
Abstract
Catalytically inactive dCas9 imposes transcriptional gene repression by sterically precluding RNA polymerase activity at a given gene to which it was directed by CRISPR (cr)RNAs. This gene silencing technology, known as CRISPR interference (CRISPRi), has been employed in various bacterial species to interrogate genes, mostly individually or in pairs. Here, we developed a multiplex CRISPRi platform in the pathogen Legionella pneumophila capable of silencing up to ten genes simultaneously. Constraints on precursor-crRNA expression were overcome by combining a strong promoter with a boxA element upstream of a CRISPR array. Using crRNAs directed against virulence protein-encoding genes, we demonstrated that CRISPRi is fully functional not only during growth in axenic media, but also during macrophage infection, and that gene depletion by CRISPRi recapitulated the growth defect of deletion strains. By altering the position of crRNA-encoding spacers within the CRISPR array, our platform achieved the gradual depletion of targets that was mirrored by the severity in phenotypes. Multiplex CRISPRi thus holds great promise for probing large sets of genes in bulk in order to decipher virulence strategies of L. pneumophila and other bacterial pathogens.
Similar content being viewed by others

Introduction
Clustered regularly interspaced short palindromic repeats (CRISPR)-Cas gene editing technologies have recently arisen as a mechanism for both fast and targeted gene manipulation in a variety of systems1,2,3. CRISPR-Cas-based genetic tools are derived from components of naturally occurring CRISPR systems found in 87% of archaea and 45% of bacteria genomes surveyed (Crisprfinder; http://crispr.i2bc.paris-saclay.fr/Server/). In these organisms, CRISPR regions serve as an adaptive immune system where fragments of foreign DNA elements such as viruses, transposons, or plasmids are incorporated into the chromosome as unique spacers (S) separated by identical repeats (R) to serve as a immunological memory of past infections1,4,5. RNA transcribed from these repeat/spacer regions, known as the precursor-CRISPR (cr)RNA, is processed into short fragments by RNase III to make individual crRNAs6. Upon re-infection, crRNAs, together with a trans-activating crRNA (tracrRNA), specifically direct, through base-pairing with the target, a protein (or contingent of proteins) with oligonucleotide cleavage capabilities to the foreign DNA or RNA elements for destruction7,8,9. Importantly, the spacers themselves are protected from self-targeting by the crRNAs that they encode as they are missing the protospacer-adjacent motif (PAM)10, a short DNA element found directly downstream of the complementary target sequence on the non-target strand that is required by the surveillance complex to cut.
In the simplest CRISPR-Cas system, Type II, only a single protein known as Cas9 is required for crRNA-guided DNA cleavage, lending it to be the most developed as a genetic tool11,12,13. In bacteria, this system has been adapted for gene silencing by making two simple changes: First, the gene encoding Cas9 was replaced with a catalytically inactive variant of Cas9, called deactivated Cas9 (dCas9), in which the two nuclease domains, RuvC-like and HNH, have been mutated, thus preventing DNA cleavage14,15; and second, by designing arrays in which the spacer sequence(s), that are typically directed against invading DNA elements, have been replaced with sequences complementary to the bacterium’s own genes. These self-targeting crRNAs form a surveillance complex together with the tracrRNA and with dCas9 which, in the absence of cleavage, imposes transcriptional gene repression by sterically precluding RNA polymerase activity at the gene to which the complex was directed, a technology referred to as CRISPR interference (CRISPRi)14,16. Gene silencing is advantageous to the study of essential genes that are otherwise intolerable to deletion, as well as for interrogating genes of interest without laborious null strain construction17. The recent development of mobile-CRISPRi has allowed for gene silencing to be performed in a number of gamma proteobacteria and Bacillales Firmicutes18, yet this and earlier studies, which implemented CRISPRi19, nearly always silenced only one gene or pairs of genes, with few exceptions20,21,22,23. To our knowledge, no group has fully exploited or probed the natural multiplex capability of CRISPR repeat/spacer arrays as a gene silencing tool in bacterial systems.
Here, we established an adaptable multiplex CRISPRi platform capable of silencing up to ten genes simultaneously (10-plex). We demonstrate that this platform can be applied to study bacterial virulence genes not only in axenic media but, importantly, also under disease-relevant conditions such as the infection of macrophages by the intracellular pathogen Legionella pneumophila, thus adding CRISPRi to our toolbox for efficiently studying genetically less tractable human pathogens.
Results
Equipping L. pneumophila with a CRISPRi system
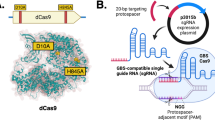
To study the potential of multiplex CRISPRi, we selected the model organism L. pneumophilia, the causative agent of Legionnaires’ pneumonia or a milder form of the disease called Pontiac fever24,25. The common laboratory strain L. pneumophila Philadelphia-1 (Lp02) does not bear an intrinsic CRISPR-Cas system (though some environmental L. pneumophila isolates do26). Thus, we exogenously introduced the genes encoding the three main components of a CRISPRi platform: a Cas protein, crRNAs, and the tracrRNA (Fig. 1a). The Streptococcus pyogenes dCas9-encoding sequence was inserted into the chromosomal thyA locus (lpg2868) as this L. pneumophila gene was already disrupted by a mutation in this strain background27, creating Lp02(dcas9). Furthermore, dcas9 was placed under the control of the tetracycline-inducible tet promoter (Ptet) rather than its native S. pyogenes promoter, allowing the degree of gene repression to reflect the level of anhydrous tetracycline (aTC) added to the system to induce dcas9, as others have done before14. The crRNA-encoding repeat/spacer arrays were provided on plasmids, as was the target-independent (invariable) tracrRNA-encoding sequence. Construction of these single CRISPRi constructs is described in the Methods section and depicted in Supplementary Fig. S1a. Although single guide (sg)RNAs, chimeras of the tracrRNA and crRNA, are convenient for silencing individual genes as to circumvent the need for processing precursor-crRNA into a mature crRNAs6, the tracrRNA-encoding sequences would become repetitive within longer CRISPR arrays. Instead, the task of processing crRNA precursors was seemingly executed efficiently by the intrinsic L. pneumophila RNAse III (lpg1869) in our system (see below).

a Schematic of the L. pneumophila single-plex CRISPRi platform. Chromosome-encoded dCas9 and plasmid-expressed tracrRNA and crRNA assemble into a CRISPRi complex. The crRNA directs the complex to the target gene through base pairing. Gene expression is repressed through sterically precluding RNA polymerase. R indicates repeats and S indicates spacers of the crRNA-encoding repeat/spacer array. b Immunoblot analysis showing the protein level of dCas9 in response to aTC-dependent induction of Ptet-dcas9, leading to decreased protein levels of the crRNAlidA target. Isocitrate dehydrogenase (ICDH) serves as a loading control. aTC concentrations are 0, 0.15, 0.3, 0.63, 1.23, 2.5, 5, 10, 20, and 80 ng/mL. c Immunoblots reveal the efficiency of target repression at different crRNA target sites (arrow heads A-D) for two genes, sidM and vipD (+40 ng/mL aTC). ICDH serves as a loading control. d, e Strains expressing crRNAsidM (site C), crRNAvipD (site C), and crRNAlidA were surveyed for CRISPRi specificity at both the protein (d) and mRNA (e) level (+40 ng/mL aTC). ICDH serves as an immunoblot loading control. qPCR was used to measure mRNA levels of sidM, vipD, lidA and four randomly chosen control genes in Lp02(dcas9) and Lp02 strains. Fold repression was determined using ΔΔCT. A value of 1 indicates that the gene level was the same in the Lp02(dcas9) and Lp02 strains.
