
Abstract
Regulatory T cells (Tregs) are critical mediators of immune homeostasis. The co-stimulatory molecule CD27 is a marker of highly suppressive Tregs, although the role of the CD27-CD70 receptor-ligand interaction in Tregs is not clear. Here we show that after prolonged in vitro stimulation, a significant proportion of human Tregs gain stable CD70 expression while losing CD27. The expression of CD70 in expanded Tregs is associated with a profound loss of regulatory function and an unusual ability to provide CD70-directed co-stimulation to TCR-activated conventional T cells. Genetic deletion of CD70 or its blockade prevents Tregs from delivering this co-stimulatory signal, thus maintaining their regulatory activity. High resolution targeted single-cell RNA sequencing of human peripheral blood confirms the presence of CD27−CD70+ Treg cells. These findings have important implications for Treg-based clinical studies where cells are expanded over extended periods in order to achieve sufficient treatment doses.
Similar content being viewed by others


Introduction
CD4+FOXP3+ regulatory T cells (Tregs) have a critical role in the maintenance of immunological self-tolerance and immune homeostasis. Tregs are also the subject of investigation as a cellular therapy for the therapeutic induction of tolerance to alloantigens or autoantigens1,2,3. Treg function is dependent on their stable expression of the transcription factor FOXP34, largely determined by epigenetic demethylation of the Treg-specific demethylation region (TSDR)5. In scurfy mice, loss-of-function mutations in Foxp3 result in the development of a fatal autoimmune disorder6 while in humans FOXP3 mutations lead to an X-linked disorder, IPEX (immunodysregulation polyendocrinopathy enteropathy X-linked)7,8,9. In humans, high CD25 cell surface expression in combination with low or absent CD127 expression is a useful marker for the distinction of Tregs from effector CD4+ T cells10,11.
Whether Tregs are a phenotypically and functionally stable population is under debate12,13,14. Elegant experiments using tracer mice containing FOXP3-GFP-Cre crossed with Rosa26-YFP have demonstrated instability in FOXP3 expression in a substantial percentage of Tregs when transferred into T cell-deficient mice15,16. However, others have demonstrated stable FOXP3 expression under homeostatic and autoimmune inflammatory conditions using similar tracer mice approach17, suggesting that the stability of the Treg linage may be regulated by environmental cues. For example, pro-inflammatory cytokines have been shown to cause Tregs to adopt a phenotype more characteristic of effector CD4+ T cells, downregulating FOXP3, losing their suppressive function and secreting pro-inflammatory cytokines18,19. Clinical examples of this plasticity are also evident, with IL-17-expressing Tregs increased in psoriasis, inflammatory bowel disease and rheumatoid arthritis, compared to healthy individuals20,21,22,23,24,25,26.
An important regulatory mechanism for Treg stability is co-stimulation, since it provides important signaling for Treg activation, survival, expansion, and acquisition of effector functions upon antigen recognition27,28. The CD27 co-stimulatory receptor is constitutively expressed on a small proportion of natural killer (NK) cells, memory B cells and resting CD8+ and CD4+ T cells, including CD4+FOXP3+ Tregs29. CD27 is expressed on thymocytes as early as the double positive stage of development30, and a role for CD27 in rescuing Tregs from apoptosis during clonal deletion in the thymic medulla has been reported31. In T cells, CD27 expression is increased on activation but then downregulated after prolonged stimulation32. CD27 expression is also lost on fully differentiated effector T cells, although central memory T cells retain CD27 expression32,33. Expression of CD70, the unique ligand for CD27, is tightly controlled and upregulated exclusively upon activation on T cells, B cells and certain subsets of dendritic cells (DCs)34,35. CD70 expression is very limited in the steady state, although increased levels of CD70 have been reported to be associated with inflammatory conditions such as chronic viral infection, cancer, or autoimmune disease36,37,38,39,40,41,42,43.
Ligation of CD27 on T cells with CD70 on antigen presenting cells (APCs) promotes T cell activation44, influences CD4+ T cell subset differentiation31,45,46 and is essential for the generation of antigen-specific T cell immunity by enhancing the survival of activated T cells47,48,49,50. Highlighting the importance of this interaction, genetic mutations in CD27 or CD70 in humans can result in persistent symptomatic EBV infection and EBV-associated lymphoproliferative disorders51,52,53,54,55,56. It has been previously shown that CD27 expression on human Tregs correlates closely with suppressive potency57,58,59,60. The mechanisms for this are unclear, although it has been proposed that CD27 on Tregs ligates CD70 on DCs, reducing the access of conventional T cells (Tconv) to this co-stimulatory molecule61. More recently, CD27 signaling has been shown to impair the conversion of tissue resident mouse Tregs into Th17 cells22. Moreover, several transcriptomic studies have demonstrated an increased expression of CD70 mRNA in human Tregs compared with conventional CD4+ T cells62,63.
CD27 signaling is sufficient to activate T cells when combined with T cell receptor (TCR) stimulation47,49,64. However, it is unclear how CD70 expressed on other cell types, such as Tregs, affects T cell function. In this study, we examine the role of the CD27/CD70 co-signaling axis in Treg function and how it is altered after prolonged Treg stimulation. We assess the relationship between CD27/CD70 and Treg fitness, showing that selection of CD27+CD70− cells after Treg expansion enables the enrichment of potently suppressive cells with a transcriptome characteristic of Tregs. We also show that the prolonged expansion of human Tregs leads to stable gain of CD70 expression with associated loss of CD27, which in turn provides co-stimulatory signaling to CD27-expressing Tconv cells. Moreover, we show that the targeting of CD70 using monoclonal antibodies or genetic editing tools preserves the function of expanded Tregs, providing a method that may be incorporated into future cell therapy production protocols.
Results
CD27 and CD70 define distinct human Treg subpopulations
We assessed expression levels of CD27 and CD70 on flow-sorted CD4+CD25+CD127low/− human Tregs isolated at high purity levels from healthy donor PBMCs. The majority of these ex vivo Tregs were CD27+CD70− (64.4–92.7%), with a variable, but minor, subpopulation expressing CD70 (Fig. 1a and Supplementary Data 1). Distinct CD27 and CD70 expression patterns were associated with different levels of FOXP3 expression (Fig. 1b and Supplementary Data 1). When normalized to the total Treg population, CD27+CD70+ and CD27−CD70+ subsets displayed a higher intensity of FOXP3 expression, while CD27−CD70− and CD27+CD70− subsets had a lower or similar level of FOXP3 expression, respectively (Fig. 1b and Supplementary Data 1).

a Expression of CD27 and CD70 was analyzed on freshly isolated CD4+CD25+CD127−/low Tregs. Left panel: Representative FACS plots from one donor. Figures display percentages. Right panel: n = 19 independent donors. Data were analyzed using a Kruskal–Wallis test with Dunn’s post-test for multiple comparisons and are represented as mean ± SD. b FOXP3 expression was measured within the total Treg population or within different Treg subsets defined by CD27 and CD70 expression. Left panel: Representative FACS plots from one donor. Right panel: FOXP3 fluorescence intensity normalized to the total Treg population in different Treg subsets (n = 16 independent donors). Mean ± SD, Kruskal–Wallis test with Dunn’s post-test for multiple comparisons. c, d: c Dot plots for one representative donor in which expression of CD27 and CD70 was measured at different time points on Tregs stimulated with anti-CD3/anti-CD28 coated beads at a ratio of 1:5 beads to cells in the presence of rhIL-2 (250U/ml). Figures show percentages. d CD27 and CD70 fluorescence intensity was analyzed within the CD27+CD70− or the CD27−CD70+ Treg populations, respectively, at different time points and normalized to levels prior stimulation. Data represented as mean ± SD. (n = 6 independent donors with 1–3 replicates per donor). Data were analyzed using a Friedman test with Dunn’s post-test for multiple comparisons. e–g Expression of CD27 and CD70 was analyzed on freshly isolated ex vivo Tregs or Tregs expanded for 2 or 4 weeks with anti-CD3/anti-CD28 coated beads at a ratio of 1:1 beads to cells in the presence of rhIL-2 (1000 U/ml). e Data show n = 11 independent donors analyzed using a Friedman test with Dunn’s post-test for multiple comparisons. f Representative plots for one donor. Figures show percentages. g Figures display percentage and fluorescence intensity of CD70+ cells for one representative donor. ns, non-significant.
Upon activation, there is dynamic expression of CD27 and CD70 in Tconv cells32. To assess whether this is also a feature in Tregs, we activated sorted CD4+CD25+CD127low/− Tregs with anti-CD3/anti-CD28 coated beads and recombinant human IL-2 (rhIL-2) over a short period of time. Analysis of expression patterns revealed a reciprocal relationship between CD27 and CD70 on activated Tregs (Fig. 1c). Both CD27 and CD70 expression was increased after 3 or 5 days of stimulation (Fig. 1d and Supplementary Data 1). By 7 days, the frequency of CD70-expressing cells increased even further, forming a distinct subpopulation (Fig. 1c).
