
Abstract
Ancient DNA is transforming our ability to reconstruct historical patterns and mechanisms shaping modern diversity and distributions. In particular, molecular data from extinct Holocene island faunas have revealed surprising biogeographic scenarios. Here, we recovered partial mitochondrial (mt) genomes for 1300–1400 year old specimens (n = 2) of the extinct “horned” crocodile, Voay robustus, collected from Holocene deposits in southwestern Madagascar. Phylogenetic analyses of partial mt genomes and tip-dated timetrees based on molecular, fossil, and stratigraphic data favor a sister group relationship between Voay and Crocodylus (true crocodiles). These well supported trees conflict with recent morphological systematic work that has consistently placed Voay within Osteolaeminae (dwarf crocodiles and kin) and provide evidence for likely homoplasy in crocodylian cranial anatomy and snout shape. The close relationship between Voay and Crocodylus lends additional context for understanding the biogeographic origins of these genera and refines competing hypotheses for the recent extinction of Voay from Madagascar.
Similar content being viewed by others


Introduction
New methods to recover genomic data from extinct lineages have helped to clarify previously enigmatic phylogenetic relationships and enabled rigorous tests of biogeographic and evolutionary hypotheses1,2,3,4. In some cases, molecular data from extinct Holocene island faunas have revealed surprising biogeographic scenarios5,6,7,8. Additional ancient DNA studies, including recent analyses of our own family, Hominidae9,10, canids11 and elephants12 have yielded dramatic insights into the role of admixture between divergent lineages in evolutionary history. Genomic data for extinct species could also yield insights into extinction mechanisms that operated in the recent past13,14.
The arrival of modern humans in Madagascar between ~9000 and ~2500 YBP15,16,17,18,19,20 preceded the extinction of much of the island’s vertebrate megafauna including giant tortoises (Aldabrachelys spp.), elephant birds that ranged to enormous size (Aepyornis, Mullerornis, Vorombe), dwarf hippos (Hippopotamus lemerlei, H. madagascariensis), and several lemur species (Megaladapis, Archaeoindris, Palaeopropithecus, Pachylemur)6,13,21,22,23. One lesser-known extinction that occurred during this period was the demise of an endemic “horned” crocodile, Voay robustus (Fig. 1). Early explorers to Madagascar noted that Malagasy peoples consistently referred to two types of extant crocodiles on the island, a large robust crocodile and a more gracile form with a preference for rivers24. This suggests that both types persisted until very recently24,25, but only the gracile form, now recognized as an isolated population of the Nile crocodile (Crocodylus niloticus), currently is found on the island26.

A skull of Voay robustus collected at Ampoza (44° 42.3’ E, 22° 18.9’ S, 570 m elevation) during the joint Mission Franco-Anglo-American expedition from 1927–1930 (White, 1930).
Despite nearly 150 years of investigation, the phylogenetic position of the extinct horned crocodile of Madagascar remains controversial. In 1872, the earliest description of the species by Grandidier and Vaillant27 noted differences between sub-fossil cranial and postcranial material excavated from Holocene deposits near Amboulisatre and extant crocodiles (C. niloticus) in Madagascar. Based on the robustness of available skeletal features, including vertebral, dental and cranial elements and snout shape, (Fig. 1), Grandidier and Vaillant named the extinct form Crocodylus robustus27. They suggested a possible affinity between the subfossil material and Crocodylus niger, now recognized as the dwarf crocodile (Osteolaemus tetraspis) that is native to west-central Africa. In the same year, Grandidier28 further contrasted the relatively stout features of C. robustus with those of the more gracile C. niloticus that currently inhabits the island. Barbour29 and Boettger30 however, suggested that the extinct robust species simply represented an aged C. niloticus. In 1910, Vaillant and Grandidier24 and later Mook31,32 examined subfossil material from additional sites and upheld C. robustus as clearly distinct from extant C. niloticus. Mook32 noted that C. robustus was instead more similar to the extant saltwater crocodile, C. porosus.
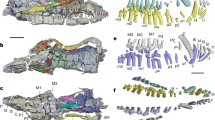
After conducting a detailed morphological study of available subfossil material representing C. robustus, Brochu33 noted that C. robustus lacks many of the distinguishing features of the genus Crocodylus. In his detailed cladistic analysis, the extinct Malagasy species grouped with extant dwarf crocodiles (Osteolaemus spp.)33 of west and central Africa. Several phenotypic characters allied C. robustus with the genus Osteolaemus, some of which might relate to overall skull shape with a relatively short and deep snout in both taxa. Based on this evidence, Brochu erected a new monotypic genus, Voay (the modern Malagasy word for extant crocodiles) within Osteolaeminae, resulting in the current species name Voay robustus33. Subsequent phylogenetic analyses of morphology34,35,36,37,38,39 as well as total evidence analyses of morphology and molecules37,39,40,41 have consistently clustered V. robustus and Osteolaemus to the exclusion of other crocodylian genera, with Crocodylus distantly related to Voay (Fig. 2).

Four representative phylogenetic hypotheses are shown (A–D). Both morphology (A, B) and combined analyses of morphology plus molecules (C, D) place Voay with Osteolaemus and extinct African osteolaemines to the exclusion of Crocodylus (true crocodiles). The tip-dated tree in (C) is from a Bayesian reanalysis of morphological data from Brochu (2013) in combination with DNA sequence data (Lee and Yates, 2018). Support scores at nodes are parsimony bootstrap percentages (A) or Bayesian posterior probabilities (B–D). Robust support for Voay + Osteolaemus is highlighted in red.
Here, we use mitochondrial (mt) capture and whole genome enrichment (WGE) of ancient DNA (aDNA) to recover mt sequences from two subfossil specimens of Voay robustus. We employ separate and combined analyses of mtDNA and morphological data to test competing hypotheses for the phylogenetic placement of Voay relative to living and extinct crocodylians. Total evidence analyses that merge molecular, fossil, and stratigraphic data yield timetrees for Crocodylidae that we utilize to better characterize the biogeographic history of the clade, the timing of Crocodylus origins, and the extinction of Voay.
Results
Carbon dating
Voay sample AMNH FR-3101 yielded AMS 14C dates of 1450 ± 30 (1422–1307) 14C yr BP, while AMNH FR-3103 yielded dates of 1380 ± 30 (1364–1280) 14C yr BP. Newly derived 14C dates are slightly younger than those recovered from vertebrates from the same deposits ca. 1,800 14C yr BP and 2,430 14C yr BP42, confirming the relatively recent age of the specimens.
Recovery of partial mt genomes for Voay robustus
Whole genome enrichment (WGE) and targeted mt capture approaches yielded partial mt genomes from two Holocene specimens of Voay robustus - AMNH FR-3101 and AMNH FR-3103 (Supplementary Data 1). WGE produced both mtDNA and nuclear sequences from Voay samples, but coverage for single-copy nuclear genes was low. We therefore focused phylogenetic analyses on mt reads derived from both enrichment procedures. Authenticity of the mtDNA data for Voay was evidenced by DNA-damage patterns, low sequence divergence between individuals, clean negative controls, and consistent phylogenetic placement of sequences from replicated processing of both specimens (Supplementary Figs. 1–2; Supplementary Table 1 and Data 3; Supplementary Data 2).
Four reconstructions of the Voay mt genome were assembled by mapping short reads from these two specimens to two reference mt genomes (Osteolaemus and C. porosus) using the EAGER pipeline (see Materials and Methods). The most complete reconstruction, Voay AMNH FR-3101 C. porosus ref., had 18% missing data relative to the reference genome sequence for mt ribosomal DNAs (rDNAs) and protein-coding genes. Reconstructions using Osteolaemus as the reference and/or Voay specimen AMNH FR-3103 yielded mt datasets with more missing data relative to reference genomes (45–72% missing). Average sequencing coverage for the four mt genome builds was as follows: Voay AMNH FR-3101 Osteolaemus ref. (5X); Voay AMNH FR-3103 Osteolaemus ref. (3X); Voay AMNH FR-3101 C. porosus ref. (5X); Voay AMNH FR-3103 C. porosus ref. (4X). Pairwise comparisons among the four Voay builds from two different specimens show minor divergence from each other at the nucleotide level. Short reads for each Voay specimen were deposited at NCBI (short read archive Bioproject PRJNA681754).