Targeting individual genes with single crRNA arrays served as proof-of-concept for introducing a foreign CRISPR-Cas9 platform into our model organism L. pneumophila. These initial experiments were performed on L. pneumophila strains grown in axenic culture while individually targeting three virulence factor, or “effector”, -encoding genes with crRNAs towards lidA (lpg0940; crRNAlidA), sidM (lpg2464; crRNAsidM), and vipD (lpg2831; crRNAvipD), since antibodies directed against their encoded gene products were available. First, we confirmed that a gradual enhancement in the concentration of the inducer aTC led to increased levels of dCas9 and, consequently, decreased levels of LidA, the product of the target lidA, indeed creating a CRISPRi system capable of tunable gene silencing (Fig. 1b; note, all uncropped immunoblots are shown in Supplementary Fig. S8). Next, we assessed how the location of the crRNA annealing site on the target gene (strand, proximity to start codon) affects gene silencing efficiency. In agreement with other studies12,14,28, we found that targeting a 30 base pair sequence near the start of the gene on the non-template strand provided the most efficient knockdown (Fig. 1c, arrowheads C and D). All target sequences were upstream of a PAM sequence (“NGG” on the template strand29).
Lastly, to confirm that crRNAs designed in this manner did not indiscriminately affect gene expression, we monitored protein (Fig. 1d) and mRNA (Fig. 1e) levels of unrelated genes. Using immunoblot analyses and quantitative polymerase chain reaction (qPCR), we found that, while the intended target genes’ protein and mRNA levels were dramatically reduced by CRISPRi, none of the other surveyed genes chosen at random were repressed by crRNA expression. Together, these results confirmed that the aTC-inducible dcas9 and the plasmid-expressed crRNA and tracrRNA formed a functional CRISPRi surveillance complex in L. pneumophila that was tunable, efficient, and target-specific.
A multiplex CRISPRi platform allows for the simultaneous repression of several genes
Most processes in bacteria, including pathogenesis, rely on the combined activity of multiple gene products. With over 300 putative effectors, L. pneumophila encodes one of the largest known virulence arsenals of any bacterial pathogen30,31. We thus explored if our CRISPRi platform could be adapted to simultaneously repress more than one gene at a time. In this new multiplex CRISPRi format, expression of S. pyogenes dcas9 from the L. pneumophila chromosome and the expression of the S. pyogenes tracrRNA from the plasmid backbone remained unchanged. Alterations were made to the method of crRNA expression: First, several unique crRNA-encoding spacer sequences, labeled S1 (for the most proximal spacer) to S10 (for the most distal), were sequentially placed between identical repeat sequences intrinsic to the S. pyogenes CRISPSR-Cas9 system (Fig. 2a). In nature, the number of spacers within a CRISPR array can vary, ranging from a few to several dozen or even hundreds32. Second, expression of the repeats and spacers was placed under the regulation of Ptet, the same aTC-inducible promoter used to express dcas9 from the L. pneumophila chromosome. We had found in experiments on Lp02(dcas9) grown in axenic culture that using the native S. pyogenes promoter (Pnative) to express the multiplex CRISPR (MC) repeat/spacer arrays proved insufficient in repressing more than three to four targets, possibly due to fewer incidences of transcription initiation at a weaker promoter and therefore, fewer opportunities for RNA polymerase to complete transcription of the entire array before termination (Fig. 2b). Incorporation of Ptet fostered expression of the array up to at least spacer S9, as confirmed by immunoblot analysis of the protein levels of LidA whose encoding gene was targeted by crRNAs expressed from the most distal spacer positions (Fig. 2b).

a Schematic representation of the theory of multiplex CRISPRi. A series of repeats, R, and spacers, S1-S10, are expressed as a single precursor-crRNA. Upon processing, individual crRNAs come together with a tracrRNA and dCas9 to simultaneously target ten unique genes for silencing. b Multiplex CRISPR (MC) repeat/spacer array constructs of increasing length were placed under the control of the native S. pyogenes promoter, Pnative, or Ptet. Efficiency of lidA targeting by crRNAs encoded by the terminal spacers of each array was assessed by monitoring LidA protein levels in Lp02(dcas9) and Lp02 by immunoblot. ICDH serves as a loading control. c A Ptet-MC construct, MC-I, capable of expressing ten unique crRNAs was designed. crRNAs encoded by S1, S3, S5, S6, S8, or S10 target genes in which antibodies directed against their encoded gene products are available. Bacteria pellets were collected from axenic cultures containing MC-I or the empty vector (+40 ng/mL aTC) and multiple immunoblots were run to accommodate the range of targets. ICDH serves as a loading control. Replicates are given in Supplementary Fig. S2.
To examine the ability of our multiplex CRISPRi platform to target a variety of genes simultaneously, we first made a synthetic MC array, called MC-I, composed of ten spacers (10-plex) under the regulation of Ptet. The four spacers in position S1, S3, S6, and S10 encoded crRNAs to target the effector-encoding genes lidA, sidM, vipD, and ravN (lpg1111), while the two spacers at position S5 and S8 target dotD (lpg2674) and dotO (lpg0456), two components of the type IV secretion system (T4SS) of L. pneumophila. Construction of MC constructs is described in the Methods section and depicted in Supplementary Fig. S1b. After growth of Lp02(dcas9) containing MC-I in axenic culture, we found that, indeed, the protein levels of all six targets were notably decreased based on immunoblot analysis as compared to a control strain bearing an empty vector (with tracrRNA but no repeat/spacer array) (Fig. 2c and Supplementary Fig. S2). This initial result showed that it is possible to simultaneously silence several unique genes in L. pneumophila using an array of repeats and spacers under the control of a strong promoter.
Spacer position influences gene silencing efficiency in Ptet -MC arrays
Encouraged by the finding that our MC-I 10-plex construct had silenced multiple target genes, we determined how silencing efficiency varied dependent on the spacer position within the CRISPR array. To that end, we designed five additional 10-plex constructs, MC-II through MC-VI (Fig. 3a), in which the aforementioned spacers encoding crRNAlidA, crRNAvipD, crRNAsidM, crRNAdotO, crRNAdotD, and crRNAravN were gradually shifted from proximal positions into more distal positions within the array, occupying either odd- or even-numbered positions. crRNAlpg2793 and crRNAlpg0208 were also encoded by spacers at various positions as these crRNAs had emerged in preliminary studies to silence very efficiently or not at all, respectively.

a Additional Ptet-MC constructs, MC-II through MC-VI, were made to test the effect of spacer position and environment on gene silencing. Each spacer is present in either position S1, S3, S5, S7, and S9 or S2, S4, S6, S8, and S10. b RNA was extracted from Lp02(dcas9) bearing either the empty vector or an MC construct after axenic culture (+40 ng/mL aTC). qPCR was used to measure mRNA levels of target genes. Fold repression was determined using ΔΔCT. Bars indicate the mean of multiple replicates shown as individual data points. A value of 1 indicates that the mRNA level was the same in both strains. c Immunoblot analyses were performed on pellets collected from the same cultures. The blot shown is a representative of three replicates, with bands rearranged corresponding to spacer position. Original immunoblots and ICDH loading controls are shown in Supplementary Fig. S3. d RNA was extracted from Lp02(dcas9) bearing either the vector or the MC-I construct after axenic culture (+40 ng/mL aTC). qPCR was used to measure mRNA levels of target genes. Fold repression was determined using ΔΔCT and plotted in comparison to data first shown in b.
