
Abstract
Modern societies are experiencing an increasing trend of reduced sleep duration, with nocturnal sleeping time below the recommended ranges for health. Epidemiological and laboratory studies have demonstrated detrimental effects of sleep deprivation on health. Sleep exerts an immune-supportive function, promoting host defense against infection and inflammatory insults. Sleep deprivation has been associated with alterations of innate and adaptive immune parameters, leading to a chronic inflammatory state and an increased risk for infectious/inflammatory pathologies, including cardiometabolic, neoplastic, autoimmune and neurodegenerative diseases. Here, we review recent advancements on the immune responses to sleep deprivation as evidenced by experimental and epidemiological studies, the pathophysiology, and the role for the sleep deprivation-induced immune changes in increasing the risk for chronic diseases. Gaps in knowledge and methodological pitfalls still remain. Further understanding of the causal relationship between sleep deprivation and immune deregulation would help to identify individuals at risk for disease and to prevent adverse health outcomes.
Similar content being viewed by others

Introduction
Sleep is an active physiological process necessary for life and normally occupying one-third of our lives, playing a fundamental role for physical, mental, and emotional health1. Sleep patterns and need are influenced by a complex interplay between chronological age, maturation stage, genetic, behavioral, environmental, and social factors2,3,4,5,6. Adults should sleep a minimum of 7 h per night to promote optimal health7,8.
Besides medical problems including obstructive sleep apnea and insomnia, factors associated mostly with the modern 24/7 society, such as work and social demands, smartphone addiction, and poor diet9,10,11, contribute to cause the current phenomenon of chronic sleep deprivation, i.e., sleeping less than the recommended amount or, better to say, the intrinsic sleep need12.
Sleep deprivation may be categorized as acute or chronic. Acute sleep deprivation refers to no sleep or reduction in the usual total sleep time, usually lasting 1–2 days, with waking time extending beyond the typical 16–18 h. Chronic sleep deprivation is defined by the Third Edition of the International Classification of Sleep Disorders as a disorder characterized by excessive daytime sleepiness caused by routine sleeping less than the amount required for optimal functioning and health maintenance, almost every day for at least 3 months13.
Population studies reported a stably increasing prevalence of adults sleeping less than 6 h per night over a long period12,14,15, also affecting children and adolescents16,17. Sleep duration decline is present not only in high-income and developed countries18 but also in low-income or racial/ethnic minorities19, thus representing a worldwide problem.
In addition to fatigue, excessive daytime sleepiness, and impaired cognitive and safety-related performance, sleep deprivation is associated with an increased risk of adverse health outcomes and all-cause mortality20,21,22,23,24. Indeed, epidemiological and experimental data support the association of sleep deprivation with the risk of cardiovascular (CV) (hypertension and coronary artery disease) and metabolic (obesity, type 2 diabetes (T2DM)) diseases24,25,26,27. In the United States, sleep deprivation has been linked to 5 of the top 15 leading causes of death including cardio- and cerebrovascular diseases, accidents, T2DM, and hypertension28. Data also point to a role for sleep deprivation in the risk of stroke, cancer, and neurodegenerative diseases (NDDs)26,29,30. Sleep deprivation is also associated with psychopathological and psychiatric disorders, including negative mood and mood regulation, psychosis, anxiety, suicidal behavior, and the risk for depression31,32,33,34,35,36.
Both too short or too long sleep durations have been found to be associated with adverse health outcomes and all-cause mortality with an U-shaped relationship37,38,39. Although the relation of long sleep duration to adverse health outcomes may be confounded by poor health conditions occurring in older adults37, the causal association of sleep deprivation with negative health effects is substantiated by experimental evidence providing biological plausibility24,40,41.
Sleep profoundly affects endocrine, metabolic, and immune pathways, whose dysfunctions play a determinant role in the development and progression of chronic diseases42,43,44. Specifically, in many chronic diseases, a deregulated/exacerbated immune response shifts from repair/regulation towards unresolved inflammatory responses45.
Regular sleep is crucial for maintaining immune function integrity and favoring a homeostatic immune defense to microbial or inflammatory insults46,47. Sleep deprivation may result in deregulated immune responses with increased pro-inflammatory signaling, thus contributing to increase the risk for the onset and/or worsening of infection, as well as inflammation-related chronic diseases.
Here we reviewed the evidence regarding the impact of sleep deprivation on immune-related diseases by discussing the major points as follows: (1) the immune–sleep relationship; (2) the association of sleep deprivation with the development and/or progression of immune-related chronic diseases; and (3) the immune consequences of sleep deprivation and their implications for diseases. Finally, possible measures to reverse sleep deprivation-associated immune changes were discussed.
Basic immune mechanisms of sleep regulation
The discovery of muramyl peptide, a bacterial cell wall component that is able to activate the immune system and induce the release of sleep-regulatory cytokines, primary regulators of the inflammatory system, provided the first molecular link between the immune system and sleep48. Thereafter, other microbial-derived factors such as the endotoxin lipopolysaccharide (LPS)49, as well as mediators of inflammation, such as the cytokines interleukin (IL)-1 and tumor necrosis factor (TNF)-α, prostaglandins (PGs), growth hormone-releasing hormone (GHRH), and growth factors, were recognized as sleep-regulating factors50.
Along this line, most animal studies have consistently shown a role in particular for IL-1, TNF-α, and PGD2 in the physiologic, homeostatic non-rapid eye movement (NREM) sleep regulation, so that the inhibition of their biological action resulted in decreased spontaneous NREM sleep, whereas their administration enhanced NREM sleep amount and intensity, and suppressed rapid eye movement (REM) sleep51,52,53. Moreover, the circulating levels of IL-1, IL-6, TNF-α, and PGD2 are highest during sleep54. Their effects are dose- and time-of-day-dependent so that, for instance, low doses of IL-1 enhance NREMS, whereas high doses inhibit sleep55. Reciprocal effects may be involved in sleep regulation: for instance, the effects of systemic bacterial products such as LPS may also involve TNF-α49. Links exist between IL-1β and GHRH/growth hormone (GH) in promoting sleep so that IL-1 induced GH release via GHRH56, and hypothalamic γ-aminobutyric acid (GABA)ergic neurons (promoting sleep) are responsive to both GHRH and IL-1β57. Instead, anti-inflammatory cytokines, including IL-4, IL-10, and IL-13, inhibited NREM sleep in animal models58.
Through these substances, the immune system may signal to the brain and interact with other factors involved in sleep regulation such as neurotransmitters (acetylcholine, dopamine, serotonin, norepinephrine, and histamine), neuropeptides (orexin), nucleosides (adenosine), the hormone melatonin, and the hypothalamus-pituitary axis (HPA) axis. Signaling mechanisms to the brain also involve vagal afferents: for instance, vagotomy attenuates intraperitoneal TNF-α-enhanced NREMS responses59.
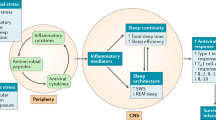
Cytokines are produced by a vast array of immune cells, including those resident in the central nervous system (CNS), and non-immune cells, e.g., neurons, astrocytes and microglia, and peripheral tissue cells60,61. Cytokines interact with the brain through humoral, neural, and cellular pathways, and form a brain cytokine network (Fig. 1) able to produce cytokines, their receptors, and amplify cytokine signals50. Peripheral cytokines reach the brain through different non-exclusive mechanisms, including blood–brain barrier (BBB) disruption62, penetration of peripheral immune cells, and via afferent nerve fibers, such as the vagus nerve, a bundle of parasympathetic sensory fibers that conveys information from peripheral organs to the CNS63.

CCL-2: C-C motif chemokine ligand-2; CXCL-10: C-X-C motif chemokine ligand 10; IL-1: interleukin-1; mNTS: medial nucleus tractus solitarius; PGE2: prostaglandin E2; TNF-α: tumor necrosis factor-α.
In the CNS, cytokines mediate a multiplicity of immunological and nonimmunologic biological functions64, such as synaptic scaling, synapse formation and elimination, de novo neurogenesis, neuronal apoptosis, brain development, cortical neuron migration65, circuit homeostasis and plasticity66, and cortical neuron migration65, and complex behaviors, sleep, appetite, aging, learning and memory65, and mental health status67,68.
A common experimental finding is that after damage to any brain area, if the animal or human survives, sleep always ensues69. Recent evidence indicates that sleep is a self-organizing emergent neuronal/glial network property of any viable network regardless of size or location, whether in vivo or in vitro53,70,71,72,73. Several sleep-regulatory substances, e.g., TNF, IL-1, nitric oxide, PGs, and adenosine are all produced within local cell circuits in response to cell use74,75.
From this point of view, TNF-α and IL-1 are closely interconnected and play a similar role in the regulation of sleep76,77,78,79,80,81. IL-1β and TNF-α self-amplify and increase each other’s mRNA expression in the brain82. In rats, IL-183 and TNF-α84 mRNAs show diurnal variations in different brain areas, with the highest concentrations recorded during increased sleep propensity and peaks occurring at time of sleep period onset in rats and mice85.
Sleep-like states in mixed cultures of neurons and glia are dependent in part on the IL-1 receptor accessory protein (AcP)69,86. In the brain, there is an AcP isoform, neuron-specific (AcPb)87, whose mRNA levels increase with sleep loss88,89. AcPb is anti-inflammatory, whereas AcP is pro-inflammatory87,88.
TNF signaling promotes sleep, whereas reverse TNF-α signaling (the soluble TNF receptor) promotes waking90. The brain production of TNF-α is neuron activity-dependent91. Afferent activity into the somatosensory cortex enhances TNF expression92, and in vitro optogenetic stimulation enhances neuronal expression of TNF immunoreactivity93.
Peripheral immune activation following acute or chronic infection or inflammatory diseases is marketed by altered cytokine concentrations and profiles, and is transmitted to the CNS initiating specific adaptive responses. Among these, a sleep response is induced and has been hypothesized to favor recovery from infection and inflammation, supposedly via the timely functional investment of energy into the energy-consuming immune processes54,94. Accordingly, acute mild immune activation enhances NREM sleep and suppresses REM sleep, whereas severe immune response with an upsurge of cytokine levels causes sleep disturbance with the suppression of both NREM and REM sleep49,95,96,97,98. This sleep change correlates to the course of the host immune response as observed in bacterial and Trypanosoma infections97,99. Supportively, the increase in NREM sleep was a favorable prognostic factor for rabbits during infectious diseases96.
Immune regulators also mediate the complex interrelation between sleep and the circadian systems74. Circadian rhythms in behavior and physiology are generated by a molecular clockwork located in the suprachiasmatic nucleus, i.e., the master circadian pacemaker, and peripheral tissues, and involving the so-called clock genes (Clock, Bmals, Npas2, Crys, Pers, Rors, and Rev-erbs)100. Cytokines, including TNF-α, IL-1β101,102, and LPS103,104,105, suppress the peripheral and hypothalamic expression of core clock genes and clock-controlled genes, resulting in reduced locomotor activity accompanied by prolonged rest time101.
Sleep deprivation and immune-related disease outcomes
In the following section, the association between sleep deprivation and risk or outcomes of immune-related disorders, as observed in human studies (mostly observational) and animal experimentations, will be examined. In this context, considering the sleep–immunity relationship, research has also begun to explore whether and how immune deregulation and inflammation may link sleep deprivation with adverse health outcomes.
Infection
A breakdown of host defense against microorganisms has been found in sleep-deprived animals, as shown by the increased mortality after septic insult in sleep-deprived mice compared with control mice106, or by systemic invasion by opportunistic microorganisms leading to increased morbidity and lethal septicemia in sleep-deprived rats107. There is growing evidence associating longer periods of sleep with a substantial reduction in parasitism levels108 and reduced sleep quality with increased risk of infection and poor infection outcome109,110. Accordingly, patients with sleep disorders exhibited a 1.23-fold greater risk of herpes zoster than did the comparison cohort111. Furthermore, sleep-deprived humans, as those with habitual short sleep (≤5 h) compared with 7–8 h sleep, are more vulnerable to respiratory infections in cross-sectional and prospective studies112,113, and after an experimental viral challenge109,114. Similarly, compared with long sleep duration (around 7 h), short sleep duration (around 6 h) is associated with an increased risk of common illnesses, including cold, flu, gastroenteritis, and other common infectious diseases, in adolescents115.
Compared with non-sleep-deprived mice, REM-sleep-deprived mice failed to control Plasmodium yoelii infection and, consequently, presented a lower survival rate110. This was correlated to an impaired T-cell effector activity, characterized by a reduced differentiation of T-helper cells (Th) into Th1 phenotype and following production of pro-inflammatory cytokines, such as interferon (IFN)-γ and TNF-α, and compromised differentiation into T-follicular helper cells (Tfh), essential to B-cell maturation, which therefore resulted to be reduced110. Accordingly, both Maf, a Tfh differentiation factor, and T-bet, a pro-Th1 transcription factor, were reduced in the REM-sleep-deprived group110. The combination of REM-sleep deprivation and P. yoelii infection resulted in an additive effect on glucocorticoid synthesis, and chemical inhibition of this exacerbated glucocorticoid synthesis reduced parasitemia, death rate, and restored CD4 T-cell, Tfh, and plasma B-cell differentiation in infected sleep-deprived mice110, suggesting a role of HPA axis hyperactivation in impairing host immune response under sleep deprivation.
Seep deprivation may exert detrimental effects on sepsis-induced multi-organ damage. Sleep deprivation (3 days) after LPS administration increased the levels of pro-inflammatory cytokines (IL-6 and TNF-α) in the plasma and organs (lung, liver, and kidney), which could be abrogated by subdiaphragmatic vagotomy or splenectomy 14 days prior to LPS administration116. Gut microbiota-vagus nerve axis and gut microbiota-spleen axis may play essential roles in post-septic sleep deprivation-induced aggravation of systemic inflammation and multi-organ injuries116.