Phylogenetic analyses and evolutionary inferences
All phylogenetic analyses of our mtDNA datasets (Fig. 3; Supplementary Table 1 and Data 3; Supplementary Data 2) corroborate relationships among the nine extant genera of Crocodylia that have been consistently supported by molecular data since 200843. The genus Crocodylus (true crocodiles) is sister to a clade composed of Mecistops (African slender-snouted crocodiles) and Osteolaemus (dwarf crocodiles) within Crocodylidae (Fig. 3). Crocodylidae groups with Gavialidae (true and false gavials), and this combined clade is sister to Alligatoridae (alligators and caimans). However, our mt trees contradict previous numerical phylogenetic analyses of morphology and combined data that robustly cluster Voay with osteolaemines (Fig. 233,34,35,36,37,38,39,40,41,44). Our mtDNA trees instead reflect a closer association with Crocodylus as hypothesized by earlier authors29,30,32,45 (Fig. 3). Parsimony and maximum likelihood (ML) analyses of partial mt genomes (two rDNAs and 13 protein-coding genes) uniformly support a sister group relationship between Voay and a monophyletic Crocodylus, as well as a clade composed of Osteolaemus and Mecistops (Fig. 3). These relationships are robustly supported by all 64 analyses of the molecular dataset (Supplementary Table 1 and Data 3; Supplementary Data 2). ML phylograms show that Voay branches from the stem lineage of extant Crocodylus at about the midpoint of this long internal branch, with limited divergence among the four partial mt genome builds reconstructed from the two Voay specimens (Supplementary Fig. 2).

The tree shown is based on ML analysis (partitioned by gene) and includes data from all four builds of the Voay mt genome. Bootstrap scores at each node are (from top to bottom): all four builds of Voay mt genome with partitioned ML analysis, Voay AMNH FR-3101 C. porosus reference build with partitioned ML analysis, Voay AMNH FR-3101 Osteolaemus reference build with partitioned ML analysis, Voay AMNH FR-3103 C. porosus reference build with partitioned ML analysis, Voay AMNH FR-3103 Osteolaemus reference build with partitioned ML analysis, and all four builds of Voay mt genome with equally-weighted parsimony analysis. Bootstrap scores for the two internodes that bound the branching point of Voay are highlighted in red. All trees were rooted with bird, turtle, and lizard outgroups (not shown). Higher level taxa are delimited by brackets to the right of species names. Paintings of crocodylians are by C. Buell, and photo of Voay (AMNH FR-3101) is by E. Hekkala.
In all six tip-dated timetrees (Supplementary Table 1 and Data 3; Supplementary Data 2), Voay again groups with Crocodylus, and Osteolaemus clusters with Mecistops (Fig. 4). Some extinct taxa that were coded for just phenotypic characters are unstable in these combined data analyses, but for each Bayesian tip-dating tree, the relationships of Voay to extant crocodylid genera are consistent in 100% of the trees in posterior distributions and congruent with all 64 analyses of mtDNA alone (Fig. 3; Supplementary Data 2). In terms of topology and divergence times, our most complete timetree for Crocodylia (Fig. 4) closely matches the hypothesis proposed by Lee and Yates37, except for the conflicting position of Voay relative to Crocodylus, Osteolaemus, and Mecistops. Our tip-dated tree shows a long unbranching lineage that split from all other sampled crocodylians in the late Oligocene and terminates at the Holocene extinction of Voay on Madagascar. This divergence from the genus Crocodylus dates to ~24.9 Ma (95% highest posterior density [HPD] = 18.8–32.1 Ma), with the earliest split among Crocodylus species (crown + stem) dated at ~19.9 Ma (95% HPD = 14.7–26.2 Ma) and ~16.3 Ma for crown group Crocodylus (95% HPD = 12.5–20.5 Ma). Voay separated from the more distantly related Osteolaemus in the Eocene at ~38.6 Ma (95% HPD = 32.4–45.3 Ma) (Fig. 4). By contrast, Voay split from Osteolaemus just ~17.8 Ma in the combined data timetree of Lee and Yates37 and at ~16.4 Ma in their tip-dated analysis of morphological characters. Our six timetree hypotheses show some variation in median divergence time estimates, due to differences in character coding, ordering of character states, and taxon sampling in the two morphological datasets that were reanalyzed37,46, as well as the inclusion or exclusion of 3rd codon positions from mt protein-coding genes (Supplementary Data 2). For example, across our six tip-dated trees, the median divergence date for Crocodylus and Voay ranges from ~22.1–27.7 Ma, and the split between Voay + Crocodylus and Osteolaemus + Mecistops ranges from ~30.7–38.6 Ma. However, we suggest caution when interpreting these dates due to disagreements on specimen dating in the published literature, and the possibility that errors in published dates may affect results of these analyses.

Bayesian posterior probabilities are at nodes; support scores for the two internodes that bound the branching point of Voay are highlighted (red). Optimization of geographic regions to internal nodes (colored circles) is based on equally-weighted parsimony and implies an African ancestry for the overall clade with minimally two migrations to Australia/Asia, two to the New World, and two to Madagascar. An identical mapping of ancestral areas results for minimum area change (MAC) parsimony analysis. The Voay AMNH FR-3101 C. porosus mt genome build (partitioned by 1st, 2nd, 3rd codons) was employed in combination with morphological characters and stratigraphic data from Lee and Yates (2018). Taxa that are distantly related to Voay are pruned from the figure; for the complete timetree, see Supplementary Data 2. Paintings of crocodylians are by C. Buell; photo of Voay (AMNH FR-3101) is by E. Hekkala.
Parsimony optimizations of geographic ranges on our tip-dated timetrees consistently reconstruct an African ancestry for both Voay and Crocodylus (Fig. 4). Although alternative reconstructions are nearly as parsimonious, the ‘out of Africa’33 pattern generally holds for our maximum clade credibility (MCC) timetrees. A migration of the ancestral Voay lineage from Africa to Madagascar is inferred, but this biogeographic shift is not well-constrained temporally given the current sampling of extinct taxa.
Mapping of morphological characters on our most complete tip-dated tree (Fig. 4) implies convergent homoplasy in multiple characters that instead group Voay with Osteolaemus in previous phylogenetic analyses of morphology and combined data (Fig. 2). For the tip-dated timetree of Lee and Yates37 that did not include any molecular data for Voay, seven morphological characters are synapomorphic for a Voay + Osteolaemus clade. All seven of these cranial characters are interpreted as convergences on our tip-dated tree (Supplementary Table 1 and Data 3). Prominent squamosal “horns”, as seen in Voay (Fig. 1), also evolved convergently in four additional crocodylid taxa (Crocodylus rhombifer, C. siamensis, C. anthropophagus + C. thorbjarnarsoni, and Euthecodon brumpti). For the taxa sampled here, this trait is restricted to just Crocodylidae but is highly homoplastic (consistency index = 0.200). There is limited morphological support, just one unambiguously optimized synapomorphy, for the novel grouping of Voay sister to Crocodylus in our tip-dated tree. Transformation from a straight or gently curved prefrontal-frontal suture to an ‘L’-shaped suture optimizes to the common ancestor of Voay + Crocodylus (Supplementary Table 1 and Data 3). This labile binary character shows minimally 11 changes on the overall tree (consistency index = 0.091).
Discussion
Our combined WGE and targeted mtDNA capture recovered partial mt genomes for two Holocene specimens of Voay robustus from Madagascar (Supplementary Data 1) and enabled the first molecular phylogenetic placement of this extinct island endemic. Molecular and combined data uniformly position Voay as sister to Crocodylus and outside of the clade comprised of Osteolaemus and Mecistops (Figs. 3, 4; Supplementary Data 2). All of our trees contradict previous quantitative phylogenetic work that consistently placed Voay within Osteolaeminae, close to the genus Osteolaemus (dwarf crocodiles) based on anatomical characters and combined analyses of morphology plus molecules (Fig. 233,34,35,36,37,38,39,40,41,44).
For Crocodylia, prior molecular phylogenetic work suggested that morphological features are commonly characterized by high levels of homoplasy40,47,48 that may be driven by convergent ecological and functional pressures49,50,51,52,53,54,55,56,57. Morphological data provide minimal character support for grouping Voay as the sister group to Crocodylus. Just one morphological character change unequivocally maps to the last common ancestor of the clade, and this grouping implies convergent homoplasy in a host of anatomical features shared by Voay and Osteolaemus (Supplementary Table 1 and Data 3). Our results further highlight ongoing conflicts between morphological and molecular characters in crocodylian phylogenetics with the caveat that the mt genome is generally interpreted as a single non-recombining locus in Crocodylia43. Corroboration from independent nuclear loci would solidify support for our novel phylogenetic hypotheses (Figs. 3, 4).