Efficiency of gene silencing by MC-II through VI after two days of induction during axenic growth was assessed for all targets at both the mRNA level and protein level and compared to that of the Lp02(dcas9) control strain containing the empty vector. qPCR analyses indeed revealed a polarity in gene silencing along the length of the repeat/spacer arrays, as has been noted for other CRISPRi platforms before21,33, with spacers in the proximal and distal positions causing the most- or least-efficient gene silencing, respectively (Fig. 3b). Some crRNAs did silence their target genes very well (crRNAsidM, crRNAravN, and crRNAlpg2793) even when encoded from distal spacer positions. In fact, ravN was as efficiently silenced by crRNAravN encoded by the S10 spacer (MC-IV) as lidA was by crRNAlidA encoded by the S1 spacer (Fig. 3b). crRNAlpg2793, which showed continuously high levels of repression, experienced greater than an order of magnitude repression even with the crRNA encoded by spacer S10. Conversely, crRNAlpg0208 did not work well regardless of the encoding spacer position. Notably, all genes except lpg0208 experienced at least an order of magnitude repression when targeted by crRNAs encoded from spacers at positions as distal as S4, and five of them showed at least an order of magnitude silencing up to spacer position S6. Immunoblot analysis of the six targets for which antibodies were available confirmed that protein levels correlated very well with the mRNA abundance (Fig. 3c and Supplementary Fig. S3), with depletion being near or below the detection limit. dotD showed better than expected gene silencing with the most distal S9 spacer (MC-IV) for reasons that remain to be determined.
Gene silencing efficiency by CRISPRi is independent of spacer environment
crRNAs within the various MCs (MC-II through MC-VI), although transcribed from spacers in different positions, were always flanked by the same pair of neighboring spacers. For example, the spacer encoding the less efficient crRNAlidA was always surrounded at the 3’ and 5’ ends by spacers encoding crRNAlpg0208 and crRNAvipD, respectively. Those neighboring spacers may have inadvertently affected transcription or processing of crRNAlidA. To test if the spacer environment had an effect on crRNA production and therefore gene silencing efficiency, which has been proposed in the past34, we compared target gene repression by crRNAs that were encoded by spacers at identical positions but surrounded by different spacers. crRNAlidA, crRNAsidM, crRNAdotD, and crRNAlpg2793 are encoded from the same position in MC-I and MC-II (spacers S1, S3, S5, and S7), while crRNAlpg0208, crRNAvipD, crRNAdotO, and crRNAravN originate from position S4, S6, S8, and S10 in both MC-I and MC-IV, yet they are flanked by entirely different sets of spacers (Fig. 3a). Upon expression in Lp02(dcas9), crRNAs encoded by MC-II or MC-IV, despite being encoded from different spacer environments, consistently achieved the same level of gene repression as those encoded by MC-I (Fig. 3d). Together, these results demonstrate that spacer position along the array has a much greater impact on multiplex CRISPRi-mediated gene silencing than the spacer environment.
boxA elements reduce polarity of spacer transcription to promote maximum gene silencing
While the addition of the strong Ptet promoter did enhance gene silencing by distal spacers (Fig. 2b), gene silencing polarity within the MC arrays was still not fully overcome (Fig. 3), indicating that additional factors likely influenced crRNA production. BoxA elements are often found upstream of long non-coding RNAs, like the 16 S RNA35, where they are bound by the Nus complex to improve RNA polymerase processivity by multiple mechanisms36. Recently Stringer et. al reported that boxA elements may be evolutionarily conserved upstream of naturally occurring CRISPR arrays where they appear to promote expression of downstream spacers within long arrays37.
To investigate whether a boxA element could promote expression of our synthetic repeat/spacer array, we surveyed the levels of precursor-crRNA expression in strains bearing constructs with and without boxA in the absence of tracrRNA (to limit precursor-crRNA processing). In deciding the boxA sequence to incorporate, we compared the E. coli boxA consensus sequence with that of the Coxiella 16 S boxA and the boxA found upstream of the naturally occurring L. pneumophila sp. Lens CRISPR array (Fig. 4a). Interestingly, a boxA element was not readily identified upstream of the L. pneumophila Philadelphia-1 16 S RNA encoding region. We chose to first mutate the leader sequence of the MC-II and MC-IV constructs to include the 11 base pair L. pneumophila sp. Lens boxA sequence (5′-GTTCTTTAAAA-3′), and the five flanking base pairs on either side, at both a distance of -58 (boxA(-58)) and -90 (boxA(-90)) base pairs upstream of the first repeat while maintaining the overall length of the leader sequence (Fig. 4b, see Supplementary Fig. S4 for sequence detail). RNA extracted from axenic cultures induced with aTC was analyzed by qPCR using primer pairs that probed three different regions along the length of the anticipated precursor-crRNA. We found that boxA containing constructs do in fact express on average ~10-fold more precursor-crRNA than boxA-less strains throughout the entire length of the synthetic repeat/spacer array (Fig. 4c), suggesting that boxA does promote array transcription.

a Sequence alignment of the boxA elements identified in E. coli (boxA consensus), Coxiella (16 S RNA encoding sequence), and L. pneumophila sp. Lens (CRISPR array). b The boxA motif from the L. pneumophila sp. Lens CRISPR array was added to the leader sequence of the Ptet-MC constructs either -58 (boxA(-58)) or -90 (boxA(-90)) base pairs upstream of the repeat/spacer array. boxA is depicted as a pink square. c Constructs lacking the tracrRNA-encoding sequence were used to probe precursor-crRNA expression from boxA-containing vs. boxA-less constructs in axenic cultures of Lp02(dcas9) (+40 ng/mL aTC). Precursor-crRNA levels were measured by qPCR using primer pairs that annealed to three different regions of the precursor-crRNA. Fold increased expression vs. boxA-less constructs was determined using ΔΔCT. Data are ordered by spacer position in constructs MC-II or MC-IV. d Constructs with the tracrRNA-encoding sequence were used to probe gene repression by qPCR of RNA from axenic cultures of Lp02(dcas9) bearing boxA constructs to that with the empty vector (+40 ng/mL aTC). Fold repression was determined using ΔΔCT. For comparison, boxA-less qPCR data from Fig. 3 is shown again. For visualization of protein levels, pellets from boxA-less and boxA(-58) and boxA(-90) strains were collected and processed for immunoblot analysis. The bands were reordered corresponding to their spacer position. Original immunoblots and ICDH loading controls are shown in Supplementary Fig. S5.
With evidence that boxA does increase precursor-crRNA levels, we next asked how gene silencing was affected. Upon introducing boxA in either the -58 or -90 position for all MC constructs (now with tracrRNA present to promote crRNA processing into individual gene targeting units), mRNA and protein analysis of construct-bearing Lp02(dcas9) strains revealed that this boxA element greatly resolved gene silencing polarity for the majority of the target genes tested (Fig. 4d and Supplementary Fig. S5). The presence of boxA in either the -58 or -90 position maximized silencing of sidM, dotO, dotD, ravN, and lpg2793 by crRNAs encoded from all spacer positions, even S9 or S10, presumably by allowing transcription of the repeat/spacer array to proceed throughout the entire length of the array. sidM, dotD, ravN, and lpg2793 displayed nearly two orders of magnitude gene repression from crRNAs encoded from all spacer positions, a vast improvement over the diminishing repression observed by boxA-less constructs. In all cases, the degree of mRNA depletion was mirrored by the reduction in protein levels. Minor discrepancies in dotD and DotD levels suggest slower protein turnover rates/increased protein stability of DotD compared to other proteins probed. Silencing of lidA, lpg0208, and vipD was not altered by the addition of boxA, suggesting that other factors such as intrinsic gene regulation and genomic environment were contributing to the expression of the target genes and were possibly inhibiting silencing efficiency. In conclusion, the processivity of transcription of long repeat/spacer arrays is the major driving factor of achieving multiplex gene silencing even from distal spacer positions.
CRISPRi is functional during L. pneumophila intracellular growth
While CRISPRi has been performed in a variety of bacteria during growth in axenic culture, gene silencing had yet to be accomplished in any pathogen during infection. We confirmed that our platform was functional in L. pneumophila undergoing intracellular growth and that, upon repression of specific targets, we could reproduce previously reported intracellular growth phenotypes.