Considering the association between sleep deprivation and immune response to infections, vaccination studies allow to assess the impact of sleep and sleep loss on ongoing immune response and the clinical outcome. Studies in which sleep deprivation (one or few nights) was applied to healthy humans during (mostly after) the immunological challenge of vaccination demonstrate that sleep deprivation reduced both the memory and effector phases of the immune response, as indexed by suppressed antigen-specific antibody and Th cell response compared with undisturbed sleep117.
Congruently, habitual (and hence chronic) short sleep duration (<6 h) compared with longer sleep duration was associated with reduced long-term clinical protection after vaccination against hepatitis B118. Sleep deprivation did not exert any impairing effect on mice already immunized119. From these studies, it seems that sleep supports—and sleep deprivation impedes—the formation of the immunological memory. Potential mechanisms involved in the beneficial effect of normal sleep on the vaccination response include: (i) the sleep-induced reduction in circulating immune cells that most likely accumulate into lymphatic tissues, increasing the probability to encounter antigens and trigger the immune response; (ii) the sleep-associated profile of inflammatory activation towards Th1 cytokines (increased IL-2, IFN-γ, etc.), which may favor macrophage activation, antigen presentation, and T-cell and B-cell activation; (iii) the effect of sleep stage on the formation of immunological memory through specific immune-active hormones: indeed, during slow wave sleep-rich early sleep, the profile of immune-active hormones, characterized by minimum concentrations of cortisol, endowed with anti-inflammatory activity, and high levels of GH, prolactin, and aldosterone, which support Th1 cell-mediated immunity, may facilitate the mounting of an effective adaptive immune response to a microbial challenge54.
Cancer
Sleep deprivation has increasingly been recognized as a risk factor for impaired anti-tumor response. Epidemiological studies suggest, albeit not consistently120, a significant association between short sleep duration and the risk for several cancers, including breast, colorectal, and prostate cancer29,121,122,123. Potential mechanisms underlying this association include a shorter duration of nocturnal secretion of melatonin (putatively due to increased light exposure at night)124, which exerts anti-cancer properties through antimitotic, antioxidant, apoptotic, anti-estrogenic, and anti-angiogenic mechanisms125. Melatonin also plays immunomodulatory and anti-inflammatory effects with relevance for its anti-cancer activity, being able to inhibit the pro-inflammatory nuclear factor-κB (NF-κB)/NLRP3 inflammasome pathways, and to support T/B-cell activation and macrophage function126. However, besides melatonin, impaired anti-tumor immune response has been invoked in the sleep deprivation-associated risk for cancer development. A reduced cytotoxic activity of natural killer (NK) cells, which are immune cells with anti-tumor effect, has been reported in 72 h sleep-deprived mice compared with control mice, accompanied by reduced numbers of the cytotoxic cells such as CD8 T cells and NK cells in the tumor microenvironment after chronic sleep deprivation (for 18 h/day during 21 days) in an animal model of experimental pulmonary metastasis127,128. In this model, the reduced anti-tumor immunity of sleep-deprived animals was also indexed by the reduced number of antigen-presenting cells (dendritic cells) in the lymph nodes, as well as by the decreased effector CD4 T-cell numbers and corresponding cytokine profile (decreased IFN-γ), resulting in lowered Th1 response of Th cells, i.e., the most effective immune response against tumors. Therefore, an immunosuppressive environment develops with sleep deprivation, which could translate into an early onset and increased growth rate of cancer128 or increased mortality129.
An integrated meta-analysis of transcriptomic data showed that circadian rhythm-related genes are downregulated and upregulated in the cortex and hypothalamus samples of mice with sleep deprivation, respectively, with downregulated genes associated with the immune system and upregulated genes associated with oxidative phosphorylation, cancer, and T2DM130. Several circadian rhythm-related genes were common to both T2DM and cancer, and seem to associate with malignant transformation and patient outcomes130.
Hence, although these sleep deprivation-induced immune-mediated mechanisms in cancer warrant further confirmation in humans, the importance of the immune function in the anti-tumor host defense is well recognized131, thus suggesting that the impaired immune response after sleep deprivation may represent a plausible mediator of the associated increased risk for cancer as described in animal models and in humans.
Neurodegenerative diseases
NDDs are aging-related diseases that selectively target different neuron populations in the CNS, and include Alzheimer’s disease, multiple sclerosis, Parkinson’s disease, Huntington’s disease, and amyotrophic lateral sclerosis. One prevailing hypothesis is that altered sleep habits and specifically sleep deprivation may be a consequence and frequently a marker of the disease132,133,134. However, human and animal studies have also suggested a causative or contributing role for sleep deprivation in the development and/or worsening of neurodegenerative processes132,133,134.
Potential pathophysiological mechanisms involve, among others, neuro-immune dysregulation. Indeed, a common feature –and a potential therapeutic target- of NDDs is the chronic activation of the immune system, where aspects of peripheral immunity and systemic inflammation integrate with the brain’s immune compartment, leading to neuroinflammation and neuronal damage135. Neuroinflammation following sleep deprivation has been studied as a pathogenic mechanism potentially mediating the association between sleep deprivation and neurodegenerative processes. Low-grade neuroinflammation as indexed by heightened levels of pro-inflammatory mediators (e.g., TNF-α, IL-1β, and COX-2) and activation of astrocytes and microglia, main immune cells in the brain, was observed in the hippocampus and piriform cortex regions of the brain of chronic sleep-deprived rats along with neurobehavioral alterations (anxiety, learning, and memory impairments)136. The sleep deprivation pro-inflammatory milieu was accompanied by oxidative stress in the brain137 and BBB disruption with consequent increased permeability to blood components138. After acute sleep deprivation, there was a significant increased recruitment of B cells in the mouse brain, which could be important given evidence of B cells involvement in NDDs139.
Progressive and chronic aggregations of unique proteins in the brain and spinal cord are hallmarks of NDDs140 and trigger inflammatory responses, gradual loss of physiological functions of the nerve cells, and cell death141. Impaired autophagy in humans, a catabolic process of cytoplasmic components, contributes to the aggregation and accumulation of β-amyloid (Aβ), cytoskeleton-related protein τ, and synuclein in neuronal cells and tissues140. Sleep plays an important role in the clearance of metabolic waste products accumulated during wakefulness and neural activity. Indeed, the Aβ protein is predominantly cleared from the brain during sleep, possibly through the glymphatic pathway. Congruently, acute and chronic experimental sleep deprivation in animals142,143 and humans144 resulted in brain Aβ accumulation and plaque formation, a typical pathological change in Alzheimer’s disease process, the most common type of dementia. Imaging studies have revealed that healthy humans with self-reported short sleep were more prone to have cerebral Aβ plaque pathology145 and disruption of deep sleep (slow wave sleep) increases Aβ in human cerebrospinal fluid (CSF)146. Likewise, patients with insomnia present higher CSF levels of Aβ147.
This pathological Aβ accumulation might reflect disrupted balance of Aβ production and clearance after sleep deprivation. On the one hand, sleep deprivation results in reduced clearance as suggested by clinical studies showing that Aβ levels in CSF are the highest before sleep and the lowest after wakening, whereas Aβ clearance from CSF was impaired by sleep deprivation148. Impaired clearance might also derive from disrupted peripheral Aβ transport, as suggested by the sleep deprivation-induced downregulation of low-density lipoprotein receptor-related protein-1 (LRP-1), which promotes Aβ efflux from the brain to the peripheral circulation across the BBB, and elevations of receptor of advanced glycation end products (RAGE), which promotes on the contrary the influx of peripheral Aβ into the brain, thus preventing Aβ clearance149. On the other hand, apart from impairing Aβ and τ interstitial fluid clearance, sleep deprivation may also have a role in increasing Aβ and τ exocytosis, thereby increasing CSF Aβ and τ levels150. In animals, sleep deprivation also leads to upregulation of β-secretase 1 (BACE-1), the most important enzyme regulating Aβ generation in the brain142,143,149, thus opening the hypothesis of increased Aβ production by sleep deprivation. Sleep deprivation-induced neuroinflammatory mediators correlate and could lead to disturbed Aβ clearance and stimulated amyloidogenic pathway143, being pro-inflammatory cytokines able to suppress the expression of LRP-1 and to increase RAGE151 and BACE-1 levels152. Likewise, oxidative stress induced by sleep deprivation may also contribute to the neuroinflammatory burden and the increased expression of BACE-1153. Furthermore, patients with insomnia, compared with healthy controls, showed decreased serum levels of neurotrophins, including brain-derived neurotrophic factor (BDNF), proteins especially relevant in neuroplasticity, memory and sleep, and this reduction was significantly related to the insomnia severity154.
Sleep deprivation is associated with a rapid decline in circulatory melatonin levels, which may be linked to rapid consumption of melatonin as a first-line defense against the sleep deprivation-associated rise in oxidative stress155. Melatonin is a potent antioxidant, interacts with BDNF156, and promotes neurogenesis and inhibits apoptosis157. The neuroprotective potential of melatonin can target events leading to Alzheimer’s disease development including Aβ pathology, τ hyperphosphorylation, oxidative stress, glutamate excitotoxicity, and calcium dyshomeostasis150,158. Accordingly, melatonin treatment could restore the autophagy flux, thereby preventing tauopathy and cognitive decline in Alzheimer’s disease mice159.
Patients with Alzheimer’s disease have an increased incidence of sleep-disordered breathing160. In addition, sleep-disordered breathing is associated with an increased risk of mild cognitive impairment or dementia and with earlier onset of Alzheimer’s disease161. Sleep-disordered breathing is also associated with altered levels of Alzheimer’s disease biomarkers in CSF, including decreased levels of Aβ and elevated levels of phosphorylated τ162. Sleep-disordered breathing possibly via hypoxia, inflammation, and sleep disruption/deprivation could contribute to Alzheimer’s disease processes, e.g., increase of Aβ production and aggregation, suppression of glymphatic clearance of Alzheimer’s disease pathogenic proteins (τ, Aβ) and oxidative stress, inflammation, and synaptic damage134,163.
To summarize, the sleep deprivation-associated risk for Alzheimer’s disease could be linked to the induction of inflammation in the brain and disorders of systemic innate and adaptive immunity164. However, the relationship of sleep deprivation to inflammation in Alzheimer’s disease is mostly speculative and needs to be confirmed.
Similar to Aβ in Alzheimer’s disease, abnormal levels of α-synuclein are common to Parkinson’s disease, the second most common NDDs165. Sleep disturbances are not only a common comorbidity in Parkinson’s disease, but often precede the onset of classic motor symptoms166. The main pathological features of Parkinson’s disease are the reduction of dopaminergic neurons in the extrapyramidal nigrostriatal body and the formation of Lewy bodies formed by the aggregation of α-synuclein and its oligomers surrounded by neurofilaments. Due to the degeneration of the dopaminergic neurons, affected people show muscle stiffness, resting tremors, and posture instability; other pathways involved in sleep, cognition, mental abnormalities, and other non-motor symptoms are also affected167. Epidemiological studies also suggest that disturbed sleep may increase the risk of Parkinson’s disease168,169. Such disease-modifying mechanisms may include activation of inflammatory and immune pathways, abnormal proteostasis, changes in glymphatic clearance, and altered modulation of specific sleep neural circuits that may prime further propagation of α-synucleinopathy in the brain169. Melatonin could reduce neurotoxin-induced α-synuclein aggregation in mice. Furthermore, melatonin pretreatment reduced neurotoxin-induced loss of axon and dendritic length in dopaminergic neurons through suppression of autophagy activated by CDK5 and α-synuclein aggregation, thereby reducing dyskinesia symptoms in Parkinson’s disease animal models170. A few reports have shown that melatonin exerts protective effects in several experimental models of Parkinson’s disease171.
However, although animal experimentations suggest a link between sleep deprivation and immune dysfunction in neurodegenerative processes, no human investigations have yet confirmed the mediating role of immune dysregulation in the association between sleep deprivation and risk or outcomes of NDDs.
Autoimmune diseases
Sleep disturbances are frequently reported in autoimmune diseases, and immunotherapy in patients with autoimmune pathologies results in sleep improvement172. However, knowledge of the immunopathology of autoimmune diseases have disclosed new concepts on the impact of sleep deprivation on autoimmune disease process, showing that sleep deprivation can promote a breakdown of immunologic self-tolerance. Human cohort studies found that non-apnea sleep disorders, including insomnia, were associated with a higher risk of developing autoimmune diseases such as rheumatoid arthritis, ankylosing spondylitis, systemic lupus erythematosus, and systemic sclerosis (adjusted hazard ratio: 1.47, 95% confidence interval (CI) 1.41–1.53)173 Similarly, in relatives of systemic lupus erythematosus patients, and hence at increased risk for systemic lupus erythematosus, self-reported short sleep duration (<7 h/night) was associated with transitioning to systemic lupus erythematosus (adjusted odds ratio: 2.0, 95% CI 1.1–4.2), independent of early preclinical features that may influence sleep duration such as prednisone use, depression, chronic fatigue, and vitamin D deficiency174. This role of sleep deprivation as a risk factor for autoimmune diseases is corroborated by animal studies. In mice genetically predisposed to develop systemic lupus erythematosus175, chronic sleep deprivation, applied at an age when animals were yet clinically healthy, caused an early onset of the disease, as indexed by the increased number of antinuclear antibodies, without affecting disease course or severity, according to data on proteinuria, a surrogate marker of autoimmune nephritis, and longevity. Several mechanisms have been postulated to explain the link between sleep deprivation and autoimmune disease risk. Sleep deprivation can accelerate disease development through mechanisms including sleep deprivation-induced increased production of several pro-inflammatory cytokines44,54, as better discussed below. Indeed, cytokines are synergistically involved in the pathogenesis of autoimmunity, such as IL-6, whose abnormal production results in polyclonal B-cell activation and the occurrence of autoimmune features176, and IL-17 and the related Th17-cell response177, which require IL-6 for activation178 and can cause greater amounts of autoantibody production and immune complex formation, or can intensify chronic inflammation by promoting angiogenesis and recruiting of inflammatory cells at inflammation sites as well as cartilage and bone erosion179. Furthermore, experimentally sleep-deprived healthy humans showed impaired suppressive activity of CD4 regulatory T cells (Treg), which normally is highest during the night and lowest in the morning180. The suppressive function of Treg towards excessive immune response is an important homeostatic mechanism, whose impairment is implicated in autoimmune disease pathogenesis181. Hence, sleep deprivation may not be merely an early symptom or a consequence of an autoimmune disease, but may contribute directly to the pathogenesis increasing the susceptibility to develop an autoimmune disease. More studies are warranted in this field.