Recent molecular hypotheses of crown group relationships within Crocodylidae have been equivocal for interpreting both the age and biogeographic origins of the genus Crocodylus58,59,60. The “out of Africa”33 hypothesis for crown group Crocodylus was tested in a probabilistic framework by Oaks who found stronger support for origin of the group in Australia/Asia58. The mt genome analysis of Meredith et al. supported monophyly as opposed to paraphyly of Crocodylus spp. from Australia and Asia59. For their tree, parsimony optimization of geography implies an African origin for Crocodylus, with a recent dispersal from Africa to the New World and another dispersal to Australia/Asia, which agrees with earlier morphological work. More recently, Nicolaï and Matzke61 partitioned geographic areas more finely and reconstructed an Asian origin for Crocodylus. Like Oaks and Meredith et al., this study did not directly consider the extensive fossil diversity of Crocodylus and more generally, Crocodylidae (e.g., 33,34,35,36).
Our biogeographic reconstructions that include fossils instead suggest an African origin for Crocodylus. In tip-dated MCC timetrees, extinct taxa that are closely related to Crocodylus (Rimasuchus, Brochuchus, Euthecodon, “Crocodylus” megarhinus) are predominantly African, as are extant outgroup taxa, Osteolaemus and Mecistops (e.g., Fig. 4). Taken together with the placement of Voay from Madagascar as the sister taxon to Crocodylus, our timetrees hint at an African origin for the genus62. However the unstable affinities of various extinct Crocodylus spp. in our timetrees complicate interpretation and beg for more comprehensive analyses in the future that incorporate the full complement of extinct geographic diversity and a broader survey of informative characters. Crown group Crocodylus initially diversified at ~16.3–17.7 Ma according to our four tip-dated MCC timetrees that sample all extant species in the genus. However, in these, as in other analyses58,60, broad 95% HPDs limit interpretation (e.g., 12.5–20.5 Ma for the Voay AMNH FR-3101 C. porosus ref. alignment).
The inferred migration or vicariance event that isolated the Voay evolutionary lineage on Madagascar is not well-constrained according to tip-dated timetrees. In our most comprehensive hypothesis (Fig. 4), Voay diverged from its sistergroup, Crocodylus, at ~24.9 Ma (95% HPD = 18.8–32.1 Ma), and there is no evidence for speciation or extinction in the V. robustus lineage up until the final demise of this single species in historical times. Movement of Voay to Madagascar therefore may have occurred between the late Oligocene and the first known occurrences of Voay in the Pleistocene, ~10,000 years ago42,63. Over this time span, Madagascar was fully isolated from Africa and other continental landmasses, so any dispersals were necessarily trans-oceanic13,64,65. Recent prevailing winds and ocean currents oppose overwater dispersal from Africa to Madagascar due to north or south-southwest flow, but some paleo-oceanographic models reconstruct intermittent and rare eastward flow in the Eocene and Oligocene66. The salt tolerance of extant Crocodylus spp. has been suggested as a driver for the relatively recent range expansion of this genus59,61, but the reconstruction of salt tolerance in extinct species, such as Voay, is ambiguous given the distribution of this trait in extant crocodylians67,68,69,70,71. Overall, our phylogenetic hypotheses (Fig. 4) broadly delimit the timing of biogeographic events, but future paleontological discoveries, in particular extinct taxa that branched from the long ~24.9 MY Voay lineage, are required to further refine this timeframe.
Several explanations have been proposed for the extinction of megafauna in Madagascar during the transition to the Anthropocene17,72,73,74. Currently viable hypotheses include environmental change or over-exploitation and habitat alteration by humans that together may have acted as synergistic drivers of megafaunal collapse across the island17,18. Bickelmann and Klein75 argued that, given the absence of evidence for direct human exploitation, competition with the Nile crocodile, C. niloticus, was the more likely driver of Voay’s extinction. Closely related species of similar body size often share similar ecologies76,77,78,79, and the molecular evidence for a more recent common ancestry with Crocodylus spp. (Figs. 3, 4) relative to the previous consensus that grouped Voay with Osteolaemus (Fig. 2) perhaps lends additional support to the competition hypothesis. Relaxed clock estimates of the Nile crocodile’s arrival in Madagascar suggest a very recent invasion ca. 2,000–3,000 YBP60 that implies temporal overlap with Voay, but the earliest documented Nile crocodile material in Madagascar dates to just ~310–460 years ago80.
A more speculative extinction scenario also requires a temporal and geographic overlap between Voay and C. niloticus in Madagascar. The mixing of genes between differentiated evolutionary lineages (‘phylogenetic species’) is well documented between some species in the genus Crocodylus81. It is therefore at least possible that introgressive hybridization with the recently invading C. niloticus contributed to decline of Voay through genetic swamping or ‘extinction via hybridization’82. Ancient mtDNA sequences from Voay, however, do not provide any compelling evidence for hybridization with the Nile crocodile. Recent introgression of mtDNA would be expressed as a clustering of these two species in mt trees as has been found in C. acutus and C. rhombifer83,84, which is not supported (Fig. 3). Future ancient DNA work that focuses on recovery of Voay nuclear DNA promises a more rigorous test of gene flow hypotheses.
Given the concurrent extinction of megafauna on Madagascar, it is perhaps more plausible that Voay succumbed to a combination of direct extirpation by humans and rapid environmental change33. Unlike large mammalian taxa such as hippos and lemurs, that were likely targeted as adults by humans, Voay populations may have been impacted by exploitation of eggs, resulting in a rapid decline. Vaillant and Grandidier noted that both species of crocodiles were recognized by communities throughout Madagascar and that crocodile eggs were regularly consumed, particularly in southwestern Madagascar24. This type of impact would be largely undetectable at archeological sites through modern taphonomic measures.
Our study provides the first molecular systematic characterization of V. robustus and indicates that this recently extinct island endemic represents the sister lineage to Crocodylus (true crocodiles). Molecule-based trees (Fig. 3) and combined phylogenetic analyses of molecules and morphology (Fig. 4) contradict trees from previous studies that grouped this species and dwarf crocodiles (Osteolaemus) with high support (Fig. 2). Tip-dated timetrees suggest that Voay diverged from Crocodylus near the Oligocene/Miocene boundary (~22.1–27.7 Ma) and represents a relict lineage that survived to historical times in Madagascar but has no known close relatives, living or extinct (Fig. 4; Supplementary Data 2). Our results highlight the value of ancient DNA for uncovering novel, unexpected evolutionary relationships and providing context for new interpretations of morphological evolution, biogeographic history, and extinction patterns.
Methods
Specimens and sample processing
The paleontological collections at the American Museum of Natural History (AMNH) include a series of specimens of Voay (= Crocodylus) robustus from Ampoza, Madagascar (44° 42.3’ E, 22° 18.9’ S, 570 m elevation). These specimens were collected during the joint Mission Franco-Anglo-American expedition from 1927–193085. White’s descriptions of field excavations denote a Holocene deposition85, and subsequent C14 dating of adjacent faunal remains from Ampoza are dated from ~2500–1000 YBP63. Interpretation of specific depositional context of V. robustus material is limited. However, White’s notes and photographs from the excavation indicate a solid surface layer of limestone, below which a dark soil held diverse disarticulated skeletal elements85. As excavations proceeded, the site filled with water from subsurface layers, and field laborers extracted material from underwater85. A reconstruction of the habitat suggests a riparian stream system near a marsh86.
Two specimens were targeted as potential sources of ancient DNA. The sampling plan was designed to minimize damage to the specimens and reduce contamination. Prior to handling specimens, all tools were sterilized by UV radiation for 15 min, soaked in DNAaway (Thermo Scientific) for 5 min, and then dried in a covered sterile chamber. For each skull, a tooth was gently lifted to expose an un-erupted tooth beneath. One un-erupted tooth from each specimen was removed for genomic analysis. All surfaces of tooth samples were rinsed with 70% DNAaway for 30 s, rinsed twice with sterile water, and then dried in a covered petri dish. Each tooth was subsequently placed in a sterile 15 ml falcon tube. Parallel sample processing and negative controls were executed during the specimen sampling and all subsequent DNA extraction processes (Supplementary Fig. 1).