To investigate how prolonged stimulation may alter CD27 and CD70 expression, the phenotype of Tregs was examined following stimulation and expansion for 2–4 weeks. While initial levels of CD27 expression were high pre-expansion, after expansion there was a progressive reduction of expression (Fig. 1e, f and Supplementary Data 1). Conversely, CD70 expression levels were initially low and then increased after expansion (Fig. 1e–g and Supplementary Data 1). Following expansion, distinct Treg subpopulations could be identified by their pattern of CD27 and CD70 expression (Fig. 1f). Although transient CD70 expression upon activation has been previously described in other cell types34,35, we found that after long-term expansion Tregs maintained an unusually high and stable level of CD70 expression (Fig. 1g).
Prolonged expansion reduces the suppressive capacity of Tregs
We next assessed whether the changes in CD27 and CD70 expression were also associated with changes in Treg function. Pre-expansion, Tregs expressed high levels of FOXP3, and while this was largely maintained during the first 2 weeks of expansion, after 4 weeks there was a significant reduction in FOXP3 expression (Fig. 2a and Supplementary Data 1). Similarly, on assessment of Treg function, we found suppressive potency to be maintained after 2 weeks of expansion but reduced after 4 weeks (Fig. 2b and Supplementary Data 1).

CD4+CD25+CD127−/lowTregs were cultured in vitro with anti-CD3/anti-CD28 coated beads and rhIL-2 before analysis of a FOXP3 expression (n = 8 donors, Friedman test with Dunn’s post-test for multiple comparisons) and b in vitro Treg suppressive capacity, assessed as suppression of responder T cell (Tresp) proliferation. Left panel: percentage of VPD-stained CD4+ or CD8+ cells in the absence of Tregs (0:1) and in the presence of Tregs at a 1:1 or 1/8:1 Treg:Tresp ratios shown for one representative donor. Right panel: percentage suppression of CD4+ and CD8+Tresp calculated based on division index as described in “Methods” section. n = 5 donors (three replicate wells for each donor). Mean ± SEM, Friedman test and Dunn’s post-test for multiple comparisons. c In vitro suppressive capacity of CD27+CD70− and CD27−CD70+ sorted Tregs. Left panel: Data from one representative donor. Right panel: Percentage suppression of CD4+ and CD8+ Tresp for 4–5 independent donors with each data point showing the mean of three replicate wells for each donor; calculated based on division index. Mean ± SEM, Kruskal–Wallis test with Dunn’s post-test for multiple comparisons. Red: CD27+CD70−Tregs; Blue: CD27−CD70+Tregs, both sorted after 2 weeks of expansion. d FOXP3 was analyzed in CD27/CD70 Treg subsets after 2 weeks of expansion. Top panel: histogram displays one representative donor (gray: isotype, red: CD27+CD70− Tregs, blue: CD27−CD70+Tregs). Bottom panel: n = 16 independent donors. Mean ± SD, Mann–Whitney test. e, f DNA methylation pattern of the Treg-Specific Demethylation Region (TSDR) was assessed in CD27+CD70− and CD27−CD70+Treg subsets from matched donors, sorted from freshly isolated and 2 weeks in vitro expanded CD4+CD25+CD127−/lowTregs. e Average methylation rate for 9 CpG motifs of the TSDR (n = 6 different donors). Top panel: Each box represents the percentage of methylation for each donor. Bottom panel: each dot denotes a donor. Mean ± SD, one-way ANOVA with Tukey post-test. f Each box represents the percentage of methylation of a single CpG residue for each donor at each time point (Left panel: day 0; right panel: 2 weeks) (n = 3 donors matched for each CD27/CD70 Treg subset). Bar colors designate: red: 0% methylation; yellow: 50% methylation; blue: 100% methylation. ns, non-significant.
We hypothesized that the loss in suppressive function was secondary to the expansion-related changes in CD27 and CD70 expression. On examination of the specific function of CD27+CD70− or CD27−CD70+ Tregs after 2 weeks of expansion, we found the CD27+CD70− Tregs to be significantly more potent at high but not low ratios of Tregs to responder cells (Fig. 2c and Supplementary Data 1). This was largely unrelated to any differences in FOXP3 expression (Fig. 2d and Supplementary Data 1). We then analyzed the methylation state of the TSDR region in sorted CD27+CD70− and CD27−CD70+ Treg subsets from freshly isolated or 2-week expanded Tregs. In freshly isolated Tregs, both Treg subsets had a highly demethylated TSDR, confirming their high purity (Fig. 2e and Supplementary Data 1). Upon 2 weeks of expansion, the TSDR remained largely demethylated in CD27+CD70− Tregs, whereas CD27−CD70+ Tregs developed a hypermethylated TSDR (Fig. 2e and Supplementary Data 1). Interestingly, the methylation pattern in expanded CD27−CD70+ Tregs was not equal across different CpG motifs (Fig. 2f and Supplementary Data 2).
Importantly, although CD4+CD25+CD127−/low cells may include a small number of contaminating Tconv cells, these contaminants are unlikely to be the CD27−CD70+ cells. While activated effector T cells may transiently express FOXP3, we found significantly higher FOXP3 expression in CD27−CD70+ Tregs compared to CD27−CD70+ Tconv cells after 2 weeks of in vitro expansion (Supplementary Fig. 1), indicating that CD27−CD70+ Tregs are a separate population to effector T cells that upregulate FOXP3.
Expanded CD27+CD70− Tregs retain suppressive potency
We next investigated whether re-sorting cells based on CD27 and CD70 expression at 2 weeks, followed by further expansion would address the loss of suppressive potency that develops between weeks 2 and 4 of expansion. Sorted Tregs were therefore expanded for 2 weeks and then flow sorted into CD27+CD70− and CD27−CD70+ populations before continuing expansion for a further 2 weeks (Fig. 3a). While sorted CD27−CD70+ Tregs maintained their phenotype for the 2 subsequent weeks of expansion, CD27+CD70− Tregs upregulated CD70 (Fig. 3b and Supplementary Data 1). Moreover, in approximately half of the analyzed donors, downregulation of CD27 was observed in re-stimulated CD27+CD70− Tregs (Fig. 3b and Supplementary Data 1). FOXP3 expression was significantly higher in expanded CD27+CD70− cells than CD27−CD70+ cells (Fig. 3b and Supplementary Data 1), which correlated with the TSDR demethylation at 2 weeks of expansion (Fig. 2e). Tregs with high CD27 expression tended to retain high levels of FOXP3 expression, while CD70 expression negatively correlated with FOXP3 expression levels (Supplementary Fig. 2).

a Schematic overview of Treg expansion strategies. CD4+CD25+CD127−/low Tregs were flow sorted from healthy donor PBMCs and expanded in vitro over two rounds of 7 days stimulated with anti-CD3/anti-CD28 coated beads (at a ratio of 1 bead to 1 cell) and rhIL-2 (1000U/ml) and rested for 2 days with a low concentration of rhIL-2 (250U/ml) and without stimulating beads. Tregs expanded for 2 weeks were flow sorted into CD27+CD70− and CD27−CD70+ subsets. Total Tregs (black), flow sorted CD27+CD70− Tregs (red) and flow sorted CD27−CD70+ Tregs (blue) were then cultured for a further 16 days before analysis. Dot plots from one representative donor are shown. Figures display percentages. b Left panel: FOXP3, CD27 and CD70 expression levels on expanded and then flow sorted CD27+CD70− and CD27−CD70+ Tregs after a further 2 weeks of expansion. n = 16–18 independent donors, analyzed using a Mann–Whitney test. Data are represented as mean ± SD. Right panel: Histogram of FOXP3 expression levels for one representative donor (gray: isotype, red: CD27+CD70- Tregs, blue: CD27-CD70+ Tregs). c, d, c In vitro suppression assay of 4 week-expanded Treg populations in one representative donor at multiple Treg:Tresp ratios. Data shown are mean ± SD. d In vitro suppression assays from 14 independent donors at 1:1 (top) and 1/8:1 (bottom) ratios of Treg to Tresp. Each data point represents the mean of three replicate wells from each donor. Mean ± SEM is depicted. Black: total Treg, red: CD27+CD70−, blue: CD27−CD70+ Tregs. Data were analyzed using Kruskal–Wallis test with Dunn’s post-test. e The in vivo suppressive capacity of CD27/CD70 Treg subsets was assessed by transplanting VPD-labeled PBMCs together with 4-week expanded CD27+CD70− or CD27−CD70+ Tregs (5:1 PBMC:Treg ratio) into the peritoneal cavity of immunodeficient mice. Top: Schematic representation of the experimental plan. Bottom: The proliferation index of PBMCs was studied by flow cytometry. Data were analyzed using a Kruskal–Wallis test with Dunn’s post-test for multiple comparisons and are represented as mean ± SD. A representative experiment of 1 cell donor out of 2 (4–5 mice per group per experiment, each mouse is represented as a data point). ns, non-significant.