L. pneumophila requires the Dot/Icm T4SS to deliver effector proteins into the host cell in order to establish a replication vacuole. Strains that bear loss-of-function mutations in the genes encoding T4SS subunits are unable to establish this specialized vacuole and fail to replicate38. The ATPase DotO and the outer membrane protein DotD are components critical for the function of the T4SS transporter, and L. pneumophila mutants with disruptions in their encoding genes (lpg0456 and lpg2674, respectively) are attenuated for virulence39,40,41. Plasmids encoding crRNAs directed against dotO (crRNAdotO) or dotD (crRNAdotD) were individually introduced to Lp02(dcas9) and, after 48 h of CRISPRi, decreased protein levels of DotO and DotD were verified by immunoblot analysis (Fig. 5a). It is important to note, and well established39,40, that mutations disabling the T4SS have no influence on the general fitness of L. pneumophila outside the context of the host. Human-derived U937 macrophages42,43 were then challenged with these crRNA-expressing strains, and bacterial growth was monitored over a period of 72 h (Fig. 5a). While the Lp02(dcas9) strain containing the empty plasmid grew several orders of magnitude, the CRISPRi strains depleted of either DotO or DotD were impaired for growth at a level comparable to that of the avirulent mutant strain Lp03 which bears a chromosomal mutation in the dotA gene that encodes another critical subunit of the T4SS27.

a Immunoblots confirm decreased levels of DotO and DotD in Lp02(dcas9) strains expressing crRNAdotO or crRNAdotD. ICDH serves as a loading control. Growth of these strains in U937 macrophages was compared to that of Lp02(dcas9) bearing an empty vector and the avirulent strain Lp03, with a chromosomal mutation in dotA. Growth is plotted as CFU/well normalized to the CFU at 2 h post infection (hpi). b Growth of Lp02 and Lp02(dcas9) expressing crRNAmavN was assayed in U937 macrophages. Growth is given as the average of CFU/well of three experiments at 2 and 72 hpi. mRNA levels of mavN in Lp02(dcas9) vs. Lp02 were measured by qPCR. Fold repression was determined using ΔΔCT.
In agreement with this result, our efforts to target and repress an effector-encoding gene also proved successful. MavN is a multi-pass membrane protein from L. pneumophila that is essential for delivering metal ions across the LCV membrane into the vacuolar lumen44,45. Deletion of mavN (lpg2815) from the L. pneumophila chromosome results in a complete growth defect in A/J mouse macrophages44. Upon targeting mavN with crRNAmavN, we observed a similarly dramatic growth defect in U937 macrophages in the Lp02(dcas9) strain, while the Lp02 control strain grew robustly over the same time period (Fig. 5b). In the absence of an antibody specific to MavN, mRNA levels were analyzed by qPCR and confirmed mavN to be repressed more than ten-fold in the Lp02(dcas9) strain relative to the Lp02 control strain. These data indicate that CRISPRi is functional in L. pneumophila both during growth in axenic media and during a multi-day infection experiment.
Multiplex CRISPRi reveals gene dosage effects during intracellular growth
Next, we explored if intracellular growth of L. pneumophila can be manipulated through multiplex CRISPRi. We created dotO-specific Ptet-MC constructs (called MC-II-dotO to MC-VI-dotO; Fig. 6a) that mimicked Ptet-MC-II through MC-VI, except that all crRNA-encoding spacers besides the crRNAdotO-encoding spacer were scrambled, rendering them incapable of base pairing with their original targets. Notably, the base pair count and composition (“agtc” content) of the scrambled spacers were maintained, and all repeat sequences remained intact. As a result, these multiplex arrays should encode only crRNAdotO as functional crRNA from spacer positions S2, S4, S6, S8, or S10 for direct comparison to the single crRNAdotO-encoding CRISPRi construct (from Fig. 5). Furthermore, we created a control construct, MC-VII-dotO, in which the dotO-targeting spacer in position S2 was scrambled to provide evidence that any observed phenotypes were a direct result of silencing dotO.

a boxA-less, dotO-targeting Ptet-MC constructs, MC-II-dotO, MC-III-dotO, MC-IV-dotO, MC-V-dotO, and MC-VI-dotO, were constructed such that the sequences of all spacers, other than dotO, were scrambled. The dotO spacer can be found in the same position as in the original MC-II through MC-VI constructs. MC-VII-dotO is identical to MC-VI-dotO except that the dotO spacer is also scrambled. crRNAdotO is the same as in Fig. 5. Later, a boxA sequence was added in the -58 position to MC-V-dotO to create boxA(-58) S10. b RNA extracted from axenic cultures of Lp02(dcas9) bearing these constructs was compared to that of Lp02(dcas9) bearing the empty vector by qPCR, as in Fig. 3 (+40 ng/mL aTC). c Immunoblot analyses were performed on pellets collected from the same cultures and samples were run according to dotO spacer position. Immunoblot replicates are found in Supplementary Fig. S6. ICDH serves as a loading control. d aTC-induced Lp02(dcas9) bearing boxA-less and boxA(-58) constructs were used to infect U937 macrophages. Growth of these strains and Lp02(dcas9) and Lp03 bearing the empty vector was monitored over 48 h post infection (hpi). Colony forming unit (CFU) counts were normalized to the count at two hours post infection. Normalized counts for each experiment at each timepoint are shown. n.s. = not significant (p = 0.44, n = 3), *p = 0.033, n = 2 or 3, one-unpaired t-test. All replicates are from distinct experiments.
We examined dotO repression by the new constructs in axenic culture to establish the degree of dotO silencing efficiency. qPCR revealed that dotO silencing by MC-VI-dotO and MC-II-dotO constructs with crRNAdotO-encoding spacers in the proximal S2 and S4 positions, respectively, was robust and comparable to that of the single crRNAdotO-encoding CRISPR construct (Fig. 6b). dotO silencing efficiency by the MC-III-dotO to MC-V-dotO constructs gradually decreased with increasing distal shift of the crRNAdotO-encoding spacer in these arrays. As expected, the scrambled dotO spacer in MC-VII-dotO did not promote gene silencing, showing that the effects of MC-II-dotO to MC-VI-dotO was specific to crRNAdotO. Again, immunoblot analyses of cell pellets collected from these axenic L. pneumophila cultures showed depletion of DotO to the same extent as would be predicted from the mRNA abundance data (Fig. 6c and Supplementary Fig. S6).
We then challenged U937 macrophages with these boxA-less crRNAdotO-expressing strains and compared their growth over a period of 48 h to that of Lp02(dcas9) with the empty vector, which served as a measure of maximum growth, and Lp03, which served as a measure of maximum attenuation (Fig. 6d, top). Strikingly, the severity of the growth defect precisely mirrored the reduction in dotO mRNA levels caused by these constructs, with the most severe defect caused by crRNA expression from the S2 spacer (MC-VI-dotO construct) and lesser defects observed with the distal shift of the dotO spacer in MC-II-dotO to MC-V-dotO. The scrambled dotO spacer of MC-VII-dotO was unable to silence dotO and allowed proficient L. pneumophila growth comparable to the Lp02 control strain. Thus, the polarity of spacer expression and the corresponding decay of target repression represents a titrated gene silencing platform.