Carbon dating
Samples from each specimen (AMNH FR-3101 and AMNH FR-3103) were sent to Beta Analytic Inc, Miami Florida for radiocarbon dating. Teeth were initially decalcified and gelatinized using EDTA and HCl. Once collagen preservation was confirmed, samples were radiocarbon dated and calibrated dates reported. Calibration was calculated using one of the databases associated with the 2013 INTCAL program. Conventional Radiocarbon Ages and Sigmas are rounded to the nearest 10 years per the conventions of the 1977 International Radiocarbon Conference. When counting statistics produce Sigmas lower than ±30 years, a conservative ±30 BP is cited for the result. All work was performed under strict chain of custody and quality control under ISO/IEC 17025:2005 Testing Accreditation PJLA #59423 accreditation protocols. Sample, modern and blanks were all analyzed in the same chemistry lines by qualified professional technicians using identical reagents and counting parameters within on Beta Analytic Inc’s own particle accelerators.
DNA isolation
Subsampling of the two tooth specimens (AMNH FR-3101 and AMNH FR-3103) was done at the AMNH, and duplicate samples were shipped to the University of British Columbia (UBC). Isolation of ancient DNA was replicated in dedicated clean room facilities at the AMNH and at UBC (Supplementary Fig. 1) according to published protocols87. For the ancient DNA extractions conducted at the AMNH, between 50 and 90 mg of surface sterilized tooth was crushed and demineralized overnight at room temperature in 1 mL 0.5 M EDTA with gentle shaking. Samples were then digested in 750 µL of a sarcosyl-based proteinase K solution and purified using the MinElute PCR Purification kit (Qiagen) with two washes of 700 µL Buffer PE and eluted twice in 80 µL (2 × 40 µL) buffer EB at 0.05% Tween-20. For the ancient DNA extractions conducted at UBC, a modified version of extraction protocol Y was employed, as originally described by Gamba et al.87. Each sample was extracted in duplicate at UBC. Approximately 250 mg of each sample was ground while submerged in liquid nitrogen using a Spex 6770 freezer mill (5 min precooling, 1 min of grinding at 10x per second). Samples were demineralized in 3 mL of 0.5 M EDTA pH 8.0, 150 μL 10% SDS, and 100 μL of 20 mg/ml Proteinase K, with incubation overnight at 56 °C. The lysate was concentrated to 250 μL using Amicon Ultra-4 30 kDa tubes by centrifugation. The resulting 250 μL of lysate were mixed with 5x volume of buffer PB and added in three steps to a MinElute (Qiagen) column and centrifuged, removing the flow-through after each step. The column was washed twice with 750 μL of PE and centrifuged, allowing desalting for 5 min during the first wash. The elution was performed using 50 μL of ultra-pure water preheated to 56 °C.
Genomic DNA replicates from both laboratories were shipped on dry ice to Arbor Biosciences, (Ann Arbor, Michigan, USA) for subsequent library preparation and enrichment processing.
Library preparation
Two duplicate Illumina® libraries for each Voay specimen were prepared in ancient DNA processing facilities by Arbor Biosciences for use in downstream WGE and targeted sequence capture of mtDNA. Each library was amplified using unique P5 and P7 indexing primers, and 10 µL of each library in 40 µL reactions were quantified on a CFX96 Real-time PCR machine (BioRad). Indexed libraries were purified using MinElute (Qiagen) columns.
Whole genome enrichment (WGE) using RNA baits
We enriched for crocodylian genomes using a modified protocol wherein genomic DNA (gDNA) from closely related taxa are converted into biotinylated RNA baits3,88. Briefly, at the AMNH, gDNA was extracted from ten modern crocodylian blood samples representing six taxa [Crocodylus moreletii (n = 1), C. acutus (n = 1), C. siamensis (n = 1), C. suchus (n = 2), C. niloticus (n = 2) and Osteolaemus tetraspis (n = 3)] using a Qiagen DNeasy kit and the manufacturer’s protocols for nucleated red blood cells. Approximately 1 µg of extracted DNA from each species was sent to Arbor Biosciences (Ann Arbor, Michigan USA) for global reverse transcription (both strands) with biotinylated rUTP using their proprietary procedure3. This yielded an aqueous suspension of approximately 100 µg of mixed crocodylian RNA baits for subsequent WGE.
Enrichment of Voay robustus genomic libraries was conducted at Arbor Biosciences according to their MYcroarray capture protocol version 3 (https:// arborbiosci.com/wp-content/uploads/2017/10/MYbaits-manual-v3.pdf). Briefly, each capture reaction used 1 µg of crocodylian RNA baits, 9 µL-indexed library (described above), and the MYBaits (MYcroarray) kit protocol for enrichment. Hybridizations were done at 48 °C for 48 h. Following SPRI bead cleanup and MinElute purification, enriched eluates were amplified for 10 cycles and then again purified with MinElute columns. Approximately 9 µL of these purified products were used in another round of capture using identical conditions as the first round, except incubation occurred at 55 °C for 39 h. Reactions were again bead-cleaned and purified with MinElute columns. Purified products were then re-amplified for 5 cycles and the resulting re-amplified, doubly-enriched libraries were purified one last time using MinElute columns.
Targeted mtDNA enrichment using synthetic baits
A previously developed MYbaits kit that targets the crocodylian mt genome was used for enrichment of the ancient crocodylian DNA libraries. Each capture reaction used 1 µg of crocodylian mt capture baits, 9 µL-indexed library (described above), and the MYBaits kit protocol version 3 (described above) for enrichment. Hybridizations were done at 48 °C for 48 h. Following bead cleanup and MinElute purification, enriched eluates were amplified for 10 cycles and then purified with MinElute columns. Purified products were then re-amplified for 5 cycles and the resulting re-amplified, doubly-enriched libraries were purified one last time using MinElute columns.
DNA sequencing
For each of the two Voay robustus specimens (AMNH FR-3101 and AMNH FR-3103), two independent samples plus negative controls were extracted (A and B), two replicate libraries were produced (1 and 2) and one pooled WGE and Mito enriched library were sequenced, resulting in 10 separately processed samples. For each specimen replicate set (either 3101 or 3103), the indexed whole genome enriched library and the targeted mtDNA enriched library were pooled with a ratio of 75 (WGE library)/25 (mtDNA capture library), and sequenced using one full lane on an Illumina HiSeq® 2500 (paired-end, 150 bp reads) at the New York Genome Center (see Supplementary Figure 1 for sample AMNH FR-3101example).
Sequence analyses and mtDNA reconstruction
Preliminary mapping analyses using EAGER, an ancient genomics pipeline89, showed that crocodylian mtDNA was not recovered from the negative control libraries. Exploratory mapping of short reads also indicated that mtDNA builds derived from AMNH and UBC libraries were homogeneous for each Voay specimen and that libraries derived from the same specimen could be safely combined for final reconstruction of ancient mt genome sequences. Merged sequence reads from Voay AMNH FR-3101 and merged reads from Voay AMNH FR-3103 were analyzed separately using EAGER, which automates read processing, mapping, variant detection, and consensus genome reconstruction. Mapping against a crocodylian reference genome enables screening of non-endogenous DNA from the often complex metagenomic mixtures in ancient samples. Moreover, these reference alignments highlight erroneous base incorporations that can signify DNA damage that is a characteristic of ancient samples90.
Using the EAGER pipeline, reads were processed by clipping adapters, merging paired ends with overlapping regions, and trimming bases with phred scores lower than 20. So that the reconstructed ancient mt genomes would not be biased toward one or the other genus that were a priori hypothesized to be closely related to Voay (ref. 91), merged reads were mapped to both Crocodylus porosus (GenBank accession # DQ273698.1) and Osteolaemus tetraspis (GenBank accession # NC_009728) reference mt genomes. Merged reads of minimum length 30 were treated as single-end and aligned to the reference genomes using BWA-MEM and default settings. After removing duplicates, the UnifiedGenotyper module in the Genome Analysis Toolkit (GATK) was used to make variant and reference base calls at each position. Both variant and reference calls were required to have the support of at least two reads, a phred-scaled genotype quality score of at least 30, and a consensus SNP frequency of at least 90%. Failing these criteria at any given position in the reference resulted in the insertion of an ‘N’ ambiguity character. With alleles compiled, EAGER’s VCF2Genome module was used to generate draft genome sequences relative to the C. porosus and Osteolaemus references. To verify the reconstruction of the ancient mt genome, EAGER’s DamageProfiler module was used to quantify alignment errors resulting from ancient DNA damage. A separate mitogenomic reconstruction for each specimen relative to each reference genome resulted in four total Voay mt genome sequences (Voay AMNH FR-3101 Osteolaemus ref.; Voay AMNH FR-3103 Osteolaemus ref.; Voay AMNH FR-3101 C. porosus ref.; Voay AMNH FR-3103 C. porosus ref.) for use in downstream analyses.
