 (2005).
Martin, A. & Daniel, J. The ABC transporter Rv1272c of Mycobacterium tuberculosis enhances the import of long-chain fatty acids in Escherichia coli. Biochem. Biophys. Res. Commun. 496, 667–672 (2018).
Jenkins, B., West, J. A. & Koulman, A. A review of odd-chain fatty acid metabolism and the role of pentadecanoic acid (C15:0) and heptadecanoic acid (C17:0) in health and disease. Molecules 20, 2425–2444 (2015).
Jenkins, T. C. Lipid metabolism in the rumen. J. Dairy Sci. 76, 3851–3863 (1993).
Leng, R. A. Interactions between microbial consortia in biofilms: a paradigm shift in rumen microbial ecology and enteric methane mitigation. Anim. Prod. Sci. 54, 519–543 (2014).
Won, M. Y., Oyama, L. B., Courtney, S. J., Creevey, C. J. & Huws, S. A. Can rumen bacteria communicate to each other? Microbiome 8, 1–8 (2020).
Patra, A., Park, T., Kim, M. & Yu, Z. Rumen methanogens and mitigation of methane emission by anti-methanogenic compounds and substances. J. Anim. Sci. Biotechnol. 8, 1–18 (2017).
Wȩgrzyn, A., Taylor, K. & Wȩgrzyn, G. The cbpA chaperone gene function compensates for dnaJ in λ plasmid replication during amino acid starvation of Escherichia coli. J. Bacteriol. 178, 5847–5849 (1996).
Wahlström, A., Sayin, S. I., Marschall, H. U. & Bäckhed, F. Intestinal crosstalk between bile acids and microbiota and its impact on host metabolism. Cell Metab. 24, 41–50 (2016).
Ramírez-Pérez, O., Cruz-Ramón, V., Chinchilla-López, P. & Méndez-Sánchez, N. The role of the gut microbiota in bile acid metabolism. Ann. Hepatol. 16, S21–S26 (2017).
Immig, I. The effect of porcine bile acids on methane production by rumen contents in vitro. Arch. Anim. Nutr. 51, 21–26 (1998).
Hooper, L. V. & Gordon, J. I. Glycans as legislators of host-microbial interactions: spanning the spectrum from symbiosis to pathogenicity. Glycobiology 11, 1–10 (2001).
Hoorens, P. R. et al. Genome wide analysis of the bovine mucin genes and their gastrointestinal transcription profile. BMC Genomics 12, 140 (2011).
Aschenbach, J. R., Penner, G. B., Stumpff, F. & Gäbel, G. Ruminant nutrition symposium: Role of fermentation acid absorption in the regulation of ruminal pH. J. Anim. Sci. 89, 1092–1107 (2011).
Lee, M., Jeong, S., Seo, J. & Seo, S. Changes in the ruminal fermentation and bacterial community structure by a sudden change to a high-concentrate diet in Korean domestic ruminants. Asian-Australas. J. Anim. Sci. 32, 92–102 (2019).
Van Kessel, J. A. S. & Russell, J. B. The effect of pH on ruminal methanogenesis. FEMS Microbiol. Ecol. 20, 205–210 (1996).
Lecompte, O., Ripp, R., Thierry, J. C., Moras, D. & Poch, O. Comparative analysis of ribosomal proteins in complete genomes: an example of reductive evolution at the domain scale. Nucleic Acids Res. 30, 5382–5390 (2002).
Smith, T. F., Lee, J. C., Gutell, R. R. & Hartman, H. The origin and evolution of the ribosome. Biol. Direct 3, 1–13 (2008).
Sorensen, D., Fernando, R. & Gianola, D. Inferring the trajectory of genetic variance in the course of artificial selection. Genet. Res. 77, 83–94 (2001).
Lara, L. A. d. C., Pocrnic, I., Gaynor, R. C. & Gorjanc, G. Temporal and genomic analysis of additive genetic variance in breeding programmes. Heredity 128, 21–32 (2020).
Rowe, S. J. et al. Selection for divergent methane yield in New Zealand sheep - a ten year perspective. Proc. Assoc. Adv. Anim. Breed. Genet. 306–309 (2019).