On functional analysis, CD27+CD70− Tregs were highly efficient at inhibiting CD4+ and CD8+ T cell proliferation and were consistently more potent than CD27−CD70+ expanded cells at all ratios tested (Fig. 3c, d and Supplementary Data 1). When comparing suppressive potency with CD27/CD70 expression in 4 week-in vitro expanded Tregs, we found no significant correlation between percentage of suppression of proliferation of CD4+ or CD8+ responder T cells and CD27 or CD70 expression, although a trend was observed (Supplementary Fig. 3).
Moreover, in the majority of donors analyzed, CD27−CD70+ Tregs actively promoted proliferation of CD4+ and CD8+ T cells (Fig. 3d and Supplementary Data 1). Although being consistently more potent than CD27−CD70+ Tregs, there was some heterogeneity in the suppressive activity of expanded CD27+CD70− cells according to donor. We considered whether impaired suppression within the expanded CD27+CD70− cells was due to the presence of cells that had downregulated CD27 and upregulated CD70. In support of this, we found that re-sorted CD27highCD70− cells demonstrated significantly enhanced suppressive activity than the total population of CD27+CD70− expanded cells (Supplementary Fig. 4).
To assess the increased potency of the CD27+CD70− Treg subset in vivo, we performed a human leukocyte proliferation assay in immunodeficient mice. Violet proliferation dye (VPD)-labeled human PBMCs were transferred into mice together with human CD27+CD70− or CD27−CD70+ Tregs which were expanded in vitro for 4 weeks. CD27+CD70− Tregs, but not CD27−CD70+ Tregs, were able to suppress CD4+ and CD8+ T cell proliferation in vivo (Fig. 3e, Supplementary Fig. 5 and Supplementary Data 1).
Transcriptome differences between CD27+CD70− and CD27−CD70+ expanded Tregs
Having shown differences in the function of expanded CD27+CD70− and CD27−CD70+ Tregs, we sought to compare gene signatures between these two Treg populations. CD27+CD70− and CD27−CD70+ Tregs expanded over an extended period (as depicted in Fig. 3a) were re-sorted into CD27+CD70− and CD27−CD70+ cells and analyzed by RNA sequencing (RNA-seq) to assess for differences in the transcriptomes (four independent blood donors). Global examination of the data by principal component analysis revealed the existence of two separate clusters of sample replicates (Supplementary Fig. 6a). In total, 1568 genes were differentially expressed between both Treg subsets (absolute Log2FC ≥ 2 and adjusted p value < 0.01); 681 transcripts were upregulated and 887 downregulated in CD27+CD70− Tregs compared to CD27−CD70+ Tregs (Supplementary Data 3). Figure 4a contains the top 64 most differentially expressed genes (absolute Log2FC ≥ 6). Gene ontology (GO) term enrichment analysis of differentially expressed genes identified specific molecular pathways altered in both Treg subsets (Supplementary Fig. 6b). A large proportion of highly differentially expressed loci were related to cytokine and chemokine signaling, with signaling by interleukins, IL-4 and IL-13 signaling, IL-10 signaling, and chemokine receptor binding chemokine pathways being among the most enriched pathways. IL-17A, the effector cytokine for Th17 function, appeared as the most differentially expressed gene between CD27+CD70− and CD27−CD70+ Tregs, being expressed ten times more in the CD27−CD70+ Treg subset (Fig. 4a and Supplementary Data 3). Overexpression of IL-17A was confirmed by measuring cytokine levels in cell culture supernatant and by intracellular cytokine staining (Supplementary Fig. 7).

Thirty-two days-expanded Tregs (as shown in Fig. 3a) were re-sorted into CD27+CD70− and CD27−CD70+ Tregs prior being assessed by RNA-seq analysis. a Heatmap and hierarchical clustering from RNA-seq data with Log2FC ≥ 6. The top 64 differentially expressed genes are shown. Data were analyzed using the edgeR package. b Venn diagram showing the overlap between genes overexpressed in Tregs65 and upregulated genes in CD27+CD70− Tregs or CD27-CD70+ Tregs. c Venn diagram showing the overlap between genes overexpressed in Tconv65 and upregulated genes in CD27+CD70- Tregs or CD27−CD70+ Tregs. d, e: d HELIOS expression was measured in freshly isolated total Tregs or within different Treg subsets defined by CD27 and CD70 expression. (Left panel) Representative FACS plots of one donor (gray: isotype, black: total Tregs, red: CD27+CD70− Tregs, blue: CD27−CD70+ Tregs, green: CD27+CD70+ Tregs, brown: CD27−CD70− Tregs). (Right panel) Percentage of HELIOS+ cells in different CD27/CD70 Treg subsets. e HELIOS expression was analyzed in 2 week-expanded total Tregs or within different Treg subsets defined by CD27 and CD70 expression. Left panel: CD27/CD70 gating strategy. Figures show percentages. Middle panel: Representative FACS plots of one donor (gray: isotype, black: total Tregs, red: CD27+CD70- Tregs, blue: CD27−CD70+ Tregs, green: CD27+CD70+ Tregs, brown: CD27−CD70− Tregs). Right panel: Percentage of HELIOS+ cells in different CD27/CD70 Treg subsets. Mean ± SD is represented. n = 3–4 independent donors. Kruskal–Wallis test with Dunn’s post-test for multiple comparisons was conducted. Only significant statistical differences are shown. f CD25, GITR, CD39, TIGIT, CD40L, and HLA-DR protein expression was analyzed in 4 weeks expanded CD27+CD70− Tregs or CD27-CD70+ Tregs. N = 6–13 independent donors. Data were analyzed by Mann–Whitney test and represented as mean ± SD. ns, non-significant.
We then compared the transcriptome profile of CD27/CD70 Treg subsets with a previously identified Treg-specific gene signature65. Ferraro et al. have identified 368 genes differentially expressed between Tregs and Tconv, with 187 and 181 genes upregulated in Tregs or Tconv, respectively65. From 187 genes specifically upregulated in Tregs, we found 31 genes that were overexpressed in CD27+CD70− Tregs (and included FOXP3, IKZF2, TIGIT, FCRL3, UTS2) and 12 genes in CD27−CD70+ Tregs (Fig. 4b and Supplementary Data 4). Of the 181 genes upregulated in Tconv, 18 were overexpressed in CD27+CD70− Tregs and 37 were overexpressed in CD27−CD70+ Tregs, including CD40L, IL7R, THEMIS, and TMEM71 (Fig. 4c). Interestingly, other genes of the so called “Treg-specific gene signature”65,66, such as CTLA-4, IL2RA, GITR and ENTPD1 (CD39) were not differentially expressed between CD27/CD70 Treg subsets. These results indicate that expanded CD27+CD70− Tregs have a gene profile more closely related to the previously identified Treg gene signature than CD27−CD70+ Tregs, with some differences.
Canonical Treg-signature genes in the CD27+CD70− cell population included typical markers associated with Treg biology such as FOXP3, IKZF2 (HELIOS), and TIGIT (Fig. 4b). In freshly isolated Tregs, HELIOS protein expression was significantly lower in CD27−CD70+ than CD27+CD70− Tregs (Fig. 4d and Supplementary Data 1). The same trend in HELIOS protein expression was observed in Tregs expanded for 2 weeks (Fig. 4e and Supplementary Data 1). Despite the fact that differences in TSDR demethylation and HELIOS expression were apparent after 2 weeks of expansion (Figs. 2e, f, 4e), FOXP3 expression and suppressive function were mostly unaltered between CD27+CD70− Tregs and CD27−CD70+ Tregs at two weeks (Fig. 2c, d). Since it is only after 4 weeks of expansion that functional changes are apparent (Fig. 3e–d), we studied the expression of Treg-associated markers in these long-term expanded cells. Significant differences were found in CD25, CD40L, and HLA-DR expression, but not in GITR, CD39 or TIGIT, in the different CD27/CD70 Treg subsets (Fig. 4f and Supplementary Data 1).
CD27+CD70− Treg suppressive function does not rely on CD27 signaling
We next explored the role that CD27 plays in the suppressive activity of Tregs. For these studies, we used anti-CD27 monoclonal antibodies (mAbs) where the precise function depends on the presence or absence of cross-linking, which can be provided by Fc receptors or artificially64,67. Using these antibodies cross-linked with goat anti-human IgG results in agonistic function, whereas an Fc-mutated anti-CD27 mAb in which cross-linking by Fc receptors is prevented results in antagonistic activity67 (Fig. 5a).