Metabolic and vascular diseases
Prospective epidemiological evidence associate sleep deprivation (commonly <7 h/night, often <5 h/night) with the incidence of fatal and non-fatal CV outcomes, with a 48% higher risk of coronary heart disease25, a 15% higher risk of stroke182, and a 12% increased risk of all-cause mortality37, which is mainly due to CV causes, according to some authors183. In a recent prospective cohort, a low-stable sleep pattern (<5 h sleep/night) during the 4-year follow-up had the highest risk of death and CV events184. Short sleep has also been associated with increased subclinical atherosclerotic burden, the dominant underlying cause of CV diseases185.
In addition, sleep deprivation increases the risk for obesity (about 55% higher risk)39,186, insulin resistance, T2DM (28% higher risk)38, and hypertension (21% higher risk)187, which are powerful and preventable risk factors for CV diseases. Notably, the risk for diabetes attributable to sleep deprivation is comparable to that of other established traditional cardiometabolic risk factors188, thus underscoring the clinical significance of targeting sleep deprivation in the prevention of cardiometabolic diseases. In contrast with normal nocturnal sleep and in particular NREM sleep characterized by a marked decrease in sympathetic activity, catecholamine plasma levels, and blood pressure, experimental sleep deprivation (acute or chronic) is accompanied by increased sympathetic outflow, with consequent higher blood pressure and heart rate, thus providing a pathogenic link between sleep deprivation and hypertension risk189,190,191,192.
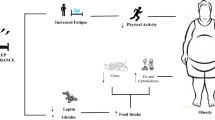
Regarding the influence of sleep deprivation on metabolic pathways, studies support a plausible causal link between sleep deprivation and the risk of overweight and obesity, possibly mediated by the effect of sleep deprivation on circulating levels of hormones (leptin, ghrelin) controlling hunger, satiety and energy balance, besides other factors intervening during sleep deprivation, including physical inactivity and overfeeding193. Furthermore, human experimental evidence with chronic sleep deprivation protocol demonstrate that sleep deprivation may alter glucose metabolism194 and insulin sensitivity195, thus increasing the risk for obesity and T2DM. The reduction in total body insulin sensitivity observed after sleep deprivation (4.5 h per night for 4 days) in healthy subjects was paralleled by impaired peripheral insulin sensitivity, as demonstrated in subcutaneous fat playing a pivotal role in energy metabolism195. Considering a more chronic sleep deprivation, reduced insulin sensitivity was reported in overweight adults after 14 days of experimental sleep deprivation (5.5 h per night) compared with 8.5 h per night of sleep196, and after habitual curtailment in sleep duration of 1.5 h (<6 h of sleep per night) in healthy young adults with a family history of T2DM197.
Although the mechanisms that underlie most associations between short sleep duration and adverse cardiometabolic outcomes are not fully understood, potential causative mechanisms involving immune-inflammatory activation have been postulated. It is indeed well established that the subclinical inflammatory status induced by sleep deprivation has pathogenic implications for metabolic and CV risk factors (glucose metabolism, diabetes, hypertension, atherogenic lipid profile, endothelial dysfunction, and coronary calcification) and outcomes (stroke and coronary heart disease)24. Accordingly, most of the markers of systemic and cellular inflammation (leukocyte counts and activation state, cytokines, acute-phase proteins, and adipose tissue-derived adipokines) found to be altered after sleep deprivation have been epidemiologically and pathogenically associated with insulin resistance, T2DM, and vascular complications198. In fact, inflammation is an early pathogenic process during the development of obesity and insulin resistance199. Many adipose tissue-released inflammatory factors with pro-atherogenic and pro-thrombotic actions have also been regarded as a molecular link between obesity and atherosclerotic CV diseases200. Furthermore, chronic inflammatory processes are firmly established as central to the development and clinical complications of CV diseases, form the initiation, promotion and progression of atherosclerotic lesions to plaque instability, and the precipitation of thrombosis, the main underlying cause of myocardial infarction or stroke. Most CV risk factors (adiposity, insulin resistance, T2DM, hypertension, and dyslipidemia) act by inducing or intensifying such underlying inflammatory processes that ultimately promote endothelial dysfunction, altered vascular reactivity, innate and adaptive immune system activation, leukocyte infiltration into the vessel wall, and thus atherogenesis201. Experimental sleep deprivation leads to endothelial dysfunction, an early marker of atherosclerosis, as indexed by impaired endothelial-dependent vasodilation or increased levels of endothelial adhesion molecules191.
Among the inflammatory markers, besides being a biomarker of future risk for CV diseases and a predictor of clinical response to statin therapy202, C-reactiove protein (CRP) has been shown to be involved in the immunologic process that triggers vascular remodeling and atherosclerotic plaque deposition202. CRP levels lack diurnal rhythm and its liver production is stimulated by cytokines including IL-6 and IL-17, which are upregulated by sleep deprivation203. As such, although limited evidence have found an elevation of circulating CRP following sleep deprivation204, CRP is a prototypical inflammatory factor with the potential to mark and—to some extent mediate—CV risk following sleep deprivation. Congruently, elevated and sustained plasma levels of CRP have been observed in healthy humans after prolonged sleep deprivation (5 or 10 nights), in concomitance with increased heart rate190,203, lymphocyte pro-inflammatory activation, and production of cytokines (e.g., IL-1, IL-6, and IL-17)203. Similarly, the increase in blood pressure and heart rate observed after acute total sleep deprivation (40 h) was accompanied and even preceded by impaired vasodilation and by increased levels of IL-6 and markers of endothelial dysfunction and activation, such as cellular adhesion molecules (E-selectin, ICAM-1, etc.)191. The sleep deprivation pro-atherogenic effect in animal model of sleep fragmentation is mediated, at least in part, by reduced hypothalamic release of hypocretin (i.e., orexin), a wake-inducing neuropeptide, which limits the production of leukocytes (monocytes and neutrophils) and atherosclerosis development, and has been inversely associated with the risk of myocardial infarction, heart failure, and obesity205. The activation of the sympathetic nervous system (SNS) may be another mechanism for the inflammatory link between sleep loss and atherosclerotic CV disease, because such activation increases the bone marrow release of progenitor cells, the production of innate immune cells (monocytes), and the levels of inflammatory cytokines, and triggers endothelial dysfunction, thereby leading to systemic and vascular inflammation and atherosclerosis206,207. Playing a key role in instigating inflammatory responses and promoting atherosclerosis208, the sleep deprivation-associated oxidative stress may also contribute to CV risk. It has also been hypothesized a role for melatonin suppression following sleep deprivation in the vascular impairment associated with sleep deprivation, given that melatonin inhibits oxidative stress and cytokine production by immune and vascular cells, and represses atherosclerotic lesion formation in vivo209.
Therefore, a significant and consistent association exists between sleep deprivation and cardiometabolic risk and clinical outcomes, with several plausible immune-mediated causative mechanisms explaining this association.
Immune mechanisms linking sleep deprivation and diseases
As shown above, sleep deprivation has been found to alter inflammatory immune processes via multiple pathways, which could lead to increased susceptibility to chronic inflammatory diseases (Fig. 2). Most of the current knowledge on immune effects of sleep deprivation come from studies using controlled experimental sleep deprivation protocols, among which chronic partial sleep deprivation, lasting 2–15 days, is that mostly resembling the human condition of chronic insufficient sleep.

Sleep deprivation, as induced experimentally or in the context of habitual short sleep, has been found to be associated with alterations in the circulating numbers and/or activity of total leukocytes and specific cell subsets, elevation of systemic and tissue (e.g., brain) pro-inflammatory markers including cytokines (e.g., interleukins [IL], tumor necrosis factor [TNF]-α), chemokines and acute phase proteins (such as C reactive Protein [CRP]), altered antigen presentation (reduced dendritic cells, altered pattern of activating cytokines, etc.), lowered Th1 response, higher Th2 response, and reduced antibody production. Furthermore, altered monocytes responsiveness to immunological challenges such as lipopolysaccharide (LPS) may contribute to sleep deprivation-associated immune modulation. Hypothesized links between immune dysregulation by sleep deprivation and the risk for immune-related diseases, such as infectious, cardiovascular, metabolic, and neurodegenerative and neoplastic diseases, are shown. The illustrations were modified from Servier Medical Art (http://smart.servier.com/), licensed under a Creative Common Attribution 3.0 Generic License. APC: antigen-presenting cells.
Some studies have observed that sleep deprivation, compared with regular nocturnal sleep, leads to increased circulating numbers of total leukocytes and specific cell subsets mainly neutrophils, monocytes, B cells, CD4 T cells, and decreased circulating numbers and cytotoxic activity of NK cells203,210,211,212,213. Other studies, however, found contrasting results, including a decrease in CD4 T cells after sleep deprivation213,214, probably due to differences in sleep deprivation protocol, sampling methodologies, and other factors. Sleep deprivation has also shown to alter circadian rhythm of circulating leukocytes215, with higher levels during the night and at awakening and a flattened rhythm210,212. Additional findings are suggestive of immune deregulation by sleep deprivation, including a decreased neutrophils phagocytic activity213, altered lymphocytes adhesion molecule expression216, and reduced stimulated production of IL-2 and IL-12, which are important for adaptive immunity211,217.
Experimental sleep deprivation has been reported to affect systemic markers of inflammation, with studies showing increased circulating pro-inflammatory molecules (IL-1, IL-6, CRP, TNF-α, and MCP-1); this associated in some studies with a subsequent homeostatic increase in endogenous inhibitors, including IL-1 receptor antagonist and TNF receptors203,218,219,220. In agreement with experimental sleep deprivation, population studies found a direct independent association between habitual short sleep duration (generally < 5 or 6 h) and elevated circulating pro-inflammatory markers, e.g., acute phase proteins (CRP and IL-6), cytokines (TNF-α, IFN-γ, IL-1, etc.), adhesion molecules, and leukocyte counts183,221,222,223,224,225. Furthermore, a reduced NK cell activity226 and a decline in naive T cells227, compatible with reduced immune competence, was reported in association with habitual short sleep. Shortening of leukocyte telomere length, a cellular senescence marker linked with inflammation, was also associated with shorter sleep duration228,229.
The reported elevation of systemic inflammation is clinically relevant, because it is suggested to specifically mediate the increased risk of mortality associated with short sleep23,230,231 and, as observed, the risk for chronic disease development.
Regarding cellular markers of inflammation, some studies found that the ex-vivo LPS-stimulated production of TNF-α232,233, IL-1β, and IL-6203,232,233,234 by human monocytes increased during sleep deprivation but decreased during regular nocturnal sleep54,203,232,233,234. However, other studies reported a decrease of TNF-α production by activated monocytes after sleep deprivation compared with regular nocturnal sleep203,235. These contrasting results need further investigations and may depend on differences in the cytokine sensitivity to different sleep deprivation protocols or sampling methods and time. For instance, it seems that partial acute sleep deprivation increased stimulated monocytic TNF-α production232,233, whereas more sustained sleep deprivation decreased it203,235.
Undisturbed sleep is predominantly characterized by a Th1 polarization of Th cells (expressing IFN-γ, IL-2, and TNF-α), and experimental sleep deprivation in humans leads to a shift from a Th1 pattern towards a Th2 pattern (expressing IL-4, IL-5, IL-10, and IL-13)217,236. Accordingly, conditions featured by disturbed sleep with specific deficit in slow wave sleep, as observed in elderly people237, alcoholic238, and insomnia239 patients, show a cytokine shift towards Th2. The balance of Th1/Th2 immunity and its shift during sleep deprivation may have crucial implications in anti-microbial and anti-tumor immune responses. Th2 over-activity is known to be involved in some forms of allergic responses, and to increase the susceptibility to infection240. Likewise, regarding the anti-tumor immune action, Th1 response supports cytotoxic lymphocytes and tumor cells destruction with the potential of elimination or control of tumor cell growth, so that a type 1 adaptive immune response (increased antigen presentation, IFN-γ signaling, and T-cell receptor signaling) may be associated with an improved survival or prognosis241,242. In contrast, Th2 over-response is thought to contribute to tumor development and progression, by limiting cytotoxic T lymphocytes proliferation and by the modulation of other inflammatory cell types241.
Several cellular and molecular signaling pathways may be involved in mediating the influence of sleep deprivation on immune and inflammatory functions (Fig. 3). Increased oxidative stress markers and/or decreased antioxidant defense have been found after sleep deprivation243,244,245. Sleep shows an antioxidant function, responsible for eliminating reactive oxygen species produced during wakefulness, and contrarily sleep deprivation may cause oxidative stress, which leads to cell senescence, unbalanced local/systemic inflammation, dysmetabolism, and immune derangements246,247.