Pszczola, M., Strabel, T., Mucha, S. & Sell-Kubiak, E. Genome-wide association identifies methane production level relation to genetic control of digestive tract development in dairy cows. Sci. Rep. 8, 1–11 (2018).
Maekawa, M., Beauchemin, K. A. & Christensen, D. A. Effect of concentrate level and feeding management on chewing activities, saliva production, and ruminal pH of lactating dairy cows. J. Dairy Sci. 85, 1165–1175 (2002).
Danielsson, R. Methane Production in Dairy Cows Impact of Feed and Rumen Microbiota (Acta Universitatis Agriculturae Sueciae, 2016).
Poehlein, A., Schneider, D., Soh, M., Daniel, R. & Seedorf, H. Comparative genomic analysis of members of the genera methanosphaera and methanobrevibacter reveals distinct clades with specific potential metabolic functions. Archaea 2018, 1–9 (2018).
Ricard, G. et al. Horizontal gene transfer from bacteria to rumen ciliates indicates adaptation to their anaerobic, carbohydrates-rich environment. BMC Genomics 7, 1–13 (2006).
Klieve, A. V. et al. Naturally occurring DNA transfer system associated with membrane vesicles in cellulolytic Ruminococcus spp. of ruminal origin. Appl. Environ. Microbiol. 71, 4248–4253 (2005).
Hess, M. K. et al. A restriction enzyme reduced representation sequencing approach for low-cost, high-throughput metagenome profiling. PLoS ONE 15, 1–18 (2020).
Duthie, C.-A. et al. Impact of adding nitrate or increasing the lipid content of two contrasting diets on blood methaemoglobin and performance of two breeds of finishing beef steers. Animal 10, 786–795 (2016).
Duthie, C. A. et al. The effect of dietary addition of nitrate or increase in lipid concentrations, alone or in combination, on performance and methane emissions of beef cattle. Animal 12, 280–287 (2018).
Somarriba, M. et al. The effects of a composite chronic stress treatment on fear responses and attention bias in beef cattle. in ISAE 2019. Proc. 53rd Congr. ISAE 53, 333 (2019).
Purcell, S. et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Matukumalli, L. K. et al. Development and characterization of a high density SNP genotyping assay for cattle. PLoS ONE 4, e5350 (2009).
Yu, Z. & Morrison, M. Improved extraction of PCR-quality community DNA from digesta and fecal samples. Biotechniques 36, 808–812 (2004).
Wood, D. E. & Salzberg, S. L. Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 15, 1–12 (2014).
Palarea-Albaladejo, J. & Martín-Fernández, J. A. ZCompositions - R package for multivariate imputation of left-censored data under a compositional approach. Chemom. Intell. Lab. Syst. 143, 85–96 (2015).
Martín-Fernández, J. A., Hron, K., Templ, M., Filzmoser, P. & Palarea-Albaladejo, J. Bayesian-multiplicative treatment of count zeros in compositional data sets. Stat. Model. 15, 134–158 (2015).
Wallace, R. J. et al. The rumen microbial metagenome associated with high methane production in cattle. BMC Genomics 16, 839 (2015).
Kanehisa, M. & Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 28, 27–30 (2000).
Gloor, G. B., Macklaim, J. M., Pawlowsky-Glahn, V. & Egozcue, J. J. Microbiome datasets are compositional: and this is not optional. Front. Microbiol. 8, 2224 (2017).
Greenacre, M. Variable selection in compositional data analysis using pairwise logratios. Math. Geosci. 51, 649–682 (2018).
Greenacre, M. Compositional Data Analysis in Practise (CRC Press, 2019).
Aitchison, J. The statistical analysis of compositional data. J. R. Stat. Soc. Ser. B (Methodol.) 44, 139–177 (1982).
Greenacre, M., Martínez-Álvaro, M. & Blasco, A. Compositional data analysis of microbiome and any-omics datasets: a revalidation of the additive logratio transformation. Front. Microbiol. 12, 727398 (2021).
Greenacre, M. Compositional data analysis. Annu. Rev. Stat. Appl. 8, 271–299 (2021).
Zeng, H., Guo, C., Sun, D., Seddik, H. E. & Mao, S. The ruminal microbiome and metabolome alterations associated with diet-induced milk fat depression in dairy cows. Metabolites 9, 154 (2019).