Phylogenetic methods
The molecular dataset included the new Voay robustus mt genome reconstructions and previously published mt genomes from 22 extant species of Crocodylia including [Genbank # in brackets]: Alligator mississippiensis [NC_001922], Alligator sinensis [NC_004448], Caiman crocodilus [NC_002744], Paleosuchus palpebrosus [NC_009729], Paleosuchus trigonatus [NC_009732], Gavialis gangeticus [NC_008241], Tomistoma schlegelii [NC_011074], Mecistops cataphractus [NC_010639], Osteolaemus tetraspis [NC_009728], Crocodylus acutus [NC_015647], Crocodylus intermedius [JF502242], Crocodylus johnstoni [NC_015238], Crocodylus mindorensis [NC_014670], Crocodylus moreletii [NC_015235], Crocodylus suchus [JF502244], Crocodylus niloticus [JF502246], Crocodylus novaeguineae [JF502240], Crocodylus palustris [NC_014706], Crocodylus porosus [DQ273698], Crocodylus rhombifer [JF502247], and Crocodylus siamensis [EF581859]. We also included three newly generated partial mt genomes from Caiman yacare ([MN885913] sample ID# C058), Caiman latirostris ([MN885912] sample ID# S234), and Melanosuchus niger ([MN885911] sample ID# 92042). Blood samples were provided by St. Augustine Alligator Farm Zoological Park (St. Augustine, Florida, USA), and protocols for DNA extraction, PCR amplification, and sequencing are outlined in Meredith et al.59. Combined, this set of taxa includes Voay and most currently recognized extant crocodylian species with the exception of recent splittings of Mecistops and Osteolaemus into multiple phylogenetic species92,93,94. One representative mt genome was included from each of these two genera (see above).
Sauropsid mt genomes were included as outgroups to root mtDNA trees. Aves generally is considered the extant sister group to Crocodylia, with Lepidosauria and Chelonia being more distantly related within the clade Sauropsida95,96. Outgroups for our phylogenetic analyses included one lizard (Anolis carolinensis [NC_010972]), two turtles (Pelodiscus_sinensis [AY962573], Chrysemys_picta [KF874616]), and five birds that represent three major divisions of Aves: Palaeognathae (Struthio camelus [NC_002785]), Galloanserae, (Anas platyrhynchos [EU755253], Gallus gallus [NC_007236], Meleagris gallopavo [NC_010195]), and Neoaves (Melopsittacus undulatus [NC_009134], Taeniopygia guttata [NC_007897]).
For the 33 extant taxa, mt genomes initially were aligned using MUSCLE97 in Geneious 8.1.998. Minor adjustments were made to the alignment using Se-Al99 and genes were delimited based on published annotations. Several pairs of genes overlap each other in the mt genomes of Crocodylia. Therefore, each overlapping region was assigned to only one of the genes for the purposes of phylogenetic analyses. To maintain reading frame in all protein-coding genes, seven autapomorphic indels (each 1 bp) were deleted from the multi-species alignment (three in Alligator sinensis, two in Crocodylus palustris, two in Pelodiscus sinensis). A 1 bp insertion in the ND3 gene shared in turtles and birds corresponds to a site that is not translated and was also excluded from the final alignment. Two rDNA genes and 13 protein-coding genes were included in the final alignment. Each reconstructed Voay mt genome build (Voay 3101 Osteolaemus ref.; Voay AMNH FR-3103 Osteolaemus ref.; Voay AMNH FR-3101 C. porosus ref.; Voay AMNH FR-3103 C. porosus ref.) was incorporated into the multispecies mtDNA alignment by inserting gaps where there were alignment gaps in the particular reference genome used as template for mapping Voay sequencing reads. The final mtDNA alignment is available in Supplementary Data 1.
Parsimony analyses were performed using PAUP* 4.0a build 161100. Gaps were treated as missing data, all character state transformations were equally weighted, and the stability of results was assessed by weighting characters by relative fit. The concavity of the weighting function, k, was set at 4, 8, and 12 in successive runs with Goloboff weighting101. Searches were heuristic with 100 random taxon addition replicates and tree-bisection reconnection (TBR) branch swapping. Relative support was assessed by nonparametric bootstrapping102 with 100–1000 pseudoreplicates, and each search included 10 random taxon addition replicates.
ML analyses with bootstrapping (1000 replicates, gaps treated as missing data, randomized MP starting trees, and the fast hill-climbing algorithm with all free parameters estimated) were performed using RAxML-HPC v.8103 on XSEDE utilizing the Cipres portal104,105. When multiple data partitions were specified (individual genes, rDNAs, stems versus loops of rDNAs, protein-coding regions, three codon positions), different GTR + Γ models were permitted for each subset of characters104. RNAalifold106 with default settings was used to predict consensus rRNA secondary structures for aligned mt rDNA sequences.
Multiple searches were performed using both parsimony and ML optimality criteria to investigate the phylogenetic placements of the four mt genome builds for Voay robustus. In our primary mt genome tree based on ML analysis (Fig. 3), the four Voay mt genome builds were included and 15 character partitions (one for each mt gene) were analyzed. Variations in taxon sampling, character sampling, character weighting, and data partitioning were explored to assess the robustness of phylogenetic results for the placement of Voay relative to the major extant lineages of Crocodylia (Supplementary Table 1 and Data 3; Supplementary File 2).
We performed six tip-dating analyses using BEAST v1.8.3107 with modified versions of xml files provided by Lee and Yates37 (Supplementary Table 1 and Data 3; Supplementary Data 2). Lee and Yates assembled two combined datasets that both utilized published DNA sequences for crocodylians37. Their primary combined dataset included 278 morphological characters for 25 extant and 92 extinct taxa as well as stratigraphic data. They also assembled a second combined dataset that included 189 morphological characters for 15 extant and 85 extinct taxa from Brochu46 and stratigraphic data. The larger dataset in their study includes several new characters as well as modifications to characters and codings used in previous analyses; our close examination of this dataset reveals incorporated errors36,108, and results should thus be treated with caution.
In our six tip-dating analyses, we replaced the molecular data in these matrices with our mt protein-coding gene alignments that included the Voay AMNH FR-3101 C. porosus ref. build or the Voay AMNH FR-3101 Osteolaemus ref. build. The mt protein-coding data were partitioned by codon (1st, 2nd, 3rd or 1st, 2nd with 3rd codons excluded), and each data partition was modeled under the GTR + G model of sequence evolution. Clock models for the different codon positions were unlinked. All model parameters for morphological data were set as in Lee and Yates37, and we utilized their xml files (“BEAST1.8.2_croc_117_9562_2ucln_NoMolecCal_NoAsc_AsPublished.xml” and “BEAST1.8.2_Brochu2013matrixPlusMolecules.xml”). For each tip-dating analysis, two to eight independent BEAST runs were implemented for 50 million generations each with sampling every 50,000 generations. In all BEAST runs, we used Tracer109 to determine burn-in and RWTY110 to test for parameter convergence. Based on these results, 20%-25% burn-in was chosen. All post-burn-in samples were combined with LogCombiner and TreeAnnotator for subsequent analyses. Tip-dating has the advantage of directly incorporating extensive fossil information in relaxed clock models, but does not account for incomplete lineage sorting, a process that can bias divergences toward older dates58.
We performed biogeographical mapping using equally weighted parsimony and minimum area change (MAC) parsimony111. Four broad geographical areas were defined (Africa, Madagascar, Australia/Asia, and the New World (Fig. 4). For our primary analysis, the BEAST MCC tree with the most taxa and molecular data for Voay was used (“BEAST1.8.2_croc_117_9562_2ucln_NoMolecCal_NoAsc_AsPublished.xml” with the Voay AMNH FR-3101 C. porosus reference build). Distantly related taxa were pruned from the overall topology (Fig. 4). Equally-weighted parsimony treated the four areas as unordered character states. MAC parsimony used a step-matrix with equal costs to gains and losses of geographic areas111. We utilized a step matrix that allows a maximum geographic range of two areas, the extreme observed for the taxa sampled in our tree. Biogeographic reconstructions for the remaining five tip-dated MCC timetrees (Supplementary Data 2) were used to assess the robustness of results.