A schematic model of potential mechanistic pathways linking sleep deprivation and inflammatory immune activation is depicted. Sleep deprivation is associated with activation of the sympathetic nervous system and release of norepinephrine and epinephrine into the systemic circulation, as well as to some extent with impaired hypothalamus-pituitary axis stimulation. These neuromediators may act along with other potential stimuli accumulated following sleep deprivation including reactive oxygen species (ROS), adenosine, metabolic waste products (e.g., β-amyloid) not cleared during normal sleep, gut microbiota dysbiosis leading to altered local and systemic pattern of metabolic products, as well as with changes in the profile of neuro-endocrine hormones, such as prolactin, growth hormone, and altered circadian rhythm of melatonin secretion. In immune cells located in the brain and the peripheral tissues, these stimuli may in concert trigger inflammatory activation, with release of cytokines, chemokines, acute phase protein, etc. via the recruitment of transcriptional regulators of pro-inflammatory gene expression, mainly nuclear factor (NF)-κB, and disturbing the circadian rhythmicity of gene expression of both clock genes and metabolic, immune and stress response genes (see text for further detail). E: epinephrine; NE: norepinephrine; TLR: Toll-like receptor. Arrows indicate stimulation; lines indicate inhibition. The illustrations were modified from Servier Medical Art (http://smart.servier.com/), licensed under a Creative Common Attribution 3.0 Generic License.
Effects of sleep deprivation on the immune response may derive from the activation of the SNS with the corresponding increase in systemic catecholamines22,248. Catecholamines signal to immune cells via adrenergic receptors, which are primarily α- and β-adrenergic in myeloid cells and β-adrenergic in lymphocytes249. The immune outcome of the sympathetic signaling is complex, and includes both stimulatory and inhibitory effects depending on cell and receptor types, cell development/activation states, and local microenvironment249,250. Some evidence suggest that β-adrenergic signaling inhibits and α-adrenergic signaling promotes excessive inflammation under endotoxemia250. Activation of α-adrenergic signaling in peripheral tissues induces the upregulation of pro-inflammatory cytokines250,251. Sympathetic activation also suppresses the transcription of type I IFNs (IFN-α and IFN-β) genes and interferon response genes, which play a key role in anti-viral immunity252, and inhibits via β-adrenergic signaling the anti-tumor cytotoxicity of T lymphocytes253. In vitro β-adrenergic stimulation repressed Th1 response and stimulated Th2 response, with varying effects found in vivo249,254. Although the specific role of SNS activation in the immune phenotype associated with sleep deprivation is not clearly established, data suggest a pro-inflammatory effect of SNS under sleep deprivation. Indeed, chemical sympathectomy has been recently shown to alleviate the inflammatory response following chronic sleep deprivation in mice255, and both α- and, to a lesser extent, β-adrenergic receptors seem to contribute to the sympathetic regulation of inflammatory responses to sleep deprivation256.
At the molecular levels, sleep deprivation led to significant gene expression changes in animal tissues257,258,259 and human blood monocytes203,233,260,261,262, with affected genes mostly related to immune and inflammatory processes (leukocyte function, Th1/Th2 balance, cytokine regulation, and TLR signaling), oxidative stress, stress response, apoptosis, and circadian system, collectively indicating immune activation and hyperinflammation.
Sleep loss and mistimed sleep also led in the blood transcriptome to alteration and reduction in the circadian rhythmicity of gene expression261,263, which is an integral part of basic biological processes and homeostasis264,265,266.
The activation of the pro-inflammatory NF-κB/Rel family of transcription factors by sleep deprivation, first demonstrated in the late 1990s in mice267, and subsequently widely confirmed233,260,261,268,269,270,271,272, is one of the most consistent findings regarding upstream transcriptional regulation. NF-κB induces the expression of genes (e.g., cytokines/chemokines, growth factors, receptors/transporters, enzymes, adhesion molecules) involved in inflammation, immunity, proliferation, and apoptosis273, circadian clock activity274, and sleep propensity275. Potential signals for NF-κB activation under sleep deprivation include increased adenosine levels, oxidative stress, altered metabolism (adiposity and decreased insulin sensitivity), brain proteins/metabolites (e.g., Aβ), melatonin suppression276, circadian clock proteins277, and catecholamine surge due to increased sympathetic activity278. Given the role of NF-κB in the pathophysiology of inflammatory diseases273, its activation under sleep deprivation may be a common pathway for the risk of morbidity and mortality.
The intestinal microbiota is also affected by sleep loss279,280,281, showing indices of dysbiosis (increased Firmicutes:Bacteroidetes ratio; decreased diversity and richness), which may affect the immune system282, and are similar to those associated with cardiometabolic diseases45.
Countermeasures for sleep deprivation: effect on immune parameters
Although the impact of strategies to improve sleep duration on neurobehavioral performance and alertness after sleep deprivation have been assessed283,284,285, sleep deprivation countermeasures to improve immune and inflammatory parameters, and, correspondingly, disease risk and outcomes have been studied to a lesser extent.
Although extension of habitual short sleep did not show to significantly counterbalance the immune consequence of sleep deprivation286,287,288, mixed results derive from nighttime recovery sleep following sleep deprivation (Table 1), with limited evidence of effectiveness for specific immune parameters210,214, and mostly after multiple consecutive nights of 8 h sleep recovery or with an extended nocturnal sleep duration212,289.
Although daytime napping (<20 min) restores alertness, and mental and physical performance without provoking sleep inertia associated with longer nap290,291,292, the effects of a short nap on immune/inflammatory parameters after sleep deprivation have yet to be firmly established. Differently form population studies293, laboratory studies found immune benefit from nap218,289,294,295. Regarding immune-related clinical outcomes, controversy exists, with studies finding no association296, inverse associations297,298 or positive association296, and a J-shaped relationship299,300,301 between napping and CV and metabolic diseases or cancer events and mortality. Whether changes in immune parameters could contribute to the associations between napping and immune-related diseases remains unclear.
Among the strategies to recover sleep deprivation-induced immune changes, cognitive behavior therapy improves sleep outcomes in insomnia and lowers cellular and systemic inflammatory markers302,303, and the risk score composed of CV and metabolic risk factors304. This highlights the potential role of targeting sleep in reducing the inflammatory risk and the associated chronic diseases.
Summary and concluding remarks
Sleep exerts immune-supportive functions and impairments of the immune-inflammatory system are a plausible mechanism mediating the negative health effects of sleep deprivation, and in particular, its role in the risk and outcomes of chronic diseases such as infections, CV, metabolic and autoimmune diseases, NDDs, and cancer. Caution should be exercised in interpreting cellular and molecular outcomes of sleep deprivation in experimental studies conducted till now as a result of an independent effect of sleep deprivation, because other factors may play a role, including extended wakefulness-associated processes, other features of sleep-wakefulness, their temporal and functional segregation or methodologies of sleep manipulation.
Randomized controlled trials assessing the effect of treatment of sleep deprivation on inflammatory immune dysfunction and/or health outcomes are needed. Knowledge of inflammatory and immunological signatures in response to sleep curtailment may inform not only on the underlying molecular links, but also contribute to refine risk profiles to be used for developing biomarkers of sleep deprivation and sleep disturbance-related health outcomes, which may also represent potential targets of interventions. Recent metabolomic305 and transcriptomic306 studies hold promise in biomarker discovery306.
These efforts may converge towards a new ground fostering interactions between the sleep research and the medical community to translate scientific knowledge into the clinic, prioritize health issues, and develop strategies and policies for subject risk stratification, to include evidence-based sleep recommendations in guidelines for optimal health and to address sleep hygiene at the individual and the population levels, as a means to prevent the negative health consequences of sleep deprivation. These actions might also foster health literacy and empowerment of individuals to actively better manage their own health and well-being throughout their life course by means of lifestyle, nutritional, and behavioral habits including sleep hygiene307.
Conclusively, in the perspective of staying healthy in this rapidly changing society, the sleep–immunity relationship raises relevant clinical implications for promoting sleep health and, as evidenced here, for improving or therapeutically controlling inflammatory response by targeting sleep. This may ultimately translate, in the era of preventive medicine, into addressing sleep as a lifestyle approach along with diet and physical activity to benefit overall public health.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All data generated or analysed during this study are included in this published article.
References
Luyster, F. S. et al. Sleep: a health imperative. Sleep 35, 727–734 (2012).
Grandner, M. A. Sleep, health, and society. Sleep. Med. Clin. 12, 1–22 (2017).
Ohayon, M. M., Carskadon, M. A., Guilleminault, C. & Vitiello, M. V. Meta-analysis of quantitative sleep parameters from childhood to old age in healthy individuals: developing normative sleep values across the human lifespan. Sleep 27, 1255–1273 (2004).
Galland, B. C., Taylor, B. J., Elder, D. E. & Herbison, P. Normal sleep patterns in infants and children: a systematic review of observational studies. Sleep. Med. Rev. 16, 213–222 (2012).
Galland, B. C. et al. Establishing normal values for pediatric nighttime sleep measured by actigraphy: a systematic review and meta-analysis. Sleep 41, https://doi.org/10.1093/sleep/zsy017 (2018).
Boulos, M. I. et al. Normal polysomnography parameters in healthy adults: a systematic review and meta-analysis. Lancet Respir. Med. 7, 533–543 (2019).
Consensus Conference, P. et al. Joint Consensus Statement of the American Academy of Sleep Medicine and Sleep Research Society on the Recommended Amount of Sleep for a Healthy Adult: methodology and discussion. J. Clin. Sleep. Med. 11, 931–952 (2015).
Consensus Conference, P. et al. Recommended amount of sleep for a healthy adult: a Joint Consensus Statement of the American Academy of Sleep Medicine and Sleep Research Society. J. Clin. Sleep. Med. 11, 591–592 (2015).
Carskadon, M. A., Vieira, C. & Acebo, C. Association between puberty and delayed phase preference. Sleep 16, 258–262 (1993).
Gulia, K. K. & Kumar, V. M. Sleep disorders in the elderly: a growing challenge. Psychogeriatrics 18, 155–165 (2018).
Garbarino, S., Lanteri, P., Sannita, W. G., Bragazzi, N. L. & Scoditti, E. Circadian rhythms, sleep, immunity, and fragility in the elderly: the model of the susceptibility to infections. Front. Neurol. 11, 558417 (2020).
Zomers, M. L. et al. Characterizing adult sleep behavior over 20 years-the Population-Based Doetinchem Cohort Study. Sleep 40, https://doi.org/10.1093/sleep/zsx085 (2017).
American Academy of Sleep Medicine. International Classification Of Sleep Disorders 3rd edn (American Academy of Sleep Medicine, 2014).
Ford, E. S., Cunningham, T. J. & Croft, J. B. Trends in self-reported sleep duration among US adults from 1985 to 2012. Sleep 38, 829–832 (2015).
Gilmour, H. et al. Longitudinal trajectories of sleep duration in the general population. Health Rep. 24, 14–20 (2013).
Matricciani, L., Olds, T. & Petkov, J. In search of lost sleep: secular trends in the sleep time of school-aged children and adolescents. Sleep. Med. Rev. 16, 203–211 (2012).
Wheaton, A. G., Jones, S. E., Cooper, A. C. & Croft, J. B. Short sleep duration among middle school and high school students - United States, 2015. MMWR Morb. Mortal. Wkly Rep. 67, 85–90 (2018).
Kocevska, D. et al. Sleep characteristics across the lifespan in 1.1 million people from the Netherlands, United Kingdom and United States: a systematic review and meta-analysis. Nat. Hum. Behav. https://doi.org/10.1038/s41562-020-00965-x (2020).
Pandi-Perumal, S. R. et al. Racial/ethnic and social inequities in sleep medicine: the tip of the iceberg? J. Natl Med. Assoc. 109, 279–286 (2017).
Irwin, M. R. Why sleep is important for health: a psychoneuroimmunology perspective. Annu. Rev. Psychol. 66, 143–172 (2015).
Vgontzas, A. N., Liao, D., Bixler, E. O., Chrousos, G. P. & Vela-Bueno, A. Insomnia with objective short sleep duration is associated with a high risk for hypertension. Sleep 32, 491–497 (2009).
Vgontzas, A. N., Fernandez-Mendoza, J., Liao, D. & Bixler, E. O. Insomnia with objective short sleep duration: the most biologically severe phenotype of the disorder. Sleep. Med. Rev. 17, 241–254 (2013).
Smagula, S. F. et al. Actigraphy- and polysomnography-measured sleep disturbances, inflammation, and mortality among older men. Psychosom. Med. 78, 686–696 (2016).
Cappuccio, F. P. & Miller, M. A. Sleep and cardio-metabolic disease. Curr. Cardiol. Rep. 19, 110 (2017).
Cappuccio, F. P., Cooper, D., D’Elia, L., Strazzullo, P. & Miller, M. A. Sleep duration predicts cardiovascular outcomes: a systematic review and meta-analysis of prospective studies. Eur. Heart J. 32, 1484–1492 (2011).
von Ruesten, A., Weikert, C., Fietze, I. & Boeing, H. Association of sleep duration with chronic diseases in the European Prospective Investigation into Cancer and Nutrition (EPIC)-Potsdam study. PLoS ONE 7, e30972 (2012).
Tobaldini, E. et al. Sleep, sleep deprivation, autonomic nervous system and cardiovascular diseases. Neurosci. Biobehav. Rev. 74, 321–329 (2017).
Kochanek, K. D., Xu, J. & Arias, E. Mortality in the United States, 2019. NCHS Data Brief No. 395, 1–8 (CDC, 2020).
Kakizaki, M. et al. Sleep duration and the risk of breast cancer: the Ohsaki Cohort Study. Br. J. Cancer 99, 1502–1505 (2008).
Ahmadian, N., Hejazi, S., Mahmoudi, J. & Talebi, M. Tau pathology of Alzheimer disease: possible role of sleep deprivation. Basic Clin. Neurosci. 9, 307–316 (2018).
Baglioni, C. et al. Insomnia as a predictor of depression: a meta-analytic evaluation of longitudinal epidemiological studies. J. Affect. Disord. 135, 10–19 (2011).