Since silencing was least efficient for the MC-V-dotO construct (S10, purple triangle in Fig. 6d, top), we added a boxA(-58) motif to the leader sequence of the array and re-examined the effect of this boxA(-58)-MC-V-dotO construct on L. pneumophila intracellular growth. Remarkably, we now observed a significant attenuation of growth in U937 macrophages over 48 h, achieving over an order of magnitude replication defect compared to Lp02(dcas9) bearing the empty vector (Fig. 6d bottom, p = 0.033, one-unpaired t-test). In fact, with the boxA(-58) motif present, the growth defect observed by expressing the crRNA from the S10 spacer was now comparable to that of expression from the S6 spacer in a boxA-less construct (green diamond in Fig. 6d, top). mRNA and protein analyses confirmed that dotO silencing by the boxA(-58)-MC-V-dotO construct mirrored that of the dotO silencing in the boxA-less MC-III-dotO construct (Figs. 6b and 6c). Together, these data demonstrate that multiplex gene silencing by long CRISPR arrays can be efficiently boosted even within infection models simply by positioning a boxA element in the leader region.
boxA-MC constructs allow for multi-gene knockdown throughout infection
At last, we tested if our multiplex CRISPRi platform, bearing both the Ptet promoter and a boxA(-58) element, is capable of targeting multiple L. pneumophila genes simultaneously throughout the course of host infection. To do so, we constructed a boxA(-58) MC 10-plex construct with ten unique spacers targeting ten unique predicted effector-encoding genes (Fig. 7a; targets in order: lpg2300, lpg1129, lpg1689, lpg0621, lpg1663, lpg2128, lpg0921, lpg2344, lpg2793, lpg0902). Targets were chosen at random and, while their protein products have not yet been characterized in detail, at least eight of the ten have been experimentally proven to be a translocated cargo of the T4SS46,47,48. Consistent with the hypothesis of high redundancy amongst L. pneumophila effectors, we did not observe differences in intracellular growth between Lp02(dcas9) bearing either the empty vector or boxA(-58) MC 10-plex upon challenging U937 macrophages for 48 h (Fig. 7b). Equal numbers of bacteria were subsequently isolated from U937 macrophages and qPCR analysis of extracted RNA revealed that multiplex CRISPRi mediated repression of the majority of targets after 48 h of infection. In fact, eight of the ten targets were repressed nearly an order of magnitude or more (Fig. 7c). These data suggest that boxA multiplex CRISPRi is a robust, multi-gene silencing technique even during host infection.

a boxA(-58) MC 10-plex was constructed with ten unique spacers for expression of crRNAs targeting ten unique predicted effector-encoding genes. b Lp02(dcas9) bearing either the empty vector or boxA(-58) MC 10-plex was used to infect U937 macrophages, and intracellular growth was determined by measuring the average of CFU/well of two to three experiments at 2 and 48 h post infection (hpi). c At 48 hpi bacteria were extracted from the U937 macrophages and harvested for RNA. mRNA levels of each of the ten target genes was measured by qPCR. Fold repression of target genes in boxA(-58) MC 10-plex compared to vector-bearing Lp02(dcas9) was determined using ΔΔCT.
Discussion
In this study, we have established a multiplex CRISPRi platform in the pathogen L. pneumophila and provide proof-of-concept for this platform to be usable not only during growth in axenic media but also during macrophage infection where it reproduced known intracellular growth phenotypes (Fig. 6). Importantly, by placing the crRNA-encoding spacer in positions further downstream within the array, the degree of gene silencing was titratable (Fig. 3). In contrast, when combined with a boxA element, our 10-plex CRISPR array had the potential to silence up to ten unique genes simultaneously (Fig. 4 and Fig. 7) making it a powerful tool to study even synergistic genetic interactions.
Our methodical approach to analyze the potential of multiplex CRISPRi not only allowed us to generate a powerful platform capable of silencing up to ten unique genes, a platform that may prove invaluable for studying the more than 300 L. pneumophila effectors in the future, but it also revealed additional surprises that could be applied to future gene interrogations. For example, some of our MC arrays encoded not just one but two crRNAs (called lidA1 and lidA2) towards the same target gene (Fig. 2b), resulting in LidA to be efficiently depleted even though the encoding spacers were located in the most distal spacer positions (S9 and S10) within the array. Notably, when only the crRNA encoded from the lidA1 spacer was used in subsequent MC constructs (Fig. 3), silencing was not nearly as efficient, suggesting that the combined action of both spacers in Ptet-MC-9/10 promoted maximum silencing. While only a single example, future users of this technology could experiment by adding more than one spacer for a given gene to enhance gene silencing. crRNAs encoded by spacers lidA1 and lidA2 target different regions of lidA, but one could hypothesize repeating the same spacer sequence could also increase gene silencing of a target by producing more of the crRNA. The malleability of our MC scaffold allows users who may not need to target ten unique genes to apply these tactics to achieve maximum silencing levels of fewer gene targets.
Our platform also provides two different strategies for titratable gene silencing. First, since both dcas9 and the CRISPR array are under the regulation of the Ptet promoter, simply altering the amount of aTC inducer presumably changes gene silencing by controlling both the quantity of dCas9 and the abundance of the crRNA (Fig. 1b). While regulating crRNA expression with a variety of inducers (e.g. aTC, arabinose, xylose, and IPTG) is common place, gene silencing in this way has been argued to be noisy49. Second, titrated gene silencing was also accomplished when Ptet-MC constructs without a boxA motif were used that were prone to diminishing transcription (Figs. 3 and 6). DotO constructs provided a clear example of how gene silencing through placing spacers at ever distant positions within the array can lead to titrated effects to the biological system (Fig. 6d). Recent efforts have been made to tailor the degree of crRNA-targeting of a gene by creating numerous variations of the spacer sequence away from the perfect match49,50. We imagine shifting the ideal spacer downward in the array would be a much simpler feat.
Using CRISPRi in L. pneumophila allowed for the systematic investigation of not only repeat/spacer arrays as a gene targeting platform, but also provided additional insight into how CRISPR arrays might function in nature. We show that with the native S. pyogenes promoter, only the most proximal spacers are transcribed (Fig. 2b). It is known that spacers acquired from most recent infections are inserted at the first position in the array51. Our data reiterate that this is an advantageous strategy as the earliest spacers in the array are the most likely to be transcribed, and therefore, will produce the greatest quantity of protective crRNAs for an ongoing viral attack. Several bacterial CRISPR systems appear to have overcome polarity of spacer expression through acquiring boxA sequences upstream of their arrays37. boxA could improve RNA polymerase processivity of these and our synthetic arrays (Fig. 4) by a number of different mechanisms, one of which may be blocking Rho-dependent transcription termination52,53,54, as hypothesized by Stringer et al.37. Still, as naturally occurring CRISPR arrays can stretch to the hundreds of spacers32, there could be additional unknown factors that regulate spacer transcription that could one day be applied to a CRISPR-based tool.
Ultimately, our dotO (Fig. 6) and MC 10-plex (Fig. 7) gene silencing experiments during host infection provided evidence that this multiplex CRISPRi technology is ready to be applied to biological investigations, especially in the study of L. pneumophila pathogenesis. As touched on above, dotO expression appears to be exquisitely tailored towards promoting maximum infection, as any deviation of expression led to growth attenuation in macrophages (Fig. 6). In what could be a nutrient limited environment, it seems L. pneumophila expresses just enough DotO in nature to achieve virulence through T4SS assembly and secretion of effectors, without being wasteful by producing an excess amount of this protein.
The Legionella community has long thought that the multitude of L. pneumophila effectors combined with the lack of growth phenotypes observed upon creating single deletion strains supports the existence of redundancy and synergy amongst the effectors55. Looking forward, the multiplex CRISPRi approach developed here holds the promise of one day probing functional overlap amongst the hundreds of L. pneumophila effectors. Not only can genes be silenced in bulk groups during infection (MC 10-plex, Fig. 7), but the mobility of our single plasmid based CRISPRi platform allows for easy transfer of MC constructs into a variety of Legionella mutant strain backgrounds to directly assess redundancy, presuming they have been equipped with a copy of dcas9. These analyses are not limited to macrophage infections, but any number of hosts. In this capacity, multiplex CRISPRi serves as an initial discovery tool that provides guidance on which proteins to focus on during future analyses. As a general rule, phenotypes observed using knockdown technologies such as this should always be followed-up by construction of true deletion strains to alleviate the possibility of off-target effects or polar effects related to genomic context (e.g., operons). Finally, when adapted for use in other microbial pathogens, the multiplex CRISPRi technology developed here has the potential to promote understanding of their biology, and possibly even foster the discovery of prospective drug targets.