Morphological synapomorphies for the placement of Voay were based on parsimony optimizations of phenotypic characters using PAUP*. Unequivocally optimized character state changes were mapped to alternative trees for Crocodylia to assess changes in character support that came with the addition of mtDNA data for Voay to the combined dataset published by Lee and Yates37. We mapped synapomorphic character state changes on our tip-dated MCC tree for the Voay + Crocodylus clade (Fig. 4) and recorded homoplastic evolution of the same characters on Lee and Yates’ tip-dated timetree. We also noted synapomorphies for the Voay + Osteolaemus clade in their tree and recorded homoplasy in these characters on our tree. Finally, the presence of squamosal “horns”, a distinctive cranial feature of Voay (Fig. 1), was optimized to infer the evolutionary history of this trait.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The paleontological collections at the American Museum of Natural History (AMNH) house the two specimens of Voay (= Crocodylus) robustus used in the study (AMNH FR-3101 and AMNH FR-3103). The short read data for each Voay specimen used in the analyses were deposited at the NCBI short read archive (SRA) under Bioproject PRJNA681754.
Change history
25 May 2021
A Correction to this paper has been published: https://doi.org/10.1038/s42003-021-02216-9
References
Willerslev, E. & Cooper, A. Ancient DNA. Proc. Biol. Sci. 272, 3–16 (2005).
Vilstrup, J. T. et al. Mitochondrial phylogenomics of modern and ancient equids. PLoS ONE 8, e55950, (2013). https://doi.org/10.1371/journal.pone.0055950.
Enk, J. M. et al. Ancient whole genome enrichment using baits built from modern DNA. Mol. Biol. Evol. 31, 1292–1294 (2014).
Grealy, A. et al. Eggshell palaeogenomics: Palaeognath evolutionary history revealed through ancient nuclear and mitochondrial DNA from Madagascan elephant bird (Aepyornis sp.) eggshell. Mol. Phylogenet. Evol. (2017). https://doi.org/10.1016/j.ympev.2017.01.005
Mitchell, K. J. et al. Ancient DNA reveals elephant birds and kiwi are sister taxa and clarifies ratite bird evolution. Science. (2009). https://doi.org/10.3356/JRR-08-18.1
Yonezawa, T. et al. Phylogenomics and morphology of extinct paleognaths reveal the origin and evolution of the ratites. Curr. Biol. (2017). https://doi.org/10.1016/j.cub.2016.10.029
Kehlmaier, C. et al. Tropical ancient DNA reveals relationships of the extinct Bahamian giant tortoise Chelonoidis alburyorum. Proc. R. Soc. B Biol. Sci. (2017). https://doi.org/10.1098/rspb.2016.2235
Grealy, A., McDowell, M., Retallick, C., Bunce, M. & Peacock, D. Novel mitochondrial haplotype of spotted-tailed quoll (Dasyurus maculatus) present on Kangaroo Island (South Australia) prior to extirpation. Holocene (2019). https://doi.org/10.1177/0959683619875805
Green, R. E. et al. A draft sequence of the Neandertal genome. Science. (2010). https://doi.org/10.1126/science.1188021
Vernot, B. et al. Excavating Neandertal and Denisovan DNA from the genomes of Melanesian individuals. Science. (2016). https://doi.org/10.1126/science.aad9416
Pilot, M. et al. Global Phylogeographic and admixture patterns in grey wolves and genetic legacy of an ancient Siberian lineage. Sci. Rep. (2019). https://doi.org/10.1038/s41598-019-53492-9
Palkopoulou, E. et al. A comprehensive genomic history of extinct and living elephants. Proc. Natl. Acad. Sci. USA (2018). https://doi.org/10.1073/pnas.1720554115
Kistler, L. et al. Comparative and population mitogenomic analyses of Madagascar’s extinct, giant “subfossil” lemurs. J. Hum. Evol. (2015). https://doi.org/10.1016/j.jhevol.2014.06.016
Kistler, L., Ware, R., Smith, O., Collins, M. & Allaby, R. G. A new model for ancient DNA decay based on paleogenomic meta-analysis. Nucleic Acids Res. (2017). https://doi.org/10.1093/nar/gkx361
Crowley, B. E. A refined chronology of prehistoric Madagascar and the demise of the megafauna. Quat. Sci. Rev. (2010). https://doi.org/10.1016/j.quascirev.2010.06.030
Goodman, S. M. & Jungers, W. L. A Brief History of Climatic Change on Madagascar since the Late Pleistocene. In Extinct Madagascar: Picturing the island’s past (2014).
Crowley, B. E. et al. Island-wide aridity did not trigger recent megafaunal extinctions in Madagascar. Ecography. (2017). https://doi.org/10.1111/ecog.02376
Godfrey, L. R. et al. A new interpretation of Madagascar’s megafaunal decline: The “Subsistence Shift Hypothesis”. J. Hum. Evol. (2019). https://doi.org/10.1016/j.jhevol.2019.03.002
Hansford, J. et al. Early Holocene human presence in Madagascar evidenced by exploitation of avian megafauna. Sci. Adv. (2018). https://doi.org/10.1126/sciadv.aat6925
Wang, L. et al. The African Humid Period, rapid climate change events, the timing of human colonization, and megafaunal extinctions in Madagascar during the Holocene: Evidence from a 2m Anjohibe Cave stalagmite. Quat. Sci. Rev. 210, 136–153 (2019).
Austin, J. J., Arnold, E. N. & Bour, R. Was there a second adaptive radiation of giant tortoises in the Indian Ocean? Using mitochondrial DNA to investigate speciation and biogeography of Aldabrachelys (Reptilia, Testudinidae). Mol. Ecol. (2003). https://doi.org/10.1046/j.1365-294X.2003.01842.x
Mitchell, K. J., Wood, J. R., Scofield, R. P., Llamas, B. & Cooper, A. Ancient mitochondrial genome reveals unsuspected taxonomic affinity of the extinct Chatham duck (Pachyanas chathamica) and resolves divergence times for New Zealand and sub-Antarctic brown teals. Mol. Phylogenet. Evol. (2014). https://doi.org/10.1016/j.ympev.2013.08.017
van der Geer, A. A. E., Lyras, G. A., Mitteroecker, P. & MacPhee, R. D. E. From Jumbo to Dumbo: Cranial shape changes in elephants and hippos during phyletic dwarfing. Evol. Biol. (2018). https://doi.org/10.1007/s11692-018-9451-1
Vaillant, L., Grandidier, A. Histoire naturelle des reptiles: premier partie: Crocodiles et tortues. Impr. Natl. 17, (1910).
Vaillant, L. Remarques sur le Crocodilus robustus, Vaillant et Grandidier, de Madagascar. Compte Rendu Acad. Sci. 97, 1081–1083 (1883).
King, F. W. & Burke, R. L. Crocodilians, tuatara, and turtle species of the world. Assoc. Syst. Coll. (1989).
Grandidier, Alfred & Vaillant, L. Sur le crocodile fossile d’Amboulistare (Madagascar). Compte Rendu Acad. Sci. 75, 150–151 (1872).
Grandidier, A. Description des quleques reptiles noveaux decouverts a Madagascar en 1870. Ann. Sci. Natur. Paris 15, 6–11 (1872).
Barbour, T. Vertebrata from Madagascar; Amphibia, Reptilia. Bull. Museum Comp. Zool. 479–489 (1918).
Boettger, O. Die Reptilien und Amphibien von Madagascar. (2011). https://doi.org/10.5962/bhl.title.9082
Mook, C. C. Individual and age variations in the skulls of recent Crocodilia. Bull. Am. Museum Nat. Hist. (1921).
Mook, C. Description of a skull of the extinct Madagascar crocodile, Crocodylus robustus Vaillant and Grandidier. Am. Museum Novit. (1921).
Brochu, C. A. Morphology, relationships, and biogeographical significance of an extinct horned crocodile (Crocodylia, Crocodylidae) from the Quaternary of Madagascar. Zool. J. Linn. Soc. 150, 835–863 (2007).
Brochu, C. A., Njau, J., Blumenschine, R. J. & Densmore, L. D. A new horned crocodile from the Plio-Pleistocene hominid sites at Olduvai Gorge, Tanzania. PLoS ONE (2010). https://doi.org/10.1371/journal.pone.0009333
Brochu, C. A. & Storrs, G. W. A giant crocodile from the Plio-Pleistocene of Kenya, the phylogenetic relationships of Neogene African crocodylines, and the antiquity of Crocodylus in Africa. J. Vertebr. Paleontol. (2012). https://doi.org/10.1080/02724634.2012.652324
Conrad, J. L. et al. New specimens of Crocodylus pigotti (Crocodylidae) from Rusinga Island, Kenya, and generic reallocation of the species. J. Vertebr. Paleontol. (2013). https://doi.org/10.1080/02724634.2013.743404
Lee, M. S. Y. & Yates, A. M. Tip-dating and homoplasy: Reconciling the shallow molecular divergences of modern gharials with their long fossil record. Proc. R. Soc. B Biol. Sci. (2018). https://doi.org/10.1098/rspb.2018.1071
Salas-Gismondi, R., Moreno-Bernal, J. W., Scheyer, T. M., Sánchez-Villagra, M. R. & Jaramillo, C. New Miocene Caribbean gavialoids and patterns of longirostry in crocodylians. J. Syst. Palaeontol. 17, (2019).