Ben Simon, E., Vallat, R., Barnes, C. M. & Walker, M. P. Sleep loss and the socio-emotional brain. Trends Cogn. Sci. 24, 435–450 (2020).
Bernert, R. A., Kim, J. S., Iwata, N. G. & Perlis, M. L. Sleep disturbances as an evidence-based suicide risk factor. Curr. Psychiatry Rep. 17, 554 (2015).
Fredriksen, K., Rhodes, J., Reddy, R. & Way, N. Sleepless in Chicago: tracking the effects of adolescent sleep loss during the middle school years. Child Dev. 75, 84–95 (2004).
Tomaso, C. C., Johnson, A. B. & Nelson, T. D. The effect of sleep deprivation and restriction on mood, emotion, and emotion regulation: three meta-analyses in one. Sleep 44, https://doi.org/10.1093/sleep/zsaa289 (2021).
Waters, F., Chiu, V., Atkinson, A. & Blom, J. D. Severe sleep deprivation causes hallucinations and a gradual progression toward psychosis with increasing time awake. Front. Psychiatry 9, 303 (2018).
Cappuccio, F. P., D’Elia, L., Strazzullo, P. & Miller, M. A. Sleep duration and all-cause mortality: a systematic review and meta-analysis of prospective studies. Sleep 33, 585–592 (2010).
Cappuccio, F. P., D’Elia, L., Strazzullo, P. & Miller, M. A. Quantity and quality of sleep and incidence of type 2 diabetes: a systematic review and meta-analysis. Diabetes Care 33, 414–420 (2010).
Bacaro, V. et al. Sleep duration and obesity in adulthood: an updated systematic review and meta-analysis. Obes. Res. Clin. Pract. 14, 301–309 (2020).
Bishir, M. et al. Sleep deprivation and neurological disorders. Biomed. Res. Int. 2020, 5764017 (2020).
Mullington, J. M., Simpson, N. S., Meier-Ewert, H. K. & Haack, M. Sleep loss and inflammation. Best. Pract. Res. Clin. Endocrinol. Metab. 24, 775–784 (2010).
Aldabal, L. & Bahammam, A. S. Metabolic, endocrine, and immune consequences of sleep deprivation. Open Respir. Med. J. 5, 31–43 (2011).
Dantzer, R. Neuroimmune interactions: from the brain to the immune system and vice versa. Physiol. Rev. 98, 477–504 (2018).
Irwin, M. R. Sleep and inflammation: partners in sickness and in health. Nat. Rev. Immunol. 19, 702–715 (2019).
Hand, T. W., Vujkovic-Cvijin, I., Ridaura, V. K. & Belkaid, Y. Linking the microbiota, chronic disease, and the immune system. Trends Endocrinol. Metab. 27, 831–843 (2016).
Irwin, M. R. & Opp, M. R. Sleep health: reciprocal regulation of sleep and innate immunity. Neuropsychopharmacology 42, 129–155 (2017).
Miller, M. A. & Cappuccio, F. P. Inflammation, sleep, obesity and cardiovascular disease. Curr. Vasc. Pharm. 5, 93–102 (2007).
Krueger, J. M., Pappenheimer, J. R. & Karnovsky, M. L. The composition of sleep-promoting factor isolated from human urine. J. Biol. Chem. 257, 1664–1669 (1982).
Mullington, J. et al. Dose-dependent effects of endotoxin on human sleep. Am. J. Physiol. Regul. Integr. Comp. Physiol. 278, R947–955 (2000).
Zielinski, M. R. & Krueger, J. M. Sleep and innate immunity. Front. Biosci. (Sch. Ed.) 3, 632–642 (2011).
Opp, M. R. Cytokines and sleep. Sleep. Med. Rev. 9, 355–364 (2005).
Urade, Y. & Hayaishi, O. Prostaglandin D2 and sleep/wake regulation. Sleep. Med. Rev. 15, 411–418 (2011).
Krueger, J. M., Majde, J. A. & Rector, D. M. Cytokines in immune function and sleep regulation. Handb. Clin. Neurol. 98, 229–240 (2011).
Besedovsky, L., Lange, T. & Haack, M. The sleep-immune crosstalk in health and disease. Physiol. Rev. 99, 1325–1380 (2019).
Opp, M. R., Obal, F. Jr. & Krueger, J. M. Interleukin 1 alters rat sleep: temporal and dose-related effects. Am. J. Physiol. 260, R52–58 (1991).
Krueger, J. M. & Obal, F. Jr. Growth hormone-releasing hormone and interleukin-1 in sleep regulation. FASEB J. 7, 645–652 (1993).
De, A., Churchill, L., Obal, F. Jr., Simasko, S. M. & Krueger, J. M. GHRH and IL1beta increase cytoplasmic Ca(2+) levels in cultured hypothalamic GABAergic neurons. Brain Res. 949, 209–212 (2002).
Kubota, T., Fang, J., Kushikata, T. & Krueger, J. M. Interleukin-13 and transforming growth factor-beta1 inhibit spontaneous sleep in rabbits. Am. J. Physiol. Regul. Integr. Comp. Physiol. 279, R786–792 (2000).
Kubota, T., Fang, J., Guan, Z., Brown, R. A. & Krueger, J. M. Vagotomy attenuates tumor necrosis factor-alpha-induced sleep and EEG delta-activity in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 280, R1213–1220 (2001).
Curfs, J. H., Meis, J. F. & Hoogkamp-Korstanje, J. A. A primer on cytokines: sources, receptors, effects, and inducers. Clin. Microbiol. Rev. 10, 742–780 (1997).
Hodo, T. W., de Aquino, M. T. P., Shimamoto, A. & Shanker, A. Critical neurotransmitters in the neuroimmune network. Front Immunol. 11, 1869 (2020).
Huang, X., Hussain, B. & Chang, J. Peripheral inflammation and blood-brain barrier disruption: effects and mechanisms. CNS Neurosci. Ther. 27, 36–47 (2021).
Grill, H. J. & Hayes, M. R. Hindbrain neurons as an essential hub in the neuroanatomically distributed control of energy balance. Cell Metab. 16, 296–309 (2012).
Kunz, N. & Kemper, C. Complement has brains-do intracellular complement and immunometabolism cooperate in tissue homeostasis and behavior? Front. Immunol. 12, 629986 (2021).
Levin, S. G. & Godukhin, O. V. Modulating effect of cytokines on mechanisms of synaptic plasticity in the brain. Biochemistry (Mosc.) 82, 264–274 (2017).
Wang, Y. et al. Astrocyte-secreted IL-33 mediates homeostatic synaptic plasticity in the adult hippocampus. Proc. Natl Acad. Sci. USA 118, https://doi.org/10.1073/pnas.2020810118 (2021).
Turnbull, A. V. & Rivier, C. Regulation of the HPA axis by cytokines. Brain Behav. Immun. 9, 253–275 (1995).
Besedovsky, H. O. & del Rey, A. The cytokine-HPA axis feed-back circuit. Z. Rheumatol. 59(Suppl 2), II/26–30 (2000).
Nguyen, J. T. et al. The neuron-specific interleukin-1 receptor accessory protein alters emergent network state properties in vitro. Neurobiol. Sleep. Circadian Rhythms 6, 35–43 (2019).
Krueger, J. M. & Obal, F. A neuronal group theory of sleep function. J. Sleep. Res. 2, 63–69 (1993).
Rector, D. M., Topchiy, I. A., Carter, K. M. & Rojas, M. J. Local functional state differences between rat cortical columns. Brain Res. 1047, 45–55 (2005).
Roy, S., Krueger, J. M., Rector, D. M. & Wan, Y. A network model for activity-dependent sleep regulation. J. Theor. Biol. 253, 462–468 (2008).
Krueger, J. M., Huang, Y. H., Rector, D. M. & Buysse, D. J. Sleep: a synchrony of cell activity-driven small network states. Eur. J. Neurosci. 38, 2199–2209 (2013).
Krueger, J. M. Sleep and circadian rhythms: evolutionary entanglement and local regulation. Neurobiol. Sleep. Circadian Rhythms 9, 100052 (2020).
Krueger, J. M., Nguyen, J. T., Dykstra-Aiello, C. J. & Taishi, P. Local sleep. Sleep. Med. Rev. 43, 14–21 (2019).
Taishi, P., Churchill, L., De, A., Obal, F. Jr. & Krueger, J. M. Cytokine mRNA induction by interleukin-1beta or tumor necrosis factor alpha in vitro and in vivo. Brain Res. 1226, 89–98 (2008).
Rockstrom, M. D. et al. Tumor necrosis factor alpha in sleep regulation. Sleep. Med. Rev. 40, 69–78 (2018).
Krueger, J. M., Walter, J., Dinarello, C. A., Wolff, S. M. & Chedid, L. Sleep-promoting effects of endogenous pyrogen (interleukin-1). Am. J. Physiol. 246, R994–999 (1984).
Imeri, L. & Opp, M. R. How (and why) the immune system makes us sleep. Nat. Rev. Neurosci. 10, 199–210 (2009).
Jewett, K. A. & Krueger, J. M. Humoral sleep regulation; interleukin-1 and tumor necrosis factor. Vitam. Horm. 89, 241–257 (2012).
Davis, C. J. et al. The neuron-specific interleukin-1 receptor accessory protein is required for homeostatic sleep and sleep responses to influenza viral challenge in mice. Brain Behav. Immun. 47, 35–43 (2015).
Churchill, L. et al. Brain distribution of cytokine mRNA induced by systemic administration of interleukin-1beta or tumor necrosis factor alpha. Brain Res. 1120, 64–73 (2006).
Taishi, P., Bredow, S., Guha-Thakurta, N., Obal, F. Jr. & Krueger, J. M. Diurnal variations of interleukin-1 beta mRNA and beta-actin mRNA in rat brain. J. Neuroimmunol. 75, 69–74 (1997).
Bredow, S., Guha-Thakurta, N., Taishi, P., Obal, F. Jr. & Krueger, J. M. Diurnal variations of tumor necrosis factor alpha mRNA and alpha-tubulin mRNA in rat brain. Neuroimmunomodulation 4, 84–90 (1997).
Floyd, R. A. & Krueger, J. M. Diurnal variation of TNF alpha in the rat brain. Neuroreport 8, 915–918 (1997).
Garlanda, C., Dinarello, C. A. & Mantovani, A. The interleukin-1 family: back to the future. Immunity 39, 1003–1018 (2013).
Smith, D. E. et al. A central nervous system-restricted isoform of the interleukin-1 receptor accessory protein modulates neuronal responses to interleukin-1. Immunity 30, 817–831 (2009).
Taishi, P. et al. Brain-specific interleukin-1 receptor accessory protein in sleep regulation. J. Appl Physiol. (1985) 112, 1015–1022 (2012).
Oles, V. et al. Sleep- and time of day-linked RNA transcript expression in wild-type and IL1 receptor accessory protein-null mice. J. Appl. Physiol. (1985) 128, 1506–1522 (2020).
Dykstra-Aiello, C. et al. A wake-like state in vitro induced by transmembrane TNF/soluble TNF receptor reverse signaling. Brain Behav. Immun. 94, 245–258 (2021).
Guan, Y. et al. Astrocytes constitute the major TNF-alpha-producing cell population in the infarct cortex in dMCAO rats receiving intravenous MSC infusion. Biomed. Pharmacother. 142, 111971 (2021).
Churchill, L. et al. Unilateral cortical application of tumor necrosis factor alpha induces asymmetry in Fos- and interleukin-1beta-immunoreactive cells within the corticothalamic projection. Brain Res. 1055, 15–24 (2005).
Jewett, K. A. et al. Tumor necrosis factor enhances the sleep-like state and electrical stimulation induces a wake-like state in co-cultures of neurons and glia. Eur. J. Neurosci. 42, 2078–2090 (2015).
Schmidt, M. H. The energy allocation function of sleep: a unifying theory of sleep, torpor, and continuous wakefulness. Neurosci. Biobehav. Rev. 47, 122–153 (2014).
Sharpley, A. L., Cooper, C. M., Williams, C., Godlewska, B. R. & Cowen, P. J. Effects of typhoid vaccine on inflammation and sleep in healthy participants: a double-blind, placebo-controlled, crossover study. Psychopharmacology (Berl.) 233, 3429–3435 (2016).
Toth, L. A., Tolley, E. A. & Krueger, J. M. Sleep as a prognostic indicator during infectious disease in rabbits. Proc. Soc. Exp. Biol. Med. 203, 179–192 (1993).
Toth, L. A. & Krueger, J. M. Alteration of sleep in rabbits by Staphylococcus aureus infection. Infect. Immun. 56, 1785–1791 (1988).
Seke Etet, P. F. et al. Sleep and rhythm changes at the time of Trypanosoma brucei invasion of the brain parenchyma in the rat. Chronobiol. Int. 29, 469–481 (2012).
Toth, L. A., Tolley, E. A., Broady, R., Blakely, B. & Krueger, J. M. Sleep during experimental trypanosomiasis in rabbits. Proc. Soc. Exp. Biol. Med. 205, 174–181 (1994).
Patke, A., Young, M. W. & Axelrod, S. Molecular mechanisms and physiological importance of circadian rhythms. Nat. Rev. Mol. Cell Biol. 21, 67–84 (2020).
Cavadini, G. et al. TNF-alpha suppresses the expression of clock genes by interfering with E-box-mediated transcription. Proc. Natl Acad. Sci. USA 104, 12843–12848 (2007).
Meier, D., Lopez, M., Franken, P. & Fontana, A. Twist1 is a TNF-inducible inhibitor of clock mediated activation of period genes. PLoS ONE 10, e0137229 (2015).