Data shown for 4 week-expanded Tregs (sorted as total Tregs, expanded for 2 weeks, sorted by CD27/CD70, then expanded for 2 weeks). a, b: a Cartoon depicting Treg CD27 targeting strategies. Tregs were pre-incubated for 1 h with Fc-mutated blocking anti-CD27 mAb or non-mutated anti-CD27 mAb, followed by 1 h cross-linking with anti-human Ig antibody, or isotype matched antibodies. b Percentage suppression of CD4+ and CD8+ T cell proliferation by Tregs pre-incubated with anti-CD27 mAbs in 4 donors. CD8+T cells did not proliferate enough to perform analysis in 2 out of 4 donors. Friedman test with Dunn’s post-test for multiple comparisons. c Top panel: percentage suppression of CD8+Tresp cell proliferation by Treg subsets pre-incubated with anti-CD70 mAb or isotype antibody. n = 5–8 independent cell donors, Wilcoxon matched-pairs test. Bottom panel: proliferation of VPD-labeled CD8+Tresp co-cultured with Tregs pre-incubated with isotype or blocking anti-CD70 mAb, one representative donor. d, e: d Cartoon depicting CD27/CD70 blocking strategies. Tregs were pre-incubated with anti-CD70 blocking mAb or matched isotype Ab. Alternatively, soluble Fc-mutated blocking anti-CD27 mAb was added to the suppression assay to block CD27 on Tresp. e Left panel: Percentage of dividing VPD-stained CD4+ or CD8+ cells in the absence of Tregs (0:1) and in the presence of CD27-CD70+ Tregs pre-incubated with isotype or anti-CD70 blocking mAb (1:1 Treg:Tresp). Right panel: Percentage suppression of proliferation of CD4+ and CD8+ Tresp. n = 7 independent donors, Friedman test with Dunn’s post-test. b, c, e Graphs calculated based on division index and formula described in “Methods” section. f–h: f VPD-labeled Tresp were co-cultured with CD27+CD70−Tregs or CD27−CD70+Tregs pre-incubated with isotype or anti-CD70 mAb in wells containing plate-bound anti-CD3 mAb (1 µg/ml). g Proliferation of CD8+ or CD4+Tresp in the absence of Tregs or when co-cultured with Treg subsets for one representative donor out of 5. h Division index of proliferating CD4+ and CD8+Tresp co-cultured with CD27−CD70+Tregs pre-incubated with isotype mAb or blocking anti-CD70 mAb. n = 7 donors, Friedman test with Dunn’s post-test. b, c, e, h Each data point represents the mean of three replicate wells for each independent cell donor. ns, non-significant.
To investigate the role of CD27 in the suppressive activity of Tregs, CD27+CD70− Tregs were pre-incubated with anti-CD27 mAb provided under either activating or blocking conditions and assessed for their capacity to suppress T cell proliferation in vitro. Tregs pre-incubated in medium alone, isotype control, agonistic anti-CD27 mAb, or blocking anti-CD27 mAb showed similar suppressive potency (Fig. 5b and Supplementary Data 1), indicating that stimulation or blocking of CD27 signaling on Tregs does not have a significant effect on Treg suppressive activity in vitro.
CD70 on Tregs provides co-stimulation to conventional T cells
In order to further explore the differences in suppressive potency between CD27+CD70− and CD27−CD70+ Tregs, we investigated whether this was a result of CD70-mediated co-stimulation. Total unsorted, CD27+CD70−, or CD27−CD70+ Tregs were pre-incubated with anti-CD70 blocking mAb or matched isotype control antibody and their suppression was assessed in vitro. The effective binding of anti-CD70 blocking mAb was confirmed by FACS using an anti-CD70-fluorochrome labeled mAb that is sterically hindered by anti-CD70 blocking mAb (Supplementary Fig. 8). Blocking CD70 inhibited the pro-stimulatory effect of CD27−CD70+ Tregs and enhanced suppressive potency of total unsorted Tregs without affecting the activity of CD27+CD70− Tregs (Fig. 5c and Supplementary Data 1). In some donors, CD27−CD70+ Tregs were only ever suppressive when CD70 was blocked (Fig. 5c).
To further demonstrate that CD70+ Tregs provide co-stimulatory signaling by binding CD27 on T cells, responder T cells were co-cultured with total Tregs or CD27−CD70+ Tregs and allogenic monocyte derived DCs (allo mo-DCs) in the presence of anti-CD27 blocking mAb (Fig. 5d). Blocking CD27 on responder T cells resulted in suppression of proliferation to a similar extent as blocking CD70 on Tregs (Fig. 5e and Supplementary Data 1).
To eliminate the possibility that anti-CD70 mAb dissociates from Tregs and subsequently inhibits T cell proliferation by binding to responder T cells or mo-DCs, we added anti-CD70 blocking mAb directly to the co-culture. In the absence of Tregs, proliferation of responder T cells was not impaired by CD70 blockade (Supplementary Fig. 9). When CD70+ Tregs were added to the cell co-culture that contained anti-CD70 blocking mAb, the pro-proliferative activity of the Tregs was neutralized (Supplementary Fig. 9). This suggests that the anti-CD70 antibody acts directly on Tregs and not on responder T cells or mo-DCs.
We next assessed whether CD70 expressed on Tregs provides sufficient co-stimulation to promote T cell proliferation in the absence of additional co-stimulatory help. Treg activity was therefore assessed in the absence of DC-driven stimulation. VPD-labeled responder T cells were co-cultured with CD27+CD70− Tregs or CD27−CD70+ Tregs in wells containing plate-bound anti-CD3 mAb (Fig. 5f). Here, only CD27−CD70+ Tregs induced T cell proliferation, with CD8+ T cells appearing more responsive to CD27−CD70+ Treg stimulation in comparison to CD4+ T cells (Fig. 5g, h, Supplementary Fig. 10 and Supplementary Data 1). Similar to the effect observed by CD27-CD70+ Tregs, stimulation using agonist anti-CD27 mAb also increased T cell proliferation, especially within the CD8+ T cell compartment (Supplementary Fig. 11a). Moreover, CD27 stimulation increased expression of the anti-apoptotic Bcl-XL molecule and reduced the frequency of cell death in CD4+ and CD8+ T cells (Supplementary Fig. 11b, c).
We next explored the specificity of this response by pre-incubation of CD27−CD70+ Tregs with blocking anti-CD70 mAb prior to the subsequent culture with responder T cells. Although some proliferation of responder T cells still took place, blocking the CD27–CD70 interaction impaired the proliferative effect induced by CD27−CD70+ Tregs. Interestingly, the effect of blocking CD70 was not complete, suggesting the presence of additional pro-inflammatory mechanisms (Fig. 5h and Supplementary Data 1).
CD70 knockout Tregs have enhanced suppressive potency
To prove the detrimental role of CD70 in Treg suppressive activity, we next produced CD70-deficient human Tregs by disrupting CD70 expression using CRISPR (clustered regularly interspaced short palindromic repeats)-associated nuclease Cas9 technology. Molecular signatures of gene editing at the CD70 locus were confirmed by PCR sequencing and ICE analysis in the total cell product (Fig. 6a and Supplementary Fig. 12). Most of the mutations comprised an insertion of 1 bp (22.9%) or deletion of 23 bp (21.46%), creating a knock out (KO)-score of 75% when analyzing the percentage of indels>20 bp or those resulting in frameshifts (Fig. 6a; bottom panel). Gene-edited Tregs were cultured in vitro for 2 weeks with anti-CD3 and anti-CD28 stimulation and in the presence of rhIL-2. Following this, we observed a significant reduction of CD70 and an increase of CD27 expression, together with the development of a CD27−CD70− population in the gene edited group (CD70-KO) (Fig. 6b and Supplementary Data 1).

a Genetic editing efficiency across the CD70 locus was confirmed by analysis using ICE algorithm. Top panel: Level of discordance between control (orange) and edited (green) genetic sequences. Sequences between control and edited samples match before the PAM sequence (denoted by a dotted line) and are discordant after the cut site. Bottom panel: Graph shows the distribution of the detected indels in the edited sample. X-axis depict the size of the indel and Y-axis shows the frequency of each deletion in the cell product. 1 sgRNA for a representative donor is shown. b Percentage of CD70 and CD27 expression in control and CD70-KO cell products. Assays were repeated in 12 independent experiments. Data were analyzed using a Wilcoxon matched-pairs test. c Treg suppressive capacity was assessed in vitro using autologous VPD-labeled CD3+CD25− T cells as responders (Tresp) and allogenic mo-DCs as stimulators. Suppressive activity of control (non-edited) and CD70-KO cell products is represented for six independent experiments. Percentage suppression of proliferation of CD4+ Tresp and CD8+ Tresp is shown as mean ± SEM. Statistical significance was calculated using a Wilcoxon matched-pairs test. d–f CD70-KO Treg cell products were sorted by magnetic isolation or flow cytometry in CD27+CD70−, CD27−CD70+ and CD27−CD70+ cells subsets before assessing d FOXP3 expression, e DNA methylation pattern of the TSDR, and f suppressive activity. Data shown for 4 independent experiments. d Left panel: histogram displays one representative donor (gray: isotype, red: CD27+CD70− Tregs, blue: CD27−CD70+ Tregs, green: CD27−CD70− Tregs). Right panel: Statistical significance was calculated using a Friedman test with Dunn’s post-test for multiple comparisons. e Average methylation rate for nine CpG motifs of the TSDR. Error bars indicate mean ± SD. Statistical significance was determined by a Wilcoxon matched-pairs test. f Treg suppressive capacity was assessed in vitro. Percentage suppression of proliferation of CD4+ Tresp and CD8+ Tresp is represented. Statistical analyses performed using Friedman test with Dunn’s post-test for multiple comparisons. ns, non-significant.