Iijima, M. & Kobayashi, Y. Mosaic nature in the skeleton of East Asian crocodylians fills the morphological gap between “Tomistominae” and Gavialinae. Cladistics (2019). https://doi.org/10.1111/cla.12372
Gatesy, J., Amato, G., Norell, M., DeSalle, R. & Hayashi, C. Combined support for wholesale taxic atavism in gavialine crocodylians. Syst. Biol. (2003). https://doi.org/10.1080/10635150390197037
Gatesy, J., Baker, R. H. & Hayashi, C. Inconsistencies in arguments for the supertree approach: Supermatrices versus supertrees of Crocodylia. Syst. Biol. (2004). https://doi.org/10.1080/10635150490423971
Muldoon, K. M. et al. Early Holocene fauna from a new subfossil site: a first assessment from Christmas River, south central Madagascar. Madagascar Conserv. Dev. (2012). https://doi.org/10.4314/mcd.v7i1.5
Gatesy, J. & Amato, G. The rapid accumulation of consistent molecular support for intergeneric crocodylian relationships. Mol. Phylogenet. Evol. (2008). https://doi.org/10.1016/j.ympev.2008.02.009
Brochu, C. A. Morphology, fossils, divergence timing, and the phylogenetic relationships of Gavialis. Syst. Biol. (1997). https://doi.org/10.2307/2413693
Mook, C. Skull characters of recent Crocodilia: With notes on the affinities of the recent genera. Am. Museum Novit. (1921).
Brochu, C. A. Phylogenetic relationships of Palaeogene ziphodont eusuchians and the status of Pristichampsus Gervais, 1853. Earth Environ. Sci. Trans. R. Soc. Edinburgh (2013). https://doi.org/10.1017/S1755691013000200
Densmore, L. D. Biochemical and immunological systematics of the Order Crocodilia. In Evolutionary Biology (1983). https://doi.org/10.1007/978-1-4615-6971-8_8
Gatesy, J. & Amato, G. D. Sequence similarity of 12S ribosomal segment of mitochondrial DNAs of gharial and false gharial. Copeia (1992). https://doi.org/10.2307/1446560
Iijima, M. Assessment of trophic ecomorphology in non-alligatoroid crocodylians and its adaptive and taxonomic implications. J. Anat. 231, 192–211 (2017).
Ballell, A., Moon, B. C., Porro, L. B., Benton, M. J. & Rayfield, E. J. Convergence and functional evolution of longirostry in crocodylomorphs. Palaeontology (2019). https://doi.org/10.1111/pala.12432
Drumheller, S. K. & Wilberg, E. W. A synthetic approach for assessing the interplay of form and function in the crocodyliform snout. Zool. J. Linn. Soc. (2019). https://doi.org/10.1093/zoolinnean/zlz081
Felice, R. N. et al. Evolutionary integration and modularity in the archosaur cranium. In Integrative and Comparative Biology (2019). https://doi.org/10.1093/icb/icz052
Godoy, P. L. Crocodylomorph cranial shape evolution and its relationship with body size and ecology. J. Evol. Biol. (2019). https://doi.org/10.1111/jeb.13540
Groh, S. S., Upchurch, P., Barrett, P. M. & Day, J. J. The phylogenetic relationships of neosuchian crocodiles and their implications for the convergent evolution of the longirostrine condition. Zool. J. Linn. Soc. (2019). https://doi.org/10.1093/zoolinnean/zlz117
Morris, Z. S., Vliet, K. A., Abzhanov, A. & Pierce, S. E. Heterochronic shifts and conserved embryonic shape underlie crocodylian craniofacial disparity and convergence. Proc. R. Soc. B Biol. Sci. (2019). https://doi.org/10.1098/rspb.2018.2389
Sookias, R. B. Exploring the effects of character construction and choice, outgroups and analytical method on phylogenetic inference from discrete characters in extant crocodilians. Zool. J. Linn. Soc. (2019). https://doi.org/10.1093/zoolinnean/zlz015
Wilberg, E. W. Phylogenetic and morphometric assessment of the evolution of the longirostrine crocodylomorphs. Univ. Iowa (2012).
Oaks, J. R. A time-calibrated species tree of Crocodylia reveals a recent radiation of the true crocodiles. Evolution 65, 3285–3297 (2011).
Meredith, R. W., Hekkala, E. R., Amato, G. & Gatesy, J. A phylogenetic hypothesis for Crocodylus (Crocodylia) based on mitochondrial DNA: evidence for a trans-Atlantic voyage from Africa to the New World. Mol. Phylogenet. Evol. 60, 183–191 (2011).
Hekkala, E. et al. An ancient icon reveals new mysteries: Mummy DNA resurrects a cryptic species within the Nile crocodile. Mol. Ecol. 20, 4199–4215 (2011).
Nicolaï, M. P. J. & Matzke, N. J. Trait-based range expansion aided in the global radiation of Crocodylidae. Glob. Ecol. Biogeogr. 28, 1244–1258 (2019).
Brochu, C. A. Phylogenetic relationships and divergence timing of Crocodylus based on morphology and the fossil record. Copeia (2000). https://doi.org/10.1643/0045-8511(2000)000[0657:pradto]2.0.co;2
Jemvall, J., Wright, P. C., Ravoavy, F. L., Simons, E. L. Report on the findings of subfossils at Amposa and Ampanihy in Southwestern Madagascar. Lemurs News 8, (2003).
deMenocal, P. B. African climate change and faunal evolution during the Pliocene-Pleistocene. Earth Planet. Sci. Lett. 220, 3–24 (2004).
Samonds, K. E. et al. Spatial and temporal arrival patterns of Madagascar’s vertebrate fauna explained by distance, ocean currents, and ancestor type. Proc. Natl. Acad. Sci. USA (2012). https://doi.org/10.1073/pnas.1113993109
Samonds, K. E. et al. Imperfect isolation: factors and filters shaping Madagascar’s extant vertebrate fauna. PLoS ONE (2013). https://doi.org/10.1371/journal.pone.0062086
Taplin, L. E. & Grigg, G. C. Historical zoogeography of the eusuchian crocodilians: a physiological perspective. Integr. Comp. Biol. 29, 885–901 (1989).
Mazzotti, F. J. & Dunson, W. A. Adaptations of Crocodylus acutus and Alligator for life in saline water. Comp. Biochem. Physiol. Part A Physiol. (1984). https://doi.org/10.1016/0300-9629(84)90462-6
Pidcock, S., Taplin, L. E. & Grigg, G. C. Differences in renal-cloacal function between Crocodylus porosus and Alligator mississippiensis have implications for crocodilian evolution. J. Comp. Physiol. B Biochem. Syst. Environ. Physiol. 167, 153–158 (1997).
Larsen, P. F., Nielsen, E. E., Williams, T. D. & Loeschcke, V. Intraspecific variation in expression of candidate genes for osmoregulation, heme biosynthesis and stress resistance suggests local adaptation in European flounder (Platichthys flesus). Heredity 101, 247–259 (2008).
Leslie, A. J. & Spotila, J. R. Osmoregulation of the Nile crocodile, Crocodylus niloticus, in Lake St. Lucia, Kwazulu/Natal, South Africa. Comp. Biochem. Physiol. A Mol. Integr. Physiol. (2000). https://doi.org/10.1016/S1095-6433(00)00215-4
MacPhee, R. D. E., Burney, D. A. & Wells, N. A. Early Holocene chronology and environment of Ampasambazimba, a Malagasy subfossil lemur site. Int. J. Primatol. https://doi.org/10.1007/BF02735571 (1985).
Burney, D. et al. Environmental change, extinction and human activity: Evidence from caves in NW Madagascar. J. Biogeogr. (2003). https://doi.org/10.1046/j.1365-2699.1997.00146.x
Burney, D. A. et al. A chronology for late prehistoric Madagascar. J. Hum. Evol. (2004). https://doi.org/10.1016/j.jhevol.2004.05.005
Bickelmann, C. & Klein, N. The late Pleistocene horned crocodile Voay robustus (Grandidier & Vaillant, 1872) from Madagascar in the Museum für Naturkunde Berlin. Foss. Rec. (2009). https://doi.org/10.1002/mmng.200800007
Chesson, P. Mechanisms of maintenance of species diversity. Annu. Rev. Ecol. Syst. 31, 343–366 (2000).