Marpegan, L., Bekinschtein, T. A., Costas, M. A. & Golombek, D. A. Circadian responses to endotoxin treatment in mice. J. Neuroimmunol. 160, 102–109 (2005).
Yamamura, Y., Yano, I., Kudo, T. & Shibata, S. Time-dependent inhibitory effect of lipopolysaccharide injection on Per1 and Per2 gene expression in the mouse heart and liver. Chronobiol. Int. 27, 213–232 (2010).
Wang, Y. et al. Endotoxin disrupts circadian rhythms in macrophages via reactive oxygen species. PLoS ONE 11, e0155075 (2016).
Friese, R. S., Bruns, B. & Sinton, C. M. Sleep deprivation after septic insult increases mortality independent of age. J. Trauma 66, 50–54 (2009).
Everson, C. A. & Toth, L. A. Systemic bacterial invasion induced by sleep deprivation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 278, R905–916 (2000).
Opp, M. R. Sleeping to fuel the immune system: mammalian sleep and resistance to parasites. BMC Evol. Biol. 9, 8 (2009).
Prather, A. A., Janicki-Deverts, D., Hall, M. H. & Cohen, S. Behaviorally assessed sleep and susceptibility to the common cold. Sleep 38, 1353–1359 (2015).
Fernandes, E. R. et al. Sleep disturbance during infection compromises Tfh differentiation and impacts host immunity. iScience 23, 101599 (2020).
Chung, W. S., Lin, H. H. & Cheng, N. C. The incidence and risk of herpes zoster in patients with sleep disorders: a population-based cohort study. Medicine (Baltimore) 95, e2195 (2016).
Patel, S. R. et al. A prospective study of sleep duration and pneumonia risk in women. Sleep 35, 97–101 (2012).
Prather, A. A. & Leung, C. W. Association of insufficient sleep with respiratory infection among adults in the United States. JAMA Intern. Med. 176, 850–852 (2016).
Cohen, S., Doyle, W. J., Alper, C. M., Janicki-Deverts, D. & Turner, R. B. Sleep habits and susceptibility to the common cold. Arch. Intern. Med. 169, 62–67 (2009).
Orzech, K. M., Acebo, C., Seifer, R., Barker, D. & Carskadon, M. A. Sleep patterns are associated with common illness in adolescents. J. Sleep. Res. 23, 133–142 (2014).
Zhang, Y., Xie, B., Chen, X., Zhang, J. & Yuan, S. A key role of gut microbiota-vagus nerve/spleen axis in sleep deprivation-mediated aggravation of systemic inflammation after LPS administration. Life Sci. 265, 118736 (2021).
Lange, T., Dimitrov, S., Bollinger, T., Diekelmann, S. & Born, J. Sleep after vaccination boosts immunological memory. J. Immunol. 187, 283–290 (2011).
Prather, A. A. et al. Sleep and antibody response to hepatitis B vaccination. Sleep 35, 1063–1069 (2012).
Renegar, K. B., Floyd, R. A. & Krueger, J. M. Effects of short-term sleep deprivation on murine immunity to influenza virus in young adult and senescent mice. Sleep 21, 241–248 (1998).
Hurley, S., Goldberg, D., Bernstein, L. & Reynolds, P. Sleep duration and cancer risk in women. Cancer Causes Control 26, 1037–1045 (2015).
Kakizaki, M. et al. Sleep duration and the risk of prostate cancer: the Ohsaki Cohort Study. Br. J. Cancer 99, 176–178 (2008).
Jiao, L. et al. Sleep duration and incidence of colorectal cancer in postmenopausal women. Br. J. Cancer 108, 213–221 (2013).
Cao, J. et al. Sleep duration and risk of breast cancer: The JACC Study. Breast Cancer Res. Treat. 174, 219–225 (2019).
Wu, A. H. et al. Sleep duration, melatonin and breast cancer among Chinese women in Singapore. Carcinogenesis 29, 1244–1248 (2008).
Wehr, T. A. The durations of human melatonin secretion and sleep respond to changes in daylength (photoperiod). J. Clin. Endocrinol. Metab. 73, 1276–1280 (1991).
Luo, J. et al. Effect of melatonin on T/B cell activation and immune regulation in pinealectomy mice. Life Sci. 242, 117191 (2020).
De Lorenzo, B. H., de Oliveira Marchioro, L., Greco, C. R. & Suchecki, D. Sleep-deprivation reduces NK cell number and function mediated by beta-adrenergic signalling. Psychoneuroendocrinology 57, 134–143 (2015).
De Lorenzo, B. H. P. et al. Chronic sleep restriction impairs the antitumor immune response in mice. Neuroimmunomodulation 25, 59–67 (2018).
Maragno-Correa, J. M. et al. Sleep deprivation increases mortality in female mice bearing Ehrlich ascitic tumor. Neuroimmunomodulation 20, 134–140 (2013).
Barbosa Vieira, T. K. et al. Correlation between circadian rhythm related genes, type 2 diabetes, and cancer: insights from metanalysis of transcriptomics data. Mol. Cell Endocrinol. 526, 111214 (2021).
Gonzalez, H., Hagerling, C. & Werb, Z. Roles of the immune system in cancer: from tumor initiation to metastatic progression. Genes Dev. 32, 1267–1284 (2018).
Palma, J. A., Urrestarazu, E. & Iriarte, J. Sleep loss as risk factor for neurologic disorders: a review. Sleep. Med. 14, 229–236 (2013).
Sabia, S. et al. Association of sleep duration in middle and old age with incidence of dementia. Nat. Commun. 12, 2289 (2021).
Sadeghmousavi, S., Eskian, M., Rahmani, F. & Rezaei, N. The effect of insomnia on development of Alzheimer’s disease. J. Neuroinflammation 17, 289 (2020).
Stephenson, J., Nutma, E., van der Valk, P. & Amor, S. Inflammation in CNS neurodegenerative diseases. Immunology 154, 204–219 (2018).
Manchanda, S., Singh, H., Kaur, T. & Kaur, G. Low-grade neuroinflammation due to chronic sleep deprivation results in anxiety and learning and memory impairments. Mol. Cell Biochem. 449, 63–72 (2018).
Xue, R. et al. Nicotinic mitigation of neuroinflammation and oxidative stress after chronic sleep deprivation. Front. Immunol. 10, 2546 (2019).
Hurtado-Alvarado, G. et al. The yin/yang of inflammatory status: blood-brain barrier regulation during sleep. Brain Behav. Immun. 69, 154–166 (2018).
Korin, B. et al. Short-term sleep deprivation in mice induces B cell migration to the brain compartment. Sleep 43, https://doi.org/10.1093/sleep/zsz222 (2020).
Luo, F. et al. Melatonin and autophagy in aging-related neurodegenerative diseases. Int. J. Mol. Sci. 21, https://doi.org/10.3390/ijms21197174 (2020).
Finkbeiner, E., Haindl, M., Raman, N. & Muller, S. SUMO routes ribosome maturation. Nucleus 2, 527–532 (2011).
Zhao, H. Y. et al. Chronic sleep restriction induces cognitive deficits and cortical beta-amyloid deposition in mice via BACE1-antisense activation. CNS Neurosci. Ther. 23, 233–240 (2017).
Liu, P. et al. Activation of inflammation is associated with amyloid-beta accumulation induced by chronic sleep restriction in rats. J. Alzheimers Dis. 74, 759–773 (2020).
Shokri-Kojori, E. et al. beta-Amyloid accumulation in the human brain after one night of sleep deprivation. Proc. Natl Acad. Sci. USA 115, 4483–4488 (2018).
Spira, A. P. et al. Self-reported sleep and beta-amyloid deposition in community-dwelling older adults. JAMA Neurol. 70, 1537–1543 (2013).
Ju, Y. S. et al. Slow wave sleep disruption increases cerebrospinal fluid amyloid-beta levels. Brain 140, 2104–2111 (2017).
Chen, D. W., Wang, J., Zhang, L. L., Wang, Y. J. & Gao, C. Y. Cerebrospinal fluid amyloid-beta levels are increased in patients with insomnia. J. Alzheimers Dis. 61, 645–651 (2018).
Ooms, S. et al. Effect of 1 night of total sleep deprivation on cerebrospinal fluid beta-amyloid 42 in healthy middle-aged men: a randomized clinical trial. JAMA Neurol. 71, 971–977 (2014).
Zhao, B. et al. Chronic sleep restriction induces abeta accumulation by disrupting the balance of abeta production and clearance in rats. Neurochem. Res. 44, 859–873 (2019).
Wu, H., Dunnett, S., Ho, Y. S. & Chang, R. C. The role of sleep deprivation and circadian rhythm disruption as risk factors of Alzheimer’s disease. Front. Neuroendocrinol. 54, 100764 (2019).
Jaeger, L. B. et al. Lipopolysaccharide alters the blood-brain barrier transport of amyloid beta protein: a mechanism for inflammation in the progression of Alzheimer’s disease. Brain Behav. Immun. 23, 507–517 (2009).
Sastre, M. et al. Nonsteroidal anti-inflammatory drugs and peroxisome proliferator-activated receptor-gamma agonists modulate immunostimulated processing of amyloid precursor protein through regulation of beta-secretase. J. Neurosci. 23, 9796–9804 (2003).
Mouton-Liger, F. et al. Oxidative stress increases BACE1 protein levels through activation of the PKR-eIF2alpha pathway. Biochim. Biophys. Acta 1822, 885–896 (2012).
Giese, M. et al. BDNF: an indicator of insomnia? Mol. Psychiatry 19, 151–152 (2014).
Tan, D. X., Manchester, L. C., Terron, M. P., Flores, L. J. & Reiter, R. J. One molecule, many derivatives: a never-ending interaction of melatonin with reactive oxygen and nitrogen species? J. Pineal Res. 42, 28–42 (2007).
Zhang, L. et al. Melatonin ameliorates cognitive impairment induced by sleep deprivation in rats: role of oxidative stress, BDNF and CaMKII. Behav. Brain Res. 256, 72–81 (2013).
Fredrich, M., Hampel, M., Seidel, K., Christ, E. & Korf, H. W. Impact of melatonin receptor-signaling on Zeitgeber time-dependent changes in cell proliferation and apoptosis in the adult murine. Hippocampus 27, 495–506 (2017).
Alghamdi, B. S. The neuroprotective role of melatonin in neurological disorders. J. Neurosci. Res. 96, 1136–1149 (2018).
Luengo, E. et al. Pharmacological doses of melatonin impede cognitive decline in tau-related Alzheimer models, once tauopathy is initiated, by restoring the autophagic flux. J. Pineal Res. 67, e12578 (2019).
Emamian, F. et al. The association between obstructive sleep apnea and Alzheimer’s disease: a meta-analysis perspective. Front. Aging Neurosci. 8, 78 (2016).
Osorio, R. S. et al. Sleep-disordered breathing advances cognitive decline in the elderly. Neurology 84, 1964–1971 (2015).
Frenkel, D. et al. Scara1 deficiency impairs clearance of soluble amyloid-beta by mononuclear phagocytes and accelerates Alzheimer’s-like disease progression. Nat. Commun. 4, 2030 (2013).
Musiek, E. S. & Holtzman, D. M. Mechanisms linking circadian clocks, sleep, and neurodegeneration. Science 354, 1004–1008 (2016).
Wang, J., Gu, B. J., Masters, C. L. & Wang, Y. J. A systemic view of Alzheimer disease - insights from amyloid-beta metabolism beyond the brain. Nat. Rev. Neurol. 13, 703 (2017).
Tanner, C. M. & Aston, D. A. Epidemiology of Parkinson’s disease and akinetic syndromes. Curr. Opin. Neurol. 13, 427–430 (2000).
Barber, A. & Dashtipour, K. Sleep disturbances in Parkinson’s disease with emphasis on rapid eye movement sleep behavior disorder. Int. J. Neurosci. 122, 407–412 (2012).
Barnett, R. Parkinson’s disease. Lancet 387, 217 (2016).
Hsiao, Y. H. et al. Sleep disorders and an increased risk of Parkinson’s disease in individuals with non-apnea sleep disorders: a population-based cohort study. J. Sleep. Res. 26, 623–628 (2017).
Bohnen, N. I. & Hu, M. T. M. Sleep disturbance as potential risk and progression factor for Parkinson’s disease. J. Parkinsons Dis. 9, 603–614 (2019).
Su, L. Y. et al. Melatonin attenuates MPTP-induced neurotoxicity via preventing CDK5-mediated autophagy and SNCA/alpha-synuclein aggregation. Autophagy 11, 1745–1759 (2015).
Srivastava, A. K., Roy Choudhury, S. & Karmakar, S. Melatonin/polydopamine nanostructures for collective neuroprotection-based Parkinson’s disease therapy. Biomater. Sci. 8, 1345–1363 (2020).
Abad, V. C., Sarinas, P. S. & Guilleminault, C. Sleep and rheumatologic disorders. Sleep. Med. Rev. 12, 211–228 (2008).
Hsiao, Y. H. et al. Sleep disorders and increased risk of autoimmune diseases in individuals without sleep apnea. Sleep 38, 581–586 (2015).
Young, K. A. et al. Less than 7 hours of sleep per night is associated with transitioning to systemic lupus erythematosus. Lupus 27, 1524–1531 (2018).
Palma, B. D., Gabriel, A. Jr., Colugnati, F. A. & Tufik, S. Effects of sleep deprivation on the development of autoimmune disease in an experimental model of systemic lupus erythematosus. Am. J. Physiol. Regul. Integr. Comp. Physiol. 291, R1527–1532 (2006).
Alarcon-Riquelme, M. E., Moller, G. & Fernandez, C. Age-dependent responsiveness to interleukin-6 in B lymphocytes from a systemic lupus erythematosus-prone (NZB x NZW)F1 hybrid. Clin. Immunol. Immunopathol. 62, 264–269 (1992).