On functional assessment, CD70-KO Tregs had enhanced suppressive potency compared to control Tregs in controlling CD4+ T cell proliferation (Fig. 6c and Supplementary Data 1). Since the KO effect was not highly effective in all donors tested, we were able to sort different Treg subsets based on CD27 and CD70 expression from the total CD70-KO cell product (see sorting gate Supplementary Fig. 13). We analyzed each subset independently for FOXP3 expression, TSDR methylation and capacity to suppress T cell proliferation in vitro. No significant differences were observed in FOXP3 expression (Fig. 6d and Supplementary Data 1) or levels of TSDR methylation (Fig. 6e and Supplementary Data 1) due to CD70 silencing, indicating that CD70 does not control stability of FOXP3 expression. As previously shown, CD27−CD70+ Tregs displayed impaired suppression compared to CD27+CD70− Tregs (Fig. 6f and Supplementary Data 1). Importantly, suppressive capacity was restored in the CD27−CD70− Treg subset (which contained the CD70-KO Tregs) (Fig. 6f).
CD70+ Tregs are present within resting CD4+ T cell populations in the peripheral blood
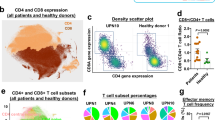
We next aimed to map CD70+ Tregs using single-cell analysis in order to determine their respective representation within resting primary CD4+ T cells in humans. For accurate phenotyping of this heterogeneous population, we used a targeted multiomics single-cell RNA sequencing (scRNAseq) system in which both gene and protein expression are simultaneously quantified, as previously described68. We reanalyzed data presented in Trzupek et al.68 with focus on CD70 expression. Briefly, CD4+ T cells from the peripheral blood of one patient with systemic lupus erythematosus (SLE; n = 9708 cells), one patient with type 1 diabetes (T1D; n = 7042 cells) and one healthy donor (HD; n = 7197 cells) were studied. Unsupervised hierarchical clustering was performed in the combined mRNA and protein expression data, yielding distinct clustering of cells across both naive/memory and effector/Treg spectra68. Analysis of CD70 expression in this dataset demonstrated restricted expression in memory and effector populations (Supplementary Fig. 14), and therefore naive cells were excluded and a novel unsupervised hierarchical clustering was performed (Fig. 7a). This approach provided detailed functional compartmentalization of the heterogeneous memory CD4+ T cell population, which is evident by the differential of genes across each numbered cluster, with clusters 0, 13, 5, and 10 expressing Treg-signature genes, including FOXP3 and IKZF2 (HELIOS; Fig. 7b). Among all clusters, CD70 expression was highest within clusters 13, 0, and 9, which displayed transcriptional profiles of activated Tregs, memory Tregs and putative cytotoxic Th1 cells, respectively (Fig. 7c and Supplementary Fig. 15a). This presence of CD70+ cells within Treg clusters 0 and 13 was consistent across patients (Fig. 7d), and although a higher frequency was observed within the SLE patient, this was associated with a general increased frequency of Tregs and therefore not a specific enrichment. On analysis of genes correlating with CD70 expression within clusters 0 and 13, we found a differential overexpression of genes related to cell cycle progression (MKI67, PCNA, HMMR, and TOP2A), suggesting that CD70+ Tregs are actively proliferating (Fig. 7e). CD70 also correlated with CCR6 expression, a marker of IL-17-producing T cells (Supplementary Fig. 15b)69. Importantly, a clear negative correlation was observed with the relative expression of CD27, which was found to be the most negatively differentially regulated gene in clusters 0 and 13 (Fig. 7e and Supplementary Fig. 15b). Together, these data indicated that CD70+ Tregs are present within resting CD4+ T cells in the peripheral blood and express a gene and protein signature consistent with our in vitro observations.

Data from primary resting CD4+ cells from the peripheral blood from one systemic lupus erythematosus (SLE) patient, one type I diabetes patient (T1D) and one healthy donor (HD) and analysed using targeted scRNA-seq and protein quantification with BD Rhapsody technology from Trzupek et al.68 were re-analysed with focus on CD70 expression. A detailed description of this methodology and analysis of these cells can be found in the ref. 68 a Unsupervised hierarchical clustering combining mRNA and protein expression data visualized as a Uniform Manifold Approximation and Projection (UMAP) plot depicting the clustering of memory/effector CD4+ T cells from all three donors integrated from two independent experiments. b Heatmap for each CD4+ cluster showing the top differentially expressed genes and proteins. c Analysis of CD70+ cell frequency within each UMAP cluster in a. The number of cells with ≥1 copy of CD70 is displayed for each cluster. The majority of CD70 mRNA-expressing cells fall within Cluster 0 (Memory Tregs) and Cluster 13 (Activated Tregs). d UMAP plots depicting relative expression of CD70 mRNA for each patient, with Treg clusters highlighted. e Correlation analysis (Spearman correlation coefficient) of CD70 and each assessed mRNA and protein target in the Treg Cluster 0 (top) and Cluster 13 (bottom), with an enhanced view of the top 12 most positively (green) or negatively (red) correlated genes.
Discussion
In this study, we show that the prolonged stimulation of Tregs results in the development of a population of cells with a divergent gene signature and pro-inflammatory activity. These cells may be characterized principally by their expression of cell surface CD70. Prolonged stimulation is a feature of current Treg cell therapy production techniques and may also occur in pathological conditions in vivo70,71. Dysfunctional Tregs are strongly associated with clinical pathology20,21,22,23,24,26, and we therefore urge caution in the therapeutic infusion of Treg cell therapies intended to be immune suppressive if these products contain a significant proportion of CD70+ Tregs.
We have previously reported that expanded naive Tregs retain higher CD27 expression than memory Tregs72, and CD27 has been shown to positively correlate with suppressive potency57,58,60. Moreover, the immunosuppressive drug rapamycin, which is currently used for ex vivo expansion of clinical grade Tregs73, has been shown to preserve CD27 expression during in vitro Treg expansion58. Here, we demonstrate that freshly isolated Tregs have high CD27 and low CD70 expression and that both markers are upregulated following activation. However, upon prolonged stimulation in a significant portion of Tregs, CD27 is downregulated while CD70 expression is upregulated as a stably expressed marker, resulting in two distinct subpopulations: CD27+CD70− and CD27−CD70+. Previous studies on ex vivo expanded human Tregs have demonstrated that Tregs maintain a regulatory phenotype and high suppressive potency after one or two cycles of expansion74. However, Hoffmann et al. showed that CD4+CD25+CD127low/− Tregs display increased methylation within the TSDR region and decreased levels of FOXP3 expression after repetitive TCR stimulation75. In agreement with these data, our work shows that prolonged in vitro stimulation and expansion of flow-sorted highly pure CD4+CD25+CD127low/− Tregs results in a final cell product that contains cells with unstable regulatory activity. Within this population, CD27+CD70− Tregs share important elements of the transcriptome with the previously identified Treg-signature transcriptome65, and retain potent suppressive activity and a demethylated TSDR upon long-term in vitro stimulation. By contrast, CD27−CD70+ Tregs have a significantly impaired suppressive capacity after prolonged in vitro stimulation and, conversely, the ability to provide co-stimulatory signals to induce T cell proliferation. While our results suggest that FOXP3 regulates CD70 expression, published data comparing Tregs and Tconv cells65,76,77 or FOXP3 transduction in CD4+ T cells78 suggest no direct interaction between FOXP3 expression and CD70. We therefore propose that CD70 has the active role in regulating Treg suppressive activity by providing direct co-stimulatory signals to responder T cells.