Rubidge, E. M., Monahan, W. B., Parra, J. L., Cameron, S. E. & Brashares, J. S. The role of climate, habitat, and species co-occurrence as drivers of change in small mammal distributions over the past century. Glob. Chang. Biol. 17, 696–708 (2011).
Dowell, S. A. & Hekkala, E. R. Divergent lineages and conserved niches: using ecological niche modeling to examine the evolutionary patterns of the Nile monitor (Varanus niloticus). Evol. Ecol. 30, (2016).
Shirley, M. H. & Austin, J. D. Did Late Pleistocene climate change result in parallel genetic structure and demographic bottlenecks in sympatric Central African crocodiles, Mecistops and Osteolaemus? Mol. Ecol. (2017). https://doi.org/10.1111/mec.14378
Mathews, J. C. & Samonds, K. E. A juvenile subfossil crocodylian from Anjohibe Cave, Northwestern Madagascar. PeerJ (2016). https://doi.org/10.7717/peerj.2296
Milián-García, Y., Ramos-Targarona, R., Pérez-Fleitas, E., Espinosa-López, G., & Russello, M. A. Genetic evidence of hybridization between the critically endangered Cuban crocodile and the American crocodile: implications for population history and in situ/ex situ conservation. Heredity (2015). https://doi.org/10.1038/hdy.2014.96
Rhymer, J. & Simberloff, D. Extinction by hybridization and introgression. Annu. Rev. Ecol. Syst. (1996). https://doi.org/10.1146/annurev.ecolsys.27.1.83
Milián-García, Y. et al. Genetic evidence supports a distinct lineage of American crocodile (Crocodylus acutus) in the Greater Antilles. PeerJ 6, e5836 (2018).
Milián-García, Y. et al. Phylogenomics reveals novel relationships among Neotropical crocodiles (Crocodylus spp.). Mol. Phylogenet. Evol. 152, 106924 (2020).
White, E. I. Fossil Hunting in Madagascar. Natural History Magazine London 209–235 (1930).
Goodman, S. M., Jungers, W. L. & Simeonovski, V. Extinct Madagascar (2019). https://doi.org/10.7208/chicago/9780226156941.001.0001
Gamba, C. et al. Comparing the performance of three ancient DNA extraction methods for high-throughput sequencing. Mol. Ecol. Resour. (2016). https://doi.org/10.1111/1755-0998.12470
Carpenter, M. L. et al. Pulling out the 1%: Whole-Genome capture for the targeted enrichment of ancient DNA sequencing libraries. Am. J. Hum. Genet. 93, 852–864 (2013).
Peltzer, A. et al. EAGER: efficient ancient genome reconstruction. Genome Biol. https://doi.org/10.1186/s13059-016-0918-z (2016).
Knapp, M. & Hofreiter, M. Next generation sequencing of ancient DNA: requirements, strategies and perspectives. Genes 1, 227–243 (2010).
Günther, T. & Nettelblad, C. The presence and impact of reference bias on population genomic studies of prehistoric human populations. PLoS Genet. (2019). https://doi.org/10.1371/journal.pgen.1008302
Eaton, M. J., Martin, A., Thorbjarnarson, J. & Amato, G. Species-level diversification of African dwarf crocodiles (Genus Osteolaemus): A geographic and phylogenetic perspective. Mol. Phylogenet. Evol. 50, 496–506 (2009).
Shirley, M. H., Vliet, K. A., Carr, A. N. & Austin, J. D. Rigorous approaches to species delimitation have significant implications for African crocodilian systematics and conservation. Proc. Biol. Sci. 281, 20132483 (2014).
Shirley, M. H., Carr, A. N., Nestler, J. H., Vliet, K. A. & Brochu, C. A. Systematic revision of the living African slender-snouted crocodiles (Mecistops Gray, 1844). Zootaxa (2018). https://doi.org/10.11646/zootaxa.4504.2.1
Gauthier, J., Kluge, A. G. & Rowe, T. Amniote phylogeny and the importance of fossils. Cladistics (1988). https://doi.org/10.1111/j.1096-0031.1988.tb00514.x
Chiari, Y., Cahais, V., Galtier, N. & Delsuc, F. Phylogenomic analyses support the position of turtles as the sister group of birds and crocodiles (Archosauria). BMC Biol. (2012) https://doi.org/10.1186/1741-7007-10-65
Edgar, R. C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. (2004). https://doi.org/10.1093/nar/gkh340
Meintjes, P. et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics (2012).
Rambaut, A. Se-Al: Sequence alignment editor, 2.0a11. (1996). http://evolve.zoo.ox.ac.uk
Swofford, D. L. Phylogenetic Analysis Using Parsimony * (and other methods). Version 4. Options (2002). https://doi.org/10.1159/000170955
Goloboff, P. A. Estimating character weights during tree search. Cladistics (1993). https://doi.org/10.1111/j.1096-0031.1993.tb00209.x
Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution (1985). https://doi.org/10.2307/2408678
Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics https://doi.org/10.1093/bioinformatics/btu033 (2014).
Miller, M. A., Pfeiffer, W. & Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In 2010 Gateway Computing Environments Workshop, GCE 2010 (2010). https://doi.org/10.1109/GCE.2010.5676129
Abadi, S., Azouri, D., Pupko, T., Mayrose, I. Model selection may not be a mandatory step for phylogeny reconstruction. Nat. Commun. (2019). https://doi.org/10.1038/s41467-019-08822-w
Bernhart, S. H., Hofacker, I. L., Will, S., Gruber, A. R., & Stadler, P. F. RNAalifold: Improved consensus structure prediction for RNA alignments. BMC Bioinformatics (2008). https://doi.org/10.1186/1471-2105-9-474
Li, W. L. S. & Drummond, A. J. Model averaging and Bayes factor calculation of relaxed molecular clocks in Bayesian phylogenetics. Mol. Biol. Evol. (2012). https://doi.org/10.1093/molbev/msr232
Cossette, A. P. et al. A new crocodylid from the middle Miocene of Kenya and the timing of crocodylian faunal change in the late Cenozoic of Africa. J. Paleontol. 94, 1165–1179 (2020).
Rambaut, A. & Drummond, A. J. Tracer v1.4. Encycl. Atmos. Sci. (2007). https://doi.org/10.1017/CBO9781107415324.004
Warren, D. L., Geneva, A. J., Lanfear, R. & Rosenberg, M. RWTY (R We There Yet): An R package for examining convergence of Bayesian phylogenetic analyses. Mol. Biol. Evol. (2017). https://doi.org/10.1093/molbev/msw279
Springer, M. S., Meredith, R. W., Janecka, J. E., & Murphy, W. J. The historical biogeography of Mammalia. Philos. Trans. R. Soc. B Biol. Sci. (2011). https://doi.org/10.1098/rstb.2011.0023
Acknowledgements
We thank K. Vliet and St. Augustine Alligator Farm Zoological Park (St. Augustine, Florida, USA) for tissue samples of extant crocodylians, and M. Lee for providing phylogenetic trees from published work. For funding we thank University of California - Riverside (to J.G.), Fordham University Faculty Fellowship and the National Science Foundation -NSF RAPID DEB1931213 (to E.H.), and NSF grants DEB-1556701, DBI-1725932 (to R.M.).
Author information
Authors and Affiliations
Contributions
J.G., R.M., A.N. and M.A.: conceptualization of analytical methodology, software management, data visualization, writing, data curation, formal analysis, project administration. E.H., S.M., M.R. and E.J.: conceptualization of laboratory methods and data collection. J.G., R.M., E.H. and G.A.: writing–review and editing, visualization, validation. E.H., C.B., M.N. and G.A: conceptualization of project, acquisition of resources, writing–original draft, review and editing, visualization, supervision, funding acquisition.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hekkala, E., Gatesy, J., Narechania, A. et al. Paleogenomics illuminates the evolutionary history of the extinct Holocene “horned” crocodile of Madagascar, Voay robustus. Commun Biol 4, 505 (2021). https://doi.org/10.1038/s42003-021-02017-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-021-02017-0
This article is cited by
-
Ancient DNA re-opens the question of the phylogenetic position of the Sardinian pika Prolagus sardus (Wagner, 1829), an extinct lagomorph
Scientific Reports (2023)
-
Cross-species chromosome painting and repetitive DNA mapping illuminate the karyotype evolution in true crocodiles (Crocodylidae)
Chromosoma (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