Yehuda, S., Sredni, B., Carasso, R. L. & Kenigsbuch-Sredni, D. REM sleep deprivation in rats results in inflammation and interleukin-17 elevation. J. Interferon Cytokine Res. 29, 393–398 (2009).
Acosta-Rodriguez, E. V., Napolitani, G., Lanzavecchia, A. & Sallusto, F. Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat. Immunol. 8, 942–949 (2007).
Volin, M. V. & Shahrara, S. Role of TH-17 cells in rheumatic and other autoimmune diseases. Rheumatology (Sunnyvale) 1, https://doi.org/10.4172/2161-1149.1000104 (2011).
Bollinger, T. et al. Sleep-dependent activity of T cells and regulatory T cells. Clin. Exp. Immunol. 155, 231–238 (2009).
Carbone, F. et al. Regulatory T cell proliferative potential is impaired in human autoimmune disease. Nat. Med. 20, 69–74 (2014).
Leng, Y. et al. Sleep duration and risk of fatal and nonfatal stroke: a prospective study and meta-analysis. Neurology 84, 1072–1079 (2015).
Ferrie, J. E. et al. Associations between change in sleep duration and inflammation: findings on C-reactive protein and interleukin 6 in the Whitehall II Study. Am. J. Epidemiol. 178, 956–961 (2013).
Wang, Y. H. et al. Association of longitudinal patterns of habitual sleep duration with risk of cardiovascular events and all-cause mortality. JAMA Netw. Open 3, e205246 (2020).
Dominguez, F. et al. Association of sleep duration and quality with subclinical atherosclerosis. J. Am. Coll. Cardiol. 73, 134–144 (2019).
Cappuccio, F. P. et al. Meta-analysis of short sleep duration and obesity in children and adults. Sleep 31, 619–626 (2008).
Meng, L., Zheng, Y. & Hui, R. The relationship of sleep duration and insomnia to risk of hypertension incidence: a meta-analysis of prospective cohort studies. Hypertens. Res. 36, 985–995 (2013).
Anothaisintawee, T., Reutrakul, S., Van Cauter, E. & Thakkinstian, A. Sleep disturbances compared to traditional risk factors for diabetes development: systematic review and meta-analysis. Sleep. Med. Rev. 30, 11–24 (2016).
Zhong, X. et al. Increased sympathetic and decreased parasympathetic cardiovascular modulation in normal humans with acute sleep deprivation. J. Appl. Physiol. (1985) 98, 2024–2032 (2005).
Meier-Ewert, H. K. et al. Effect of sleep loss on C-reactive protein, an inflammatory marker of cardiovascular risk. J. Am. Coll. Cardiol. 43, 678–683 (2004).
Sauvet, F. et al. Effect of acute sleep deprivation on vascular function in healthy subjects. J. Appl. Physiol. (1985) 108, 68–75 (2010).
Dettoni, J. L. et al. Cardiovascular effects of partial sleep deprivation in healthy volunteers. J. Appl. Physiol. (1985) 113, 232–236 (2012).
Spiegel, K., Tasali, E., Leproult, R. & Van Cauter, E. Effects of poor and short sleep on glucose metabolism and obesity risk. Nat. Rev. Endocrinol. 5, 253–261 (2009).
Buxton, O. M. et al. Adverse metabolic consequences in humans of prolonged sleep restriction combined with circadian disruption. Sci. Transl. Med. 4, 129ra143 (2012).
Broussard, J. L., Ehrmann, D. A., Van Cauter, E., Tasali, E. & Brady, M. J. Impaired insulin signaling in human adipocytes after experimental sleep restriction: a randomized, crossover study. Ann. Intern. Med. 157, 549–557 (2012).
Nedeltcheva, A. V., Kilkus, J. M., Imperial, J., Schoeller, D. A. & Penev, P. D. Insufficient sleep undermines dietary efforts to reduce adiposity. Ann. Intern. Med. 153, 435–441 (2010).
Darukhanavala, A. et al. Changes in insulin secretion and action in adults with familial risk for type 2 diabetes who curtail their sleep. Diabetes Care 34, 2259–2264 (2011).
Esser, N., Legrand-Poels, S., Piette, J., Scheen, A. J. & Paquot, N. Inflammation as a link between obesity, metabolic syndrome and type 2 diabetes. Diabetes Res. Clin. Pract. 105, 141–150 (2014).
Longo, M. et al. Adipose tissue dysfunction as determinant of obesity-associated metabolic complications. Int. J. Mol. Sci. 20, https://doi.org/10.3390/ijms20092358 (2019).
Freitas Lima, L. C. et al. Adipokines, diabetes and atherosclerosis: an inflammatory association. Front. Physiol. 6, 304 (2015).
Geovanini, G. R. & Libby, P. Atherosclerosis and inflammation: overview and updates. Clin. Sci. (Lond.) 132, 1243–1252 (2018).
Yousuf, O. et al. High-sensitivity C-reactive protein and cardiovascular disease: a resolute belief or an elusive link? J. Am. Coll. Cardiol. 62, 397–408 (2013).
van Leeuwen, W. M. et al. Sleep restriction increases the risk of developing cardiovascular diseases by augmenting proinflammatory responses through IL-17 and CRP. PLoS ONE 4, e4589 (2009).
Irwin, M. R., Olmstead, R. & Carroll, J. E. Sleep disturbance, sleep duration, and inflammation: a systematic review and meta-analysis of cohort studies and experimental sleep deprivation. Biol. Psychiatry 80, 40–52 (2016).
McAlpine, C. S. et al. Sleep modulates haematopoiesis and protects against atherosclerosis. Nature 566, 383–387 (2019).
Dutta, P. et al. Myocardial infarction accelerates atherosclerosis. Nature 487, 325–329 (2012).
Kadoya, M. & Koyama, H. Sleep, autonomic Nnervous function and atherosclerosis. Int. J. Mol. Sci. 20, https://doi.org/10.3390/ijms20040794 (2019).
Yang, X. et al. Oxidative stress-mediated atherosclerosis: mechanisms and therapies. Front. Physiol. 8, 600 (2017).
Li, H. Y., Leu, Y. L., Wu, Y. C. & Wang, S. H. Melatonin inhibits in vitro smooth muscle cell inflammation and proliferation and atherosclerosis in apolipoprotein E-deficient mice. J. Agric. Food Chem. 67, 1889–1901 (2019).
Born, J., Lange, T., Hansen, K., Molle, M. & Fehm, H. L. Effects of sleep and circadian rhythm on human circulating immune cells. J. Immunol. 158, 4454–4464 (1997).
Dimitrov, S., Lange, T., Nohroudi, K. & Born, J. Number and function of circulating human antigen presenting cells regulated by sleep. Sleep 30, 401–411 (2007).
Lasselin, J., Rehman, J. U., Akerstedt, T., Lekander, M. & Axelsson, J. Effect of long-term sleep restriction and subsequent recovery sleep on the diurnal rhythms of white blood cell subpopulations. Brain Behav. Immun. 47, 93–99 (2015).
Said, E. A. et al. Sleep deprivation alters neutrophil functions and levels of Th1-related chemokines and CD4(+) T cells in the blood. Sleep. Breath. 23, 1331–1339 (2019).
Dinges, D. F. et al. Leukocytosis and natural killer cell function parallel neurobehavioral fatigue induced by 64 hours of sleep deprivation. J. Clin. Invest. 93, 1930–1939 (1994).
Scheiermann, C., Kunisaki, Y. & Frenette, P. S. Circadian control of the immune system. Nat. Rev. Immunol. 13, 190–198 (2013).
Redwine, L., Dang, J. & Irwin, M. Cellular adhesion molecule expression, nocturnal sleep, and partial night sleep deprivation. Brain Behav. Immun. 18, 333–340 (2004).
Axelsson, J. et al. Effects of sustained sleep restriction on mitogen-stimulated cytokines, chemokines and T helper 1/ T helper 2 balance in humans. PLoS ONE 8, e82291 (2013).
Shearer, W. T. et al. Soluble TNF-alpha receptor 1 and IL-6 plasma levels in humans subjected to the sleep deprivation model of spaceflight. J. Allergy Clin. Immunol. 107, 165–170 (2001).
Hu, J. et al. Sleep-deprived mice show altered cytokine production manifest by perturbations in serum IL-1ra, TNFa, and IL-6 levels. Brain Behav. Immun. 17, 498–504 (2003).
Vgontzas, A. N. et al. Adverse effects of modest sleep restriction on sleepiness, performance, and inflammatory cytokines. J. Clin. Endocrinol. Metab. 89, 2119–2126 (2004).
Miller, M. A. et al. Gender differences in the cross-sectional relationships between sleep duration and markers of inflammation: Whitehall II study. Sleep 32, 857–864 (2009).
Patel, S. R. et al. Sleep duration and biomarkers of inflammation. Sleep 32, 200–204 (2009).
Perez de Heredia, F. et al. Self-reported sleep duration, white blood cell counts and cytokine profiles in European adolescents: the HELENA study. Sleep. Med. 15, 1251–1258 (2014).
Bakour, C. et al. Sleep duration trajectories and systemic inflammation in young adults: results from the National Longitudinal Study of Adolescent to Adult Health (Add Health). Sleep 40, https://doi.org/10.1093/sleep/zsx156 (2017).
Richardson, M. R. & Churilla, J. R. Sleep duration and C-reactive protein in US adults. South Med. J. 110, 314–317 (2017).
Fondell, E. et al. Short natural sleep is associated with higher T cell and lower NK cell activities. Brain Behav. Immun. 25, 1367–1375 (2011).
Carroll, J. E. et al. Epigenetic aging and immune senescence in women with insomnia symptoms: findings from the Women’s Health Initiative Study. Biol. Psychiatry 81, 136–144 (2017).
Jackowska, M. et al. Short sleep duration is associated with shorter telomere length in healthy men: findings from the Whitehall II cohort study. PLoS ONE 7, e47292 (2012).
James, S. et al. Sleep duration and telomere length in children. J. Pediatr. 187, 247–252 e241 (2017).
Hall, M. H. et al. Association between sleep duration and mortality is mediated by markers of inflammation and health in older adults: the Health, Aging and Body Composition Study. Sleep 38, 189–195 (2015).
Li, Y. et al. Hs-CRP and all-cause, cardiovascular, and cancer mortality risk: a meta-analysis. Atherosclerosis 259, 75–82 (2017).
Uthgenannt, D., Schoolmann, D., Pietrowsky, R., Fehm, H. L. & Born, J. Effects of sleep on the production of cytokines in humans. Psychosom. Med. 57, 97–104 (1995).
Irwin, M. R., Wang, M., Campomayor, C. O., Collado-Hidalgo, A. & Cole, S. Sleep deprivation and activation of morning levels of cellular and genomic markers of inflammation. Arch. Intern. Med. 166, 1756–1762 (2006).
Simpson, N. S. et al. Repeating patterns of sleep restriction and recovery: do we get used to it? Brain Behav. Immun. 58, 142–151 (2016).
Dimitrov, S., Besedovsky, L., Born, J. & Lange, T. Differential acute effects of sleep on spontaneous and stimulated production of tumor necrosis factor in men. Brain Behav. Immun. 47, 201–210 (2015).
Dimitrov, S., Lange, T., Tieken, S., Fehm, H. L. & Born, J. Sleep associated regulation of T helper 1/T helper 2 cytokine balance in humans. Brain Behav. Immun. 18, 341–348 (2004).
Ginaldi, L. et al. The immune system in the elderly: I. Specific humoral immunity. Immunol. Res 20, 101–108 (1999).
Redwine, L., Dang, J., Hall, M. & Irwin, M. Disordered sleep, nocturnal cytokines, and immunity in alcoholics. Psychosom. Med. 65, 75–85 (2003).
Sakami, S. et al. Coemergence of insomnia and a shift in the Th1/Th2 balance toward Th2 dominance. Neuroimmunomodulation 10, 337–343 (2002).
Moser, E. K., Field, N. S. & Oliver, P. M. Aberrant Th2 inflammation drives dysfunction of alveolar macrophages and susceptibility to bacterial pneumonia. Cell Mol. Immunol. 15, 480–492 (2018).
Disis, M. L. Immune regulation of cancer. J. Clin. Oncol. 28, 4531–4538 (2010).
Lee, H. L. et al. Inflammatory cytokines and change of Th1/Th2 balance as prognostic indicators for hepatocellular carcinoma in patients treated with transarterial chemoembolization. Sci. Rep. 9, 3260 (2019).
Villafuerte, G. et al. Sleep deprivation and oxidative stress in animal models: a systematic review. Oxid. Med. Cell Longev. 2015, 234952 (2015).
Teixeira, K. R. C. et al. Night workers have lower levels of antioxidant defenses and higher levels of oxidative stress damage when compared to day workers. Sci. Rep. 9, 4455 (2019).
Trivedi, M. S., Holger, D., Bui, A. T., Craddock, T. J. A. & Tartar, J. L. Short-term sleep deprivation leads to decreased systemic redox metabolites and altered epigenetic status. PLoS ONE 12, e0181978 (2017).
Chen, X., Song, M., Zhang, B. & Zhang, Y. Reactive oxygen species regulate T cell immune response in the tumor microenvironment. Oxid. Med. Cell Longev. 2016, 1580967 (2016).
Liguori, I. et al. Oxidative stress, aging, and diseases. Clin. Inter. Aging 13, 757–772 (2018).
Irwin, M., Thompson, J., Miller, C., Gillin, J. C. & Ziegler, M. Effects of sleep and sleep deprivation on catecholamine and interleukin-2 levels in humans: clinical implications. J. Clin. Endocrinol. Metab. 84, 1979–1985 (1999).
Nance, D. M. & Sanders, V. M. Autonomic innervation and regulation of the immune system (1987-2007). Brain Behav. Immun. 21, 736–745 (2007).