It has been suggested that CD27+ Tregs may compete for CD70 binding on DCs, resulting in endocytosis and degradation of the CD27/CD70 complex61. Consequently, DCs are unable to provide CD70-mediated co-stimulation to responder T cells61. However, our experiments in which we blocked CD27 on Tregs indicate that CD27 does not play a non-redundant role in their suppressive activity. These findings are consistent with studies where Tregs from wild type and CD27−/− mice were shown to be equally suppressive31. Moreover, Remedios et al. have shown that CD27 signaling limits the conversion of skin-resident Tregs into Th17 cells under an inflammatory environment driven by Candida albicans infection22. Indeed, in our experiments we have also shown that in humans IL-17 production is limited to the CD27−CD70+ Treg population. Interestingly, Remedios et al. found that while CD27 ligation prevented expression of Th17-associated molecules in Tregs, it did not affect FOXP3 or CD25 expression levels, suggesting that CD27 signaling does not increase Treg stability22. Here, we have shown that CD27−CD70− Tregs (isolated from CD70-KO Treg cell products after in vitro expansion) are as potent as CD27+CD70− Tregs at inhibiting T cell proliferation in vitro, further indicating that suppressive activity can be preserved in Tregs that are expanded over a prolonged period independently of CD27 expression.
Previous studies investigating CD70 co-stimulation provided by effector T cells have reported different outcomes under transient or constitutive antigen exposure in vivo. For example, in the presence of chronic viral infection, CD70 co-stimulation provides survival signals to antigen-activated T cells for their maintenance that ultimately leads to T cell exhaustion and defective memory immune responses79,80. However, during transient infections, CD70-driven co-stimulation enhances T cell immune responses without resulting in T cell exhaustion79,80. In line with these data, O’Neill et al. have described a role for T cell-derived CD70 co-stimulation restraining inflammatory T cell responses by inducing T cell apoptosis in mouse models of autoimmune inflammatory bowel disease (IBD) and GvHD81. Here, we have shown that CD70 is expressed by Tregs following activation and that CD70+ Tregs (stably expressing CD70) after prolonged in vitro stimulation, can provide sufficient co-stimulation to induce T cell activation and proliferation. In resting primary CD4+ T cells from the peripheral blood, CD70+ Tregs appear to reside within activated and memory Treg subpopulations where their gene expression signature is consistent with proliferating cells that downregulate expression of CD27. However, it is possible that CD70+ Tregs may promote different outcomes in vivo depending on the nature and precise context of the immune response. Due to the therapeutic potential of Treg cell therapy in transplantation and autoimmunity, both disorders in which there is persistent antigen presence, future work is needed to determine the role of CD70+ Tregs in vivo in both steady state and inflammatory states.
Interestingly, CD70+ Tregs primarily promoted proliferation of CD8+ T cells. CD27 co-stimulation has been identified as a key CD4+ T cell-delivered help signal to CD8+ T cells82,83,84. Here, antigen presenting-DCs upregulate CD70 upon interaction with CD4+ T cells via CD40L–CD40 interaction and transmit the help signal to trigger CD8+CD27+ T cell activation82. In our study, we show that Tregs induce CD4+ and CD8+ T cell proliferation via a CD27–CD70 interaction in the absence of DCs, indicating that CD4+CD70+ Tregs can provide direct co-stimulation to CD4+ and CD8+ T cells.
Genetic deletion or blockade of CD70 on Tregs or CD27 on T cells significantly enhanced Treg suppression, partly abolishing the pro-stimulatory effect of CD70+ Tregs. While contamination with effector T cells within CD70+ Tregs may be possible, CD27−CD70+ cells were already present within freshly isolated Tregs, had similar levels of TSDR demethylation as CD27+CD70− Tregs, and expressed high FOXP3 levels. Moreover, CD27−CD70+ Tregs suppressed T cell proliferation after 2 weeks of expansion but lost this ability after 4 weeks. In addition, within the highly pure isolated CD27+CD70− Tregs, some cells converted to CD27−CD70+ cells and the re-sorted CD27highCD70− Tregs showed increased suppressive ability. The ability to convert some CD27−CD70+ into actively suppressive cells through the simple blockade of CD70 with an antibody argues against the possibility that these cells are contaminating effector cells. The last piece of evidence that supports Treg instability comes from our genetic editing experiments disrupting CD70 expression. Here, CD27−CD70− Tregs isolated from a CD70-KO product were as suppressive as CD27+CD70− Tregs, proving that CD70+ Tregs can exert potent suppressive capacity when CD70 function is disabled. These results further support our hypothesis of CD70 being a crucial regulator of Treg activity, although it is possible that other molecules identified by our RNA sequencing as differentially expressed could also play a role.
Monoclonal antibodies that target co-stimulatory and co-inhibitory molecules have a wide-range of established roles in the treatment of immune-mediated pathologies85. For cancer, the most promising are those that inhibit the checkpoint receptors PD-1 and CTLA-4, leading to increased antitumor T cell activity86. Conversely, CD28 antagonists designed as CTLA-4-Ig fusion proteins are effective at ameliorating symptoms in autoimmune disorders and at preventing transplant rejection87. The modulation of members of the tumor necrosis factor receptor family (TNFR) including CD40, OX40, and 4-1BB is also currently a focus of investigation64,88,89,90, with amplification of CD27 signaling being explored for cancer immunotherapy67,91. Our study provides a new understanding of human Treg stability and suggests that the CD27/CD70 axis could be exploited for regulating inflammatory and regulatory T cell responses. This could be approached through the direct treatment of patients with modulators of this pathway or manipulation of Tregs ex vivo for subsequent infusion. For example, the pro-proliferative effects of CD70+ Tregs can be prevented by monoclonal antibodies that block CD70 on Tregs or CD27 on T effector cells. Additionally, CD70-genetic ablation or the selection of ex vivo Tregs based on the CD27+CD70− phenotype after expansion eliminates a major component of unstable, pro-stimulatory cells in the final product. We propose that depletion of CD70+ Tregs following in vitro expansion may be incorporated as an additional selection step into current clinical grade production methods. Moreover, with the recent introduction of genetic editing tools into clinical trials, it is possible that deletion of CD70 on Tregs may be incorporated into GMP-compliant Treg expansion protocols.
Methods
Treg flow-sorting strategies and cell culture
Peripheral blood mononuclear cells (PBMCs) were isolated from blood cones obtained from healthy donors (NHS Blood and Transplant [NHSBT] UK) by LSM1077 (PAA Laboratories, Pasching, Austria) gradient centrifugation and then incubated with CD25+ Microbeads to derive CD25+ enriched cells using LS columns (Miltenyi Biotec, Bergisch Gladbach, Germany). CD4+CD25+CD127−/low Tregs were isolated from CD25+ enriched PBMCs by FACS using a BD FACSAria I or II cell sorter.
Cells were cultured in complete media, composed of RPMI 1640 media supplemented with l-Glutamine, penicillin/streptomycin and sodium pyruvate (Sigma-Aldrich, St. Louis, MO, USA). For Treg expansion, 10% of human antibody pooled serum (Seralab, Haywards Heath, UK) (heat-inactivated for 20 min at 56 °C) was added to the medium. Tregs were expanded as described previously92. For in vitro assays, 10% of fetal calf serum (FCS, heat inactivated for 20 min at 56 °C) (Gibo, UK) was used instead of human serum. Cells were incubated at 37 °C, 5% carbon dioxide and >80% humidity. Expanded Tregs were stained with anti-CD27 PeCy7 (M-T271) and anti-CD70 APC (113-16) (BioLegend) antibodies in order to separate into CD27+CD70− and CD27−CD70+ cells (Fig. 3a). Cell sorting was performed using a BD FACSAria I or II cell sorter.
In vitro stimulation of Tregs
Sorted Tregs were activated with Dynabead Human T-activator anti-CD3/anti-CD28 beads (Thermo Fisher Scientific, MA, USA) in a 5:1 cell:bead ratio in complete media, in the presence of recombinant human IL-2 (rhIL-2) (250U/ml) (Novartis, Surrey, UK). For prolonged in vitro culture (16–36 days), Tregs were stimulated with anti-CD3/anti-CD28 beads every 7 days in a 1:1 cell:bead ratio in complete media in the presence of rhIL-2 (1000U/ml). Cells were split, and media changed as required. Tregs were rested without beads and with 250 U/ml rhIL-2 for 2 days every 14 days of culture.
Flow cytometric phenotyping
Cells were stained with 7-AAD viability dye, anti-CD4 PE-eFluor 610 (RPA-T4), anti-CD27 eFluor 450 (O323), anti-FOXP3 FITC (PCH101), anti-CD8 APC-Cy7 (SK1) (eBioscience), anti-CD25 PECy7 (M-A251), anti-CD25 APC-Cy7 (M-A251), anti-CD127 PE (HIL-7R-M2), anti-CD27 FITC (M-T271), anti-CD70 PE (Ki-24), anti-CD3 PECy7 (SK7), anti-mouse CD45 FITC (30-F11) (BD Biosciences), anti-FOXP3 Alexa Fluor 647 (259D), anti-CD27 PECy7 (M-T271), anti-CD70 APC (113-16) (BioLegend) and anti-BCL-XL (7B2.5) (Abcam, UK) specific antibodies. Intracellular staining was performed using FOXP3 staining buffers (eBioscience). For staining, antibodies were used in a 1:45 dilution. Data were acquired using a FACS Canto II and analysed using FlowJo software (Treestar).