Methods
Construction of dcas9-containing strains
dcas9 was added to the chromosome of Legionella pneumophila Philadelphia-1 Lp02 (thyA hsdR rpsL) through allelic exchange to make MML109 (Lp02(dcas9)). The tetR-dcas9 segment of pdCas9-bacteria (Addgene #44249) was amplified with BKMP108-109 and introduced into pDonorP4r-P3r (Invitrogen) to generate pMME1080. The N- and C-terminus of thyA (lpg2868) were amplified by BKMP94-95 and BKMP98-99 primers and introduced into pDonorP1-P4 and pDonorP3-P2 (Invitrogen) to generate pMME1084 and pMME1094, respectively. These three donor vectors were introduced into pNPTS138_Cm-DEST (pMME1020, constructed by placing the Gateway DEST sequence on pNPTS138_Cm (Addgene #41891) at HindIII and SpeI) by a Gateway LR reaction (Invitrogen) to generate pMME1115 (N-terminus thyA::tetR-dcas9 cassette::C-terminus thyA). pMME1115 was introduced into Lp02 by electroporation and strains containing the plasmid were selected for on CYET-Chloramphenicol (Cm) plates (CYET plates56). Cm-resistant colonies were patched on CYET plates containing 5% sucrose to remove the plasmid backbone, leaving behind the N-terminus thyA::tetR-dcas9 cassette::C-terminus thyA by homologous recombination. dcas9 incorporation into Legionella chromosome was confirmed by immunoblot analyses and whole genome sequencing. Primer sequences are listed in Supplementary Data 3.
Construction of single CRISPRi constructs and preliminary multiplex CRISPRi arrays
crRNA-expressing constructs were built using Invitrogen Gateway-compatible plasmids as depicted in Supplementary Fig. S1a. The BsaI-CRISPR segment of pCRISPR (Addgene #42875) was amplified with BKMP185-186 and introduced into pDonorP5-P2 (Invitrogen) to generate pMME1540. Specific single crRNA-encoding spacer sequences were designed as explained in Fig. 1 and added to the BsaI-CRISPR segment of pMME1540 as previously described for pCRISPR29. To make preliminary multiplex CRISPRi constructs as shown in Fig. 2b, 21 crRNA-encoding spacers separated by repeats were synthesized by GenScript and provided on a pUC57 vector (pMME1170). CRISPR backbones containing four (MC-3/4), eight (MC-7/8), or ten (MC-9/10) crRNA-encoding spacers were amplified from this plasmid using forward primers crsidF_F, crsidD_F, crsetA_F, respectively, and reverse primer crlidA2_R. PCR products were BsaI treated and ligated into pMME1540 (Pnative) or pMME1748 (Ptet) as above.
Next, the Streptococcus pyogenes tracrRNA-encoding sequence from pCas9 (Addgene #42876) was amplified with BKMP45-46 and introduced into pDonorP1-P5 (Invitrogen) to generate pMME985. The crRNA donor plasmids, for both the single CRISPRi constructs and the preliminary multiplex CRISPRi arrays, and pMME985 were introduced into pMME977 by the Gateway LR reaction to generate the final crRNA-encoding plasmids. Final plasmids were introduced to Lp02(-dcas9) or Lp02(dcas9) by electroporation and the strains containing the plasmid were selected for on CYE media. All final strains and crRNA-encoding spacer sequences are listed in Supplementary Data 2. Primer sequences are listed in Supplementary Data 3.
Construction of Ptet-MC multiplex CRISPRi constructs
Multiplex crRNA-expressing constructs were built from GenScript-synthesized plasmids as depicted in Supplementary Fig. S1b and the nucleotide scaffold for Ptet-MC is given in Supplementary Fig. S7a. MC-I through MC-VI, MC-II-dotO through MC-VII-dotO, and MC 10-plex array sequences were provided on a pUC57 vector (Supplementary Data 1). Next, the MC array within pUC57 was amplified using Gateway5-Ptet and Gateway2-T1term and introduced into pDonorP5-P2 by the Gateway BP reaction to generate donor plasmids. The MC backbone vector, containing simply the leader sequence of the multiplex CRISPRi array, was amplified from a GenScript plasmid using Gateway5-leader and Gateway2-leader and similarly introduced into pDonorP5-P2. These donor plasmids and pMME985 were introduced into pMME977 by the Gateway LR reaction to generate the multiplex CRISPRi constructs listed in Supplementary Data 2. These plasmids were then introduced to Lp02(dcas9) by electroporation and the strains containing the plasmid were selected for on CYE plates without thymidine. All final strains and crRNA-encoding spacer sequences are listed in Supplementary Data 2. Primer sequences are listed in Supplementary Data 3.
Addition of boxA to Ptet-MC multiplex CRISPRi constructs
The boxA sequence from L. pneumophila sp. Lens was introduced to the leader region of the MC-II through MC-VI, MC-V-dotO, and MC 10-plex arrays in our donor plasmids by quickchange PCR with Pfu Turbo Polymerase (Agilent #600250-52). boxA(-58) was added using primers BoxA_srtF/R and boxA(-90) was added using primers BoxA_ farF/R. Then sequence-verified donor plasmids and either pMME985 (+tracrRNA) or pMME2603 (-tracrRNA; made by adding the leader sequence of a MC construct to pDonorP1-P5 with attB1-lessTR/ attB5r-lessTR) were introduced into pMME977 by the Gateway LR reaction as before. Again, these plasmids were introduced to Lp02(dcas9) by electroporation and the strains containing the plasmid were selected for on CYE plates. The nucleotide scaffold for boxA(-58) Ptet-MC is given in Supplementary Fig. S7b. All final strains and crRNA-encoding spacer sequences are listed in Supplementary Data 2. Primer sequences are listed in Supplementary Data 3.
Axenic growth of L. pneumophila CRISPRi strains
For experiments with single CRISPRi constructs (Figs. 1 and 5), L. pneumophila were grown overnight in AYE (10 g ACES, 10 g yeast extract per liter, pH 6.9 with 0.4 mg/ml cysteine and 0.135 mg/ml ferric nitrate) under inducing conditions (by adding either 20 ng/mL or 40 ng/mL anhydrous tetracycline (aTC, Clontech #631310)). For all other experiments, L. pneumophila cultures were grown overnight in AYE under non-inducing conditions (-aTC). On the second day, cultures were sub-cultured twice (AM and PM, ~6–7 h apart) to OD600 0.2–0.3 with 2–3 mL fresh AYE containing 40 ng/mL aTC. On the third day, cultures that had reached OD600 3–5 (post-exponential growth) were collected for mRNA analyses, immunoblot analyses, and/or use in host cell infections.
Immunoblot assays and antibodies
Immunoblot assays were performed on bacteria pellets resuspended in SDS sample buffer to either 1×108 CFU or OD600 = 10. SDS-PAGE gels were run on a Protein III system and transfers were performed with the Trans-Blot Turbo Transfer System (BioRad). Protein bands were detected using a primary antibody followed by an HRP-conjugated secondary antibody (rabbit, Life technologies #G21234, or mouse, Invitrogen #G21040) and visualized via chemiluminescence using Clarity Western ECL Substrate and the ChemiDoc MP Imaging System (BioRad). Primary antibodies directed against SidM57, VipD58, RavN59, and LidA60 were described before. Primary antibody against ICDH was a kind gift of Abraham (Linc) Sonenshein (Tufts University School of Medicine). Primary antibodies against DotD and DotO were a kind gift of Joseph B. Vogel (Washington University in St. Louis). Anti-Cas9 monoclonal antibody was purchased from Active motif (Catalog #61577). All uncropped immunoblots are found in Supplementary Fig. S8.