Barnes, M. A., Carson, M. J. & Nair, M. G. Non-traditional cytokines: How catecholamines and adipokines influence macrophages in immunity, metabolism and the central nervous system. Cytokine 72, 210–219 (2015).
Grebe, K. M. et al. Cutting edge: sympathetic nervous system increases proinflammatory cytokines and exacerbates influenza A virus pathogenesis. J. Immunol. 184, 540–544 (2010).
Collado-Hidalgo, A., Sung, C. & Cole, S. Adrenergic inhibition of innate anti-viral response: PKA blockade of Type I interferon gene transcription mediates catecholamine support for HIV-1 replication. Brain Behav. Immun. 20, 552–563 (2006).
Kalinichenko, V. V., Mokyr, M. B., Graf, L. H. Jr., Cohen, R. L. & Chambers, D. A. Norepinephrine-mediated inhibition of antitumor cytotoxic T lymphocyte generation involves a beta-adrenergic receptor mechanism and decreased TNF-alpha gene expression. J. Immunol. 163, 2492–2499 (1999).
Irwin, M. R. & Cole, S. W. Reciprocal regulation of the neural and innate immune systems. Nat. Rev. Immunol. 11, 625–632 (2011).
Mishra, I. et al. Chemical sympathectomy reduces peripheral inflammatory responses to acute and chronic sleep fragmentation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 318, R781–R789 (2020).
Wheeler, N. D., Ensminger, D. C., Rowe, M. M., Wriedt, Z. S. & Ashley, N. T. Alpha- and beta- adrenergic receptors regulate inflammatory responses to acute and chronic sleep fragmentation in mice. PeerJ 9, e11616 (2021).
Cirelli, C., Gutierrez, C. M. & Tononi, G. Extensive and divergent effects of sleep and wakefulness on brain gene expression. Neuron 41, 35–43 (2004).
Mackiewicz, M., Zimmerman, J. E., Shockley, K. R., Churchill, G. A. & Pack, A. I. What are microarrays teaching us about sleep? Trends Mol. Med. 15, 79–87 (2009).
Barclay, J. L. et al. Circadian desynchrony promotes metabolic disruption in a mouse model of shiftwork. PLoS ONE 7, e37150 (2012).
Aho, V. et al. Partial sleep restriction activates immune response-related gene expression pathways: experimental and epidemiological studies in humans. PLoS ONE 8, e77184 (2013).
Moller-Levet, C. S. et al. Effects of insufficient sleep on circadian rhythmicity and expression amplitude of the human blood transcriptome. Proc. Natl Acad. Sci. USA 110, E1132–1141 (2013).
Irwin, M. R., Witarama, T., Caudill, M., Olmstead, R. & Breen, E. C. Sleep loss activates cellular inflammation and signal transducer and activator of transcription (STAT) family proteins in humans. Brain Behav. Immun. 47, 86–92 (2015).
Archer, S. N. et al. Mistimed sleep disrupts circadian regulation of the human transcriptome. Proc. Natl Acad. Sci. USA 111, E682–691 (2014).
Mure, L. S. et al. Diurnal transcriptome atlas of a primate across major neural and peripheral tissues. Science 359, https://doi.org/10.1126/science.aao0318 (2018).
Sulli, G., Manoogian, E. N. C., Taub, P. R. & Panda, S. Training the circadian clock, clocking the drugs, and drugging the clock to prevent, manage, and treat chronic diseases. Trends Pharm. Sci. 39, 812–827 (2018).
Christou, S. et al. Circadian regulation in human white adipose tissue revealed by transcriptome and metabolic network analysis. Sci. Rep. 9, 2641 (2019).
Chen, Z., Gardi, J., Kushikata, T., Fang, J. & Krueger, J. M. Nuclear factor-kappaB-like activity increases in murine cerebral cortex after sleep deprivation. Am. J. Physiol. 276, R1812–1818 (1999).
Williams, J. A., Sathyanarayanan, S., Hendricks, J. C. & Sehgal, A. Interaction between sleep and the immune response in Drosophila: a role for the NFkappaB relish. Sleep 30, 389–400 (2007).
Basheer, R., Rainnie, D. G., Porkka-Heiskanen, T., Ramesh, V. & McCarley, R. W. Adenosine, prolonged wakefulness, and A1-activated NF-kappaB DNA binding in the basal forebrain of the rat. Neuroscience 104, 731–739 (2001).
Brandt, J. A. et al. Sleep deprivation increases the activation of nuclear factor kappa B in lateral hypothalamic cells. Brain Res. 1004, 91–97 (2004).
Ramesh, V., Thatte, H. S., McCarley, R. W. & Basheer, R. Adenosine and sleep deprivation promote NF-kappaB nuclear translocation in cholinergic basal forebrain. J. Neurochem. 100, 1351–1363 (2007).
Irwin, M. R. et al. Sleep loss activates cellular inflammatory signaling. Biol. Psychiatry 64, 538–540 (2008).
Madonna, R. & De Caterina, R. Relevance of new drug discovery to reduce NF-kappaB activation in cardiovascular disease. Vasc. Pharm. 57, 41–47 (2012).
Bellet, M. M., Zocchi, L. & Sassone-Corsi, P. The RelB subunit of NFkappaB acts as a negative regulator of circadian gene expression. Cell Cycle 11, 3304–3311 (2012).
Kuo, T. H., Pike, D. H., Beizaeipour, Z. & Williams, J. A. Sleep triggered by an immune response in Drosophila is regulated by the circadian clock and requires the NFkappaB Relish. BMC Neurosci. 11, 17 (2010).
Negi, G., Kumar, A. & Sharma, S. S. Melatonin modulates neuroinflammation and oxidative stress in experimental diabetic neuropathy: effects on NF-kappaB and Nrf2 cascades. J. Pineal Res. 50, 124–131 (2011).
Narasimamurthy, R. et al. Circadian clock protein cryptochrome regulates the expression of proinflammatory cytokines. Proc. Natl Acad. Sci. USA 109, 12662–12667 (2012).
Kolmus, K., Tavernier, J. & Gerlo, S. beta2-Adrenergic receptors in immunity and inflammation: stressing NF-kappaB. Brain Behav. Immun. 45, 297–310 (2015).
Benedict, C. et al. Gut microbiota and glucometabolic alterations in response to recurrent partial sleep deprivation in normal-weight young individuals. Mol. Metab. 5, 1175–1186 (2016).
Poroyko, V. A. et al. Chronic sleep disruption alters gut mmicrobiota, induces systemic and adipose tissue inflammation and insulin resistance in mice. Sci. Rep. 6, 35405 (2016).
Gao, T. et al. Role of melatonin in sleep deprivation-induced intestinal barrier dysfunction in mice. J. Pineal Res. 67, e12574 (2019).
Belkaid, Y. & Hand, T. W. Role of the microbiota in immunity and inflammation. Cell 157, 121–141 (2014).
Belenky, G. et al. Patterns of performance degradation and restoration during sleep restriction and subsequent recovery: a sleep dose-response study. J. Sleep. Res. 12, 1–12 (2003).
Rupp, T. L., Wesensten, N. J., Bliese, P. D. & Balkin, T. J. Banking sleep: realization of benefits during subsequent sleep restriction and recovery. Sleep 32, 311–321 (2009).
Banks, S., Van Dongen, H. P., Maislin, G. & Dinges, D. F. Neurobehavioral dynamics following chronic sleep restriction: dose-response effects of one night for recovery. Sleep 33, 1013–1026 (2010).
Haack, M. et al. Increasing sleep duration to lower beat-to-beat blood pressure: a pilot study. J. Sleep. Res. 22, 295–304 (2013).
Chennaoui, M. et al. Leukocyte expression of type 1 and type 2 purinergic receptors and pro-inflammatory cytokines during total sleep deprivation and/or sleep extension in healthy subjects. Front Neurosci. 11, 240 (2017).
Swinbourne, R., Miller, J., Smart, D., Dulson, D. K. & Gill, N. The effects of sleep extension on sleep, performance, immunity and physical stress in rugby players. Sports (Basel) 6, https://doi.org/10.3390/sports6020042 (2018).
Faraut, B. et al. Benefits of napping and an extended duration of recovery sleep on alertness and immune cells after acute sleep restriction. Brain Behav. Immun. 25, 16–24 (2011).
Takahashi, M. & Arito, H. Maintenance of alertness and performance by a brief nap after lunch under prior sleep deficit. Sleep 23, 813–819 (2000).
Milner, C. E. & Cote, K. A. Benefits of napping in healthy adults: impact of nap length, time of day, age, and experience with napping. J. Sleep. Res. 18, 272–281 (2009).
Keramidas, M. E., Siebenmann, C., Norrbrand, L., Gadefors, M. & Eiken, O. A brief pre-exercise nap may alleviate physical performance impairments induced by short-term sustained operations with partial sleep deprivation - A field-based study. Chronobiol. Int. 35, 1464–1470 (2018).
Leng, Y. et al. Daytime napping, sleep duration and serum C reactive protein: a population-based cohort study. BMJ Open 4, e006071 (2014).
Faraut, B. et al. Napping reverses the salivary interleukin-6 and urinary norepinephrine changes induced by sleep restriction. J. Clin. Endocrinol. Metab. 100, E416–426 (2015).
Vgontzas, A. N. et al. Daytime napping after a night of sleep loss decreases sleepiness, improves performance, and causes beneficial changes in cortisol and interleukin-6 secretion. Am. J. Physiol. Endocrinol. Metab. 292, E253–261 (2007).
Zhong, G., Wang, Y., Tao, T., Ying, J. & Zhao, Y. Daytime napping and mortality from all causes, cardiovascular disease, and cancer: a meta-analysis of prospective cohort studies. Sleep. Med. 16, 811–819 (2015).
Naska, A., Oikonomou, E., Trichopoulou, A., Psaltopoulou, T. & Trichopoulos, D. Siesta in healthy adults and coronary mortality in the general population. Arch. Intern. Med. 167, 296–301 (2007).
Hausler, N., Haba-Rubio, J., Heinzer, R. & Marques-Vidal, P. Association of napping with incident cardiovascular events in a prospective cohort study. Heart 105, 1793–1798 (2019).
Yamada, T., Hara, K., Shojima, N., Yamauchi, T. & Kadowaki, T. Daytime napping and the risk of cardiovascular disease and all-cause mortality: a prospective study and dose-response meta-analysis. Sleep 38, 1945–1953 (2015).
Yamada, T., Shojima, N., Yamauchi, T. & Kadowaki, T. J-curve relation between daytime nap duration and type 2 diabetes or metabolic syndrome: A dose-response meta-analysis. Sci. Rep. 6, 38075 (2016).
Mohammad, Y. Siesta and risk for ischemic stroke: results from a case-control study. Medicina (Kaunas) 56, https://doi.org/10.3390/medicina56050222 (2020).
Irwin, M. R. et al. Cognitive behavioral therapy vs. Tai Chi for late life insomnia and inflammatory risk: a randomized controlled comparative efficacy trial. Sleep 37, 1543–1552 (2014).
Irwin, M. R. et al. Cognitive behavioral therapy and tai chi reverse cellular and genomic markers of inflammation in late-life insomnia: a randomized controlled trial. Biol. Psychiatry 78, 721–729 (2015).
Carroll, J. E. et al. Improved sleep quality in older adults with insomnia reduces biomarkers of disease risk: pilot results from a randomized controlled comparative efficacy trial. Psychoneuroendocrinology 55, 184–192 (2015).
Weljie, A. M. et al. Oxalic acid and diacylglycerol 36:3 are cross-species markers of sleep debt. Proc. Natl Acad. Sci. USA 112, 2569–2574 (2015).
Laing, E. E., Moller-Levet, C. S., Dijk, D. J. & Archer, S. N. Identifying and validating blood mRNA biomarkers for acute and chronic insufficient sleep in humans: a machine learning approach. Sleep 42, https://doi.org/10.1093/sleep/zsy186 (2019).
Garbarino, S. & Scoditti, E. On the role of sleep hygiene in health management during COVID-19 pandemic. Sleep. Med. 77, 74 (2020).
Ruiz, F. S. et al. Immune alterations after selective rapid eye movement or total sleep deprivation in healthy male volunteers. Innate Immun. 18, 44–54 (2012).
Carroll, J. E. et al. Sleep deprivation and divergent toll-like receptor-4 activation of cellular inflammation in aging. Sleep 38, 205–211 (2015).
Pejovic, S. et al. Effects of recovery sleep after one work week of mild sleep restriction on interleukin-6 and cortisol secretion and daytime sleepiness and performance. Am. J. Physiol. Endocrinol. Metab. 305, E890–896 (2013).
Author information
Authors and Affiliations
Contributions
E.S. reviewed the literature and wrote the manuscript draft. S.G. and P.L. contributed to writing the manuscript and revised the manuscript draft. N.L.B. and N.M. reviewed the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editor: Karli Montague-Cardoso. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Garbarino, S., Lanteri, P., Bragazzi, N.L. et al. Role of sleep deprivation in immune-related disease risk and outcomes. Commun Biol 4, 1304 (2021). https://doi.org/10.1038/s42003-021-02825-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-021-02825-4
This article is cited by
-
Sleep and allergic diseases among young Chinese adults from the Singapore/Malaysia Cross-Sectional Genetic Epidemiology Study (SMCGES) cohort
Journal of Physiological Anthropology (2024)
-
Autoimmune diseases and adverse pregnancy outcomes: an umbrella review
BMC Medicine (2024)
-
Effects of lifestyle factors on leukocytes in cardiovascular health and disease
Nature Reviews Cardiology (2024)
-
Evaluating reliability in wearable devices for sleep staging
npj Digital Medicine (2024)
-
Inverted U-shaped relationship between sleep duration and phenotypic age in US adults: a population-based study
Scientific Reports (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