TSDR DNA methylation analysis
Methylation at the Treg-Specific Demethylated Region (TSDR) was studied in CD27+CD70− and CD27−CD70+ Treg cell subsets from matched donors sorted from CD4+CD25+CD127−/low Tregs either freshly isolated or after 2 weeks of in vitro expansion. Methylation analysis was conducted by EpigenDx by pyrosequencing of bisulphite-modified DNA purified from frozen cells. Nine representative CpG residues in the TSDR were analysed using assay ADS783-FS2 for human FOXP3.
In vitro suppression assays
CD3+CD25− T responder cells (Tresp) were negatively isolated from PBMCs by incubation with CD25+ Microbeads followed by the positive selection of CD3+ T cells by CD3 Microbeads using the MACS system (Miltenyi Biotec). For monocyte-derived dendritic cell (mo-DC) generation, CD14+ monocytes were magnetic bead-isolated from PBMCs (Miltenyi Biotec) and cultured with 50U/ml IL-4 and 50 ng/ml GM-CSF in complete media supplemented with 10% FCS. After 5 days of incubation, mo-DCs were harvested and stored in liquid nitrogen for later use.
Variable numbers of Tregs were incubated with 2 × 104 allogenic monocyte-derived dendritic cells (allo mo-DCs) as stimulators and 10 × 104 autologous CD3+CD25− T cells labeled with 1 µM VPD450 (562158, BD Biosciences) proliferation dye as responders (Tresp) in triplicate wells. Labeled Tresp cultured alone in the presence of allo mo-DCs were used as a negative control for suppression. Following 80–96 h of incubation, co-cultures were stained to distinguish CD8+ and CD4+ effector T cells, and VPD dilution was analysed by flow cytometry. Percentage of suppression was calculated by using division index of co-cultures containing Tregs, Tresp and DCs compare to division index of Tresp with DCs alone, according to the following formula: (1 − (div.index Treg + Tresp/div.index Tresp)) × 100. Data were analysed with FlowJo software.
In vivo proliferation assay
BALB/c Rag2−/−cγ−/− mice were maintained in individually-ventilated cages under specific pathogen-free in the Biomedical Service Unite of the University of Oxford (Oxford, UK). Female mice were aged between 8 and 12 weeks at the time of the experimental procedure. Mice received an intraperitoneal injection of 5 × 106 cryopreserved and thawed PBMCs labeled with 1 µM VPD450 (562158, BD Biosciences) together with 1 × 106 cryopreserved and thawed 4-week expanded CD27+CD70− or CD27−CD70+ Tregs. Mice were injected with different Treg subsets or no Tregs in a blind way and allocated randomly in different cages. Cells were extracted by peritoneal lavage at day 4 after transfer, stained, and analysed by flow cytometry. VPD dilution was assessed to calculate proliferation index with FlowJo software. On analysis, lavage fluid was procured and analysed in a blinded fashion before unblinding at the final analysis.
CD70 co-stimulation test
CD3+CD25− T responder cells (Tresp) were purified from PBMCs by MACS (Miltenyi Biotec) and labeled with 1uM VPD450 (562158, BD Biosciences). T cells were cultured in plates coated with 1 µg/ml anti-CD3 mAb (OKT3, BioLegend) and in the presence of in vitro stimulated Treg subsets. Proliferation of responder CD4+ and CD8+ T cells was calculated as division index using FlowJo software.
Anti-CD27 and anti-CD70 mAbs for functional assays
Anti-CD70 blocking mAb (clone BU.69) was purchased from Abcam (UK) as mouse anti-human antibody or from Absolute Antibody Ltd as fully human Ab. Fully human anti-CD27 mAb and its Fc-mutated version were provided by Celldex Therapeutics. Secondary goat anti-human antibody for crosslinking was purchased from Abcam (UK). Isotype control antibodies were mouse IgG1 (BioLegend), fully human IgG1 (BioXcell, NH, USA) and human IgG1 Fc-silent (Absolute Antibody Ltd, UK). Where indicated, Tregs were pre-incubated with 20 µg/ml anti-CD70, anti-CD27 or control isotype antibodies for 1 h at 37 °C. Excess antibodies were washed out before assessing functionality of pre-incubated Tregs. Alternatively, 20 µg/ml anti-CD70, Fc-mutated anti-CD27 blocking mAbs or corresponding isotype antibody was added to the functional assay.
Bulk RNA-sequencing analysis
RNA sequencing (RNA-seq) analysis was performed in 4 weeks expanded CD27+CD70− and CD27−CD70+ Tregs as depicted in Fig. 3a for four independent donors. Before analysis, expanded cell products were re-sorted according to CD27 and CD70 expression to high purity. RNA was isolated from 300,000 CD27+CD70− and CD27−CD70+ Tregs cells using RNeasy Plus Mini Kit (QIAGEN) according to the manufacturer’s protocol. mRNA capture and cDNA synthesis were performed using KAPA mRNA Hyper Kit for illumine platform (KAPA) according to the manufacturer’s protocol. After quality check using Qubit High-sensitivity kit (Life Technologies), 16 cycles of PCR were carried out for amplifying each library. Each experiment starting from RNA isolation was performed in triplicate using cells from four different donors. Amplified cDNA libraries were sequenced on an Illumina HiSeq4000 machine using paired end reads (2 × 150 bp) at Novogene, China. Forty million reads per sample were gained and aligned using STAR against the human genome (UCSC hg38). The non-adjusted read counts for each gene were assessed statistically for global differential expression between the specified populations using the edgeR package. Genes that are significant at a 1% false discovery rate (calculated using a Benajmini–Hochberg adjusted p-value) were considered differentially expressed between populations as described93. Gene ontology enrichment was assessed using the topGO package and pathway enrichment was analysed on the ReactomePA package. Sequencing data is available from the NCBI GEO database under accession numbers GSE129251.
Single cell RNA sequencing reanalysis
Single cell RNA sequencing data presented in this manuscript are reanalyzed data from three blood donors presented in Trzupek et al.68. Detailed description of sorting strategy, library preparation, sequencing and data analysis and QC is presented in Trzupek et al.68. Briefly, CD4+ T cells enriched from PBMCs from one SLE patient, one T1D patient and one autoantibody negative healthy donor were stained with sorting antibodies and BD AbSeq antibodies (BD Biosciences) against 43 T-cell expressed targets, and sorted as CD127lowCD25hi Tregs, CD127hi Teffs and CD127low Teffs. Sorted subsets were barcoded with oligo-conjugated sample multiplexing tag antibodies (BD Biosciences) and mixed at 1:1:1 ratio to increase the relative distribution of less abundant Tregs and CD127low Teffs. Cells were next loaded on a BD Rhapsody cartridge (BD Biosciences) and captured as single cells using the BD Rhapsody Express system according to manufacturer’s instructions. cDNA libraries were prepared using Human Immune Response primer panel (BD Biosciences) and sequenced using HiSeq 4000 sequencer (Illumina). Data analysis and QC was performed following the BD Biosciences Rhapsody pipeline (BD Biosciences) and the R package Seurat 3.094, as previously described68. For the memory/effector-restricted T cell analysis presented here, the identified naive clusters (0, 1, and 2 in ref. 68) were excluded leading to the following number of remaining cells used in the analysis: SLE patient n = 5167, T1D patient n = 3154, healthy donor n = 2376. Uniform manifold approximation and projection (UMAP) was used for dimensionality reduction. The default number of used dimensions of PCA reduction was increased to 30 and the default resolution parameter value for clustering was increased to 1.4 to obtain more fine-grained set of clusters. Differential expression analysis was performed using a tailored hurdle model from MAST package95 used by Seurat.
CRISPR/Cas9 genome editing
A third-generation self-inactivating “terminal” lentiviral vector expressing GFP and single guide RNA (sgRNA) was generated as described previously96. The vector incorporates guide sequences into the U3 region of the 3′LTR, thereby coupling transgene expression and CRISPR effects after subsequent Cas9 delivery by electroporation. Concentrated vector preparations were produced by transient transfection of 293T cells97. Ten days in vitro expanded-CD4+CD25+CD127−/low Tregs were transduced with lentiviral vector at a MOI of 5, and CleanCap™ Streptococcus pyogenes Cas9 (spCas9) mRNA (L-7606 TriLink Biotechnologies San Diego, USA) was delivered by electroporation 3 days later at a concentration of 100 µg/ml. Lentiviral transduced Tregs that were not subjected to electroporation or that were electroporated in the absence of Cas9 mRNA were used as a control for genetic knockout (KO) effect. Successfully transduced Tregs were flow sorted based on GFP expression and expanded in vitro prior to additional analysis. Alternatively, sgRNA was complexed with spCas9 protein and delivered as ribonucleoprotein (RNP) by electroporation in 2 weeks in vitro expanded Tregs. Gene-specific sgRNAs were obtained from Integrated DNA Technologies, Inc. as CRISPR RNAs (crRNAs) and tracer RNA (tracrRNA). SpCas9 was purchased as Alt-R S.p. HiFi Cas9 Nuclease from Integrated DNA Technologies, Inc (cat: 1081061). RNP complexes were prepared according to the manufacturer’s instruction. Tregs electroporated with Cas9 without sgRNA were used as a control for KO effect.