RNA extraction and qPCR
Bacterial pellets were obtained from liquid culture or harvested from host cells. Bacteria from macrophages were harvested using digitonin extraction, as described below for intracellular growth assays, and pelleted at 10,000g for 10 min. Bacterial RNA extraction was performed on bacteria pellets using the Trizol Max Bacterial RNA Isolation Kit (Invitrogen #16096040). Contaminating DNA was removed using the Turbo DNA-free Kit (Invitrogen #AM1907) and RNA was converted to cDNA using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems #4368814). qPCR was performed using the SYBR Green Master Mix (Applied Biosystems #4367659) on a StepOnePlus Real-Time PCR System (Applied Biosystems) using comparative CT and the standard 2-hour protocol. qPCR primers (found in Supplementary Data 3) were designed using NCBI Primer-BLAST such that they amplified ~100–150 bp of sequence near the 5’ end of the gene of interest and had a melting temperature between 57 °C and 63 °C. mRNA levels from different samples were normalized to the house-keeping gene (rpsL) levels and mRNA levels in CRISPRi strains were compared to that of -dCas9 strains or the empty vector control strain using the ΔΔCT method61 to determine fold repression. Similarly, mRNA levels in boxA-bearing MC CRISPRi strains (-tracrRNA) were compared to that of boxA-less MC CRISPRi strains (-tracrRNA) using the ΔΔCT method to determine increased precursor-crRNA expression.
L. pneumophila intracellular growth assay in human derived U937 cells
U937 monocytes (ATCC CRL-1593.2) were maintained in DMEM + 10% FBS + glutamine. Three days prior to challenge with L. pneumophila, cells were plated on 24-well plates at 3×105 cells/well with 0.1 µg/mL 12-O-tetradecanoylphorbol-13-acetate (TPA, Sigma-Aldrich #P1585) to promote differentiation. L. pneumophila strains were incubated under inducing conditions, as described above, and added to differentiated U937 macrophages in DMEM + 10% FBS + glutamine containing 40 ng/mL aTC at a multiplicity of infection (MOI) of 0.05. Plates were centrifuged for five minutes at 200 x g to increase cell-cell contact. After a 2-h incubation, extracellular bacteria were removed by washing cells twice with DMEM + FBS + glutamine media containing 40 ng/mL aTC. Bacteria were collected 2, 24, 48, or 72 h post infection (hpi). To extract the bacteria from the macrophages, digitonin (0.02% final concentration) was added to each well and incubated 10 min at 37 °C. Subsequently, lysate was collected, and each well was rinsed with dH2O to ensure collection of all bacteria. Bacterial samples were serially diluted and spotted on CYE plates to determine CFU. Results are given as the CFU normalized to the CFU at 2 hpi.
Statistics and reproducibility
Statistical comparison of growth between MC-V-dotO and Lp02(dcas9) bearing the empty vector or boxA(-58)-MC-V-dotO and Lp02(dcas9) bearing the empty vector in Fig. 6 was carried out in GraphPad Prism 8 using the t-test analysis function. A one-unpaired t-test was performed without correction for multiple comparisons and without assuming a consistent SD. Changes in growth were considered significant if p < 0.05. Two to three replicates were obtained for each strain and replicates were obtained from distinct experiments. Replicate data for all other experiments of this manuscript are shown as individual data points and were similarly collected from distinct experiments.
Data availability
All data supporting the conclusions of this study are available within the article and its supplementary figures and data.
References
Hille, F. et al. The biology of CRISPR-Cas: backward and forward. Cell 172, 1239–1259 (2018).
Adli, M. The CRISPR tool kit for genome editing and beyond. Nat. Commun. 9, 1911 (2018).
Strich, J. R. & Chertow, D. S. CRISPR-Cas biology and its application to infectious diseases. J. Clin. Microbiol. 57, e01307–18 (2019).
Barrangou, R. et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science 315, 1709–1712 (2007).
Bhaya, D., Davison, M. & Barrangou, R. CRISPR-Cas systems in bacteria and archaea: versatile small RNAs for adaptive defense and regulation. Annu. Rev. Genet. 45, 273–297 (2011).
Deltcheva, E. et al. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 471, 602–607 (2011).
Brouns, S. J. et al. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 321, 960–964 (2008).
Wiedenheft, B., Sternberg, S. H. & Doudna, J. A. RNA-guided genetic silencing systems in bacteria and archaea. Nature 482, 331–338 (2012).
Zhu, Y., Klompe, S. E., Vlot, M., van der Oost, J. & Staals, R. H. J. Shooting the messenger: RNA-targetting CRISPR-Cas systems. Biosci. Rep. 38, BSR20170788 (2018).
Marraffini, L. A. & Sontheimer, E. J. Self versus non-self discrimination during CRISPR RNA-directed immunity. Nature 463, 568–571 (2010).
Jinek, M. et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 (2012).
Gasiunas, G., Barrangou, R., Horvath, P. & Siksnys, V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl Acad. Sci. USA 109, E2579–E2586 (2012).
Chylinski, K., Makarova, K. S., Charpentier, E. & Koonin, E. V. Classification and evolution of type II CRISPR-Cas systems. Nucleic Acids Res. 42, 6091–6105 (2014).
Qi, L. S. et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152, 1173–1183 (2013).
Bikard, D. et al. Programmable repression and activation of bacterial gene expression using an engineered CRISPR-Cas system. Nucleic Acids Res. 41, 7429–7437 (2013).
Larson, M. H. et al. CRISPR interference (CRISPRi) for sequence-specific control of gene expression. Nat. Protoc. 8, 2180–2196 (2013).
Vigouroux, A. & Bikard, D. CRISPR tools to control gene expression in bacteria. Microbiol. Mol. Biol. Rev. 84, e00077–19 (2020).
Peters, J. M. et al. Enabling genetic analysis of diverse bacteria with Mobile-CRISPRi. Nat. Microbiol. 4, 244–250 (2019).
Adiego-Perez, B. et al. Multiplex genome editing of microorganisms using CRISPR-Cas. FEMS Microbiol. Lett. 366, fnz086 (2019).
Peters, J. M. et al. A comprehensive, CRISPR-based functional analysis of essential genes in bacteria. Cell 165, 1493–1506 (2016).
Kaczmarzyk, D., Cengic, I., Yao, L. & Hudson, E. P. Diversion of the long-chain acyl-ACP pool in Synechocystis to fatty alcohols through CRISPRi repression of the essential phosphate acyltransferase PlsX. Metab. Eng. 45, 59–66 (2018).
Yao, L., Cengic, I., Anfelt, J. & Hudson, E. P. Multiple gene repression in cyanobacteria using CRISPRi. ACS Synth. Biol. 5, 207–212 (2016).
Reis, A. C. et al. Simultaneous repression of multiple bacterial genes using nonrepetitive extra-long sgRNA arrays. Nat. Biotechnol. 37, 1294–1301 (2019).
McDade, J. E. et al. Legionnaires’ disease: isolation of a bacterium and demonstration of its role in other respiratory disease. N. Engl. J. Med. 297, 1197–1203 (1977).
Fraser, D. W. et al. Legionnaires’ disease: description of an epidemic of pneumonia. N. Engl. J. Med. 297, 1189–1197 (1977).
Rao, C. et al. Active and adaptive Legionella CRISPR-Cas reveals a recurrent challenge to the pathogen. Cell Microbiol. 18, 1319–1338 (2016).
Berger, K. H. & Isberg, R. R. Two distinct defects in intracellular growth complemented by a single genetic locus in Legionella pneumophila. Mol. Microbiol. 7, 7–19 (1993).