Kang, S., Denman, S. & McSweeney, C. Draft genome sequence and annotation of Oribacterium sp. strain C9, isolated from a cattle rumen. Microbiol. Resour. Announc. 8, e01562–18 (2019).
Iwasawa, K. et al. Dysbiosis of the salivary microbiota in pediatric-onset primary sclerosing cholangitis and its potential as a biomarker. Sci. Rep. 8, 1–10 (2018).
Oksanen, J. et al. vegan: Community Ecology Package. R package version 2.5-7 (2020).
VanRaden, P. M. Efficient methods to compute genomic predictions. J. Dairy Sci. 91, 4414–4423 (2008).
Blasco, A. Bayesian Data Analysis for Animal Scientists: The Basics. https://doi.org/10.1007/978-3-319-54274-4 (2017).
Misztal, I. et al. Manual for BLUPF90 Family of Programs (Univ. Georgia Athens, 2018).
Spiegelhalter, D. J., Best, N. G., Carlin, B. P. & Van Der Linde, A. Bayesian measures of model complexity and fit. J. R. Stat. Soc. Ser. B Stat. Methodol. 64, 583–616 (2002).
Newton, M. A. & Raftery, Adrian E. Approximate Bayesian Inference with the weighted likelihood bootstrap. J. R. Stat. Soc. Ser. B Stat. Methodol. 58, 3–48 (1984).
van Doorn, J. et al. The JASP guidelines for conducting and reporting a Bayesian analysis. Psychon. Bull. Rev. https://doi.org/10.3758/s13423-020-01798-5 (2020).
Falconer, D. S. & Mackay, T. F. C. Introduction to Quantitative Genetics (Pearson, 1981).
Freeman, T. C. et al. Graphia: a platform for the graph-based visualisation and analysis of complex data. Preprint at bioRxiv https://doi.org/10.1101/2020.09.02.279349 (2020).
Freeman, T. C. et al. Construction, visualisation, and clustering of transcription networks from microarray expression data. PLoS Comput. Biol. 3, 2032–2042 (2007).
Schneeberger, M., Barwick, S. A., Crow, G. H. & Hammond, K. Economic indices using breeding values predicted by BLUP. J. Anim. Breed. Genet. 109, 180–187 (1992).
Acknowledgements
The authors thank Professor Ignacy Misztal and Dr. Shogo Tsuruta for making software available to us, Professor Agustín Blasco and Professor Chris Haley for their statistic advice, and Professor Michael Greenacre for his advice on compositional data analysis. We also thank Bin Zhao for his contribution to the identification and biological description of metagenomics data and Dr. Larissa Zetouni for her comments on the manuscript.
Author information
Authors and Affiliations
Contributions
M.M.-A., R.R., and M.W. conceived and designed the overall study, and M.M-A., M.W., and R.R. conceived, designed, and executed the bioinformatics analysis. M.D.A., C.-A.D., R.J.D., and M.A.C. provided essential insight into microbiology, rumen metabolism, nutrition, methane emissions and animal breeding. M.M.-A. and R.R. wrote the initial draft, and subsequently, all authors contributed intellectually to the interpretation and presentation of the results in the manuscript, which was edited and approved by all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Grum Gebreyesus and the other anonymous reviewer for their contribution to the peer review of this work. Primary handling editors: Anna Heintz-Buschart and Caitlin Karniski. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Martínez-Álvaro, M., Auffret, M.D., Duthie, CA. et al. Bovine host genome acts on rumen microbiome function linked to methane emissions. Commun Biol 5, 350 (2022). https://doi.org/10.1038/s42003-022-03293-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-022-03293-0
This article is cited by
-
Including microbiome information in a multi-trait genomic evaluation: a case study on longitudinal growth performance in beef cattle
Genetics Selection Evolution (2024)
-
Heritability and recursive influence of host genetics on the rumen microbiota drive body weight variance in male Hu sheep lambs
Microbiome (2023)
-
Combined effects of nitrate and medium-chain fatty acids on methane production, rumen fermentation, and rumen bacterial populations in vitro
Scientific Reports (2023)
-
Microbiome-driven breeding strategy potentially improves beef fatty acid profile benefiting human health and reduces methane emissions
Microbiome (2022)
-
Host genetic control on rumen microbiota and its impact on dairy traits in sheep
Genetics Selection Evolution (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