Choudhary, E., Thakur, P., Pareek, M. & Agarwal, N. Gene silencing by CRISPR interference in mycobacteria. Nat. Commun. 6, 6267 (2015).
Jiang, W., Bikard, D., Cox, D., Zhang, F. & Marraffini, L. A. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat. Biotechnol. 31, 233–239 (2013).
Burstein, D. et al. Genomic analysis of 38 Legionella species identifies large and diverse effector repertoires. Nat. Genet. 48, 167–175 (2016).
Gomez-Valero, L. et al. More than 18,000 effectors in the Legionella genus genome provide multiple, independent combinations for replication in human cells. Proc. Natl Acad. Sci. USA 116, 2265–2273 (2019).
Grissa, I., Vergnaud, G. & Pourcel, C. CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 35, W52–W57 (2007).
Luo, M. L., Mullis, A. S., Leenay, R. T. & Beisel, C. L. Repurposing endogenous type I CRISPR-Cas systems for programmable gene repression. Nucleic Acids Res. 43, 674–681 (2015).
Liao, C. et al. Modular one-pot assembly of CRISPR arrays enables library generation and reveals factors influencing crRNA biogenesis. Nat. Commun. 10, 2948 (2019).
Arnvig, K. B. et al. Evolutionary comparison of ribosomal operon antitermination function. J. Bacteriol. 190, 7251–7257 (2008).
Gaudino R. J., Morgan E. A. The Role of Boxa in Transcription of Ribosomal RNA Operons of EscherichaColi: Changes in the Processivity of RNA Polymerase. In: Post-Transcriptional Control of Gene Expression (eds McCarthy J. E. G., Tuite M. F.) (Springer, 1990).
Stringer, A. M., Baniulyte, G., Lasek-Nesselquist, E., Seed, K. D. & Wade, J. T. Transcription termination and antitermination of bacterial CRISPR arrays. Elife 9, e58182 (2020).
Vogel, J. P. & Isberg, R. R. Cell biology of Legionella pneumophila. Curr. Opin. Microbiol 2, 30–34 (1999).
Andrews, H. L., Vogel, J. P. & Isberg, R. R. Identification of linked Legionella pneumophila genes essential for intracellular growth and evasion of the endocytic pathway. Infect. Immun. 66, 950–958 (1998).
Yerushalmi, G., Zusman, T. & Segal, G. Additive effect on intracellular growth by Legionella pneumophila Icm/Dot proteins containing a lipobox motif. Infect. Immun. 73, 7578–7587 (2005).
Chetrit, D., Hu, B., Christie, P. J., Roy, C. R. & Liu, J. A unique cytoplasmic ATPase complex defines the Legionella pneumophila type IV secretion channel. Nat. Microbiol 3, 678–686 (2018).
Sundstrom, C. & Nilsson, K. Establishment and characterization of a human histiocytic lymphoma cell line (U-937). Int J. Cancer 17, 565–577 (1976).
Pearlman, E., Jiwa, A. H., Engleberg, N. C. & Eisenstein, B. I. Growth of Legionella pneumophila in a human macrophage-like (U937) cell line. Micro. Pathog. 5, 87–95 (1988).
Isaac, D. T., Laguna, R. K., Valtz, N. & Isberg, R. R. MavN is a Legionella pneumophila vacuole-associated protein required for efficient iron acquisition during intracellular growth. Proc. Natl Acad. Sci. USA 112, E5208–E5217 (2015).
Christenson, E. T. et al. The iron-regulated vacuolar Legionella pneumophila MavN protein is a transition-metal transporter. Proc. Natl Acad. Sci. USA 116, 17775–17785 (2019).
Huang, L. et al. The E Block motif is associated with Legionella pneumophila translocated substrates. Cell Microbiol 13, 227–245 (2011).
Lifshitz, Z. et al. Computational modeling and experimental validation of the Legionella and Coxiella virulence-related type-IVB secretion signal. Proc. Natl Acad. Sci. USA 110, E707–E715 (2013).
Zhu, W. et al. Comprehensive identification of protein substrates of the Dot/Icm type IV transporter of Legionella pneumophila. PLoS One 6, e17638 (2011).
Vigouroux, A., Oldewurtel, E., Cui, L., Bikard, D. & van Teeffelen, S. Tuning dCas9’s ability to block transcription enables robust, noiseless knockdown of bacterial genes. Mol. Syst. Biol. 14, e7899 (2018).
Jost, M. et al. Titrating gene expression using libraries of systematically attenuated CRISPR guide RNAs. Nat. Biotechnol. 38, 355–364 (2020).
McGinn, J. & Marraffini, L. A. Molecular mechanisms of CRISPR-Cas spacer acquisition. Nat. Rev. Microbiol 17, 7–12 (2019).
Mitra, P., Ghosh, G., Hafeezunnisa, M. & Sen, R. Rho protein: roles and mechanisms. Annu Rev. Microbiol 71, 687–709 (2017).
Torres, M., Balada, J. M., Zellars, M., Squires, C. & Squires, C. L. In vivo effect of NusB and NusG on rRNA transcription antitermination. J. Bacteriol. 186, 1304–1310 (2004).
Squires, C. L., Greenblatt, J., Li, J., Condon, C. & Squires, C. L. Ribosomal RNA antitermination in vitro: requirement for Nus factors and one or more unidentified cellular components. Proc. Natl Acad. Sci. USA 90, 970–974 (1993).
Ghosh, S. & O’Connor, T. J. Beyond paralogs: the multiple layers of redundancy in bacterial pathogenesis. Front Cell Infect. Microbiol. 7, 467 (2017).
Feeley, J. C. et al. Charcoal-yeast extract agar: primary isolation medium for Legionella pneumophila. J. Clin. Microbiol 10, 437–441 (1979).
Neunuebel, M. R. et al. De-AMPylation of the small GTPase Rab1 by the pathogen Legionella pneumophila. Science 333, 453–456 (2011).
VanRheenen, S. M., Luo, Z. Q., O’Connor, T. & Isberg, R. R. Members of a Legionella pneumophila family of proteins with ExoU (phospholipase A) active sites are translocated to target cells. Infect. Immun. 74, 3597–3606 (2006).
Lin, Y. H. et al. RavN is a member of a previously unrecognized group of Legionella pneumophila E3 ubiquitin ligases. PLoS Pathog. 14, e1006897 (2018).
Conover, G. M., Derre, I., Vogel, J. P. & Isberg, R. R. The Legionella pneumophila LidA protein: a translocated substrate of the Dot/Icm system associated with maintenance of bacterial integrity. Mol. Microbiol. 48, 305–321 (2003).
Schmittgen, T. D. & Livak, K. J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 3, 1101–1108 (2008).
Acknowledgements
We thank members of the Machner laboratory for their critical reading of the manuscript, Dr. Joseph T. Wade (New York State Department of Health, Wadsworth Center) for advice and direction on boxA motifs, and Dr. Caroline Esnault (National Institutes of Health) for instruction on qPCR procedure. This work was funded by the Intramural Research Program of the National Institutes of Health, USA (Project Number: 1ZIAHD008893-10).
Author information
Authors and Affiliations
Contributions
N.A.E., B.K. and M.P.M. contributed to experimental design; N.A.E., B.K. and J.T. performed experiments; N.A.E. and M.P.M. composed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ellis, N.A., Kim, B., Tung, J. et al. A multiplex CRISPR interference tool for virulence gene interrogation in Legionella pneumophila. Commun Biol 4, 157 (2021). https://doi.org/10.1038/s42003-021-01672-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-021-01672-7
This article is cited by
-
CRISPR and Antisense RNA Technology: Exploiting Nature’s Tool to Restrain Virulence in Tenacious Pathogens
Molecular Biotechnology (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