Electroporation was performed in 100 µl tips using a protocol consisting of three pulses at 1600 V and 10 ms on the Neon Transfection System (MPK10025, Neon System Kit, ThermoFisher Scientific) in accordance with manufacturer’s instructions.
sgRNA sequences targeting CD70 were described previously52 or designed using Benchling software (https://benchling.com/crispr). sgRNA sequences were: 1_3: GCTACGTATCCATCGTGA, 2_3: GTACACATCCAGGTGACGC, 3_3: GCAGGCTGATGCTACGGG and 1_1: TCACCAAGCCCGCGACCAAT.
KO efficiency was calculated as percentage of CD70+ cells in Tregs electroporated with Cas9 mRNA or RNPs compared to Tregs that were not electroporated or that were electroporated in the absence of Cas9 mRNA or sgRNA according to the following formula: (100 − (%positive cells in KO-Tregs * 100/%positive cells in control-Tregs)). In addition, genetic modifications at the CD70 locus were analyzed using ICE protocols (https://ice.synthego.com/). Genomic DNA extraction was performed using DNeasy Blood and Tissue Kit (69504, Qiagen, Hilden, Germany) and amplification of 700 bp around sites of predicted Cas9 scission was performed by PCR. Primers were 1_3 forward: CACCGGCTACGTATCCATCGTGA, 1_3 reverse: AAACTCACGATGGATACGTAGCC, 3_3 forward: CACCGCAGGCTGATGCTACGGG and 3_3 reverse: AAACCCCGTAGCATCAGCCTGC. PCR products were sequenced and analyzed using ICE algorithm.
Study approvals
All mouse experiments were performed using protocols approved by the Committee on Animal Care and Ethical Review at the University of Oxford and in accordance with the UK Animals (Scientific Procedures) Act 1986 and under PPL number P8869535A. Experiments using donated human cells were performed with ethical approval from the Oxfordshire Research Ethics Committee (REC B), study number 07/H0605/130. Single cell sequencing experiments were performed with ethical approval from Peterborough and Fenland Research Ethics Committee (05/Q0106/20) for SLE sample and the Royal Free Hospital & Medical School Research Ethics Committee; REC (08/H0720/25; D-GAP study) for T1D and HD samples. Informed consent was obtained from all subjects.
Statistics and reproducibility
Graphs were produced and statistical analyses performed using Prism version 5 or 7 (GraphPad Software, San Diego, CA, USA). Statistical significance was determined as indicated in the figure legends. Non-parametric tests were selected. Unpaired tests were performed to compare between cell subsets, and paired tests were used when comparing a specific cell subset over time or upon treatment with monoclonal antibodies. SD is represented in graphs showing replicates from one representative donor or in graphs with data from multiple donors, when each data point displays the data of one donor. SEM is used in graphs with data from multiple donors, when each data point represents a mean of multiple replicates for each donor. Functional assays were set up with, at least, three replicates for each sample. All source data underlying the graphs presented in the main figures are included in Supplementary Data 1 file.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The authors declare that all data supporting the findings of this study are available within the paper and its supplementary information files. Please contact the corresponding author (J.H.) for access of raw data, which is stored electronically, and will be made available upon reasonable requests. Sequencing data is available from the NCBI GEO database under accession numbers GSE129251.
References
Ferrer, I. R., Hester, J., Bushell, A. & Wood, K. J. Induction of transplantation tolerance through regulatory cells: from mice to men. Immunol. Rev. 258, 102–116 (2014).
Sakaguchi, S. Naturally arising CD4+ regulatory t cells for immunologic self-tolerance and negative control of immune responses. Annu. Rev. Immunol. 22, 531–562 (2004).
Wood, K. J. & Sakaguchi, S. Regulatory T cells in transplantation tolerance. Nat. Rev. Immunol. 3, 199–210 (2003).
Hori, S., Nomura, T. & Sakaguchi, S. Control of regulatory T cell development by the transcription factor Foxp3. Science 299, 1057–1061 (2003).
Baron, U. et al. DNA demethylation in the human FOXP3 locus discriminates regulatory T cells from activated FOXP3(+) conventional T cells. Eur. J. Immunol. 37, 2378–2389 (2007).
Ramsdell, F. & Ziegler, S. F. FOXP3 and scurfy: how it all began. Nat. Rev. Immunol. 14, 343–349 (2014).
Bennett, C. L. et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat. Genet. 27, 20–21 (2001).
Brunkow, M. E. et al. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat. Genet. 27, 68–73 (2001).
Wildin, R. S. et al. X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat. Genet. 27, 18–20 (2001).
Liu, W. et al. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J. Exp. Med. 203, 1701–1711 (2006).
Seddiki, N. et al. Expression of interleukin (IL)-2 and IL-7 receptors discriminates between human regulatory and activated T cells. J. Exp. Med. 203, 1693–1700 (2006).
Barbi, J., Pardoll, D. & Pan, F. Treg functional stability and its responsiveness to the microenvironment. Immunol. Rev. 259, 115–139 (2014).
Sawant, D. V. & Vignali, D. A. Once a Treg, always a Treg? Immunol. Rev. 259, 173–191, https://doi.org/10.1111/imr.12173 (2014).
Hori, S. Lineage stability and phenotypic plasticity of Foxp3(+) regulatory T cells. Immunol. Rev. 259, 159–172 (2014).
Zhou, X. et al. Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat. Immunol. 10, 1000–1007 (2009).
Tsuji, M. et al. Preferential generation of follicular B helper T cells from Foxp3+ T cells in gut Peyer’s patches. Science 323, 1488–1492 (2009).
Rubtsov, Y. P. et al. Stability of the regulatory T cell lineage in vivo. Science 329, 1667–1671 (2010).
Oldenhove, G. et al. Decrease of Foxp3+ Treg cell number and acquisition of effector cell phenotype during lethal infection. Immunity 31, 772–786 (2009).
Yang, X. O. et al. Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity 29, 44–56 (2008).
Bovenschen, H. J. et al. Foxp3+ regulatory T cells of psoriasis patients easily differentiate into IL-17A-producing cells and are found in lesional skin. J. Invest. Dermatol. 131, 1853–1860 (2011).
Sanchez Rodriguez, R. et al. Memory regulatory T cells reside in human skin. J. Clin. Invest. 124, 1027–1036 (2014).
Remedios, K. A. et al. The TNFRSF members CD27 and OX40 coordinately limit TH17 differentiation in regulatory T cells. Sci. Immunol. https://doi.org/10.1126/sciimmunol.aau2042 (2018).
Karlsson, F. H. et al. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature 498, 99–103 (2013).
Hovhannisyan, Z., Treatman, J., Littman, D. R. & Mayer, L. Characterization of interleukin-17-producing regulatory T cells in inflamed intestinal mucosa from patients with inflammatory bowel diseases. Gastroenterology 140, 957–965 (2011).
Voo, K. S. et al. Identification of IL-17-producing FOXP3+ regulatory T cells in humans. Proc. Natl Acad. Sci. USA 106, 4793–4798 (2009).
Pesenacker, A. M. et al. CD161 defines the subset of FoxP3+ T cells capable of producing proinflammatory cytokines. Blood 121, 2647–2658 (2013).
Golovina, T. N. et al. CD28 costimulation is essential for human T regulatory expansion and function. J. Immunol. 181, 2855–2868 (2008).
Wing, K. et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science 322, 271–275 (2008).
Hintzen, R. Q. et al. Regulation of CD27 expression on subsets of mature T-lymphocytes. J. Immunol. 151, 2426–2435 (1993).
Martorell, J., Rojo, I., Vilella, R., Martinez-Caceres, E. & Vives, J. CD27 induction on thymocytes. J. Immunol. 145, 1356–1363 (1990).
Coquet, J. M. et al. Epithelial and dendritic cells in the thymic medulla promote CD4+Foxp3+ regulatory T cell development via the CD27-CD70 pathway. J. Exp. Med. 210, 715–728 (2013).
Nolte, M. A., van Olffen, R. W., van Gisbergen, K. P. & van Lier, R. A. Timing and tuning of CD27-CD70 interactions: the impact of signal strength in setting the balance between adaptive responses and immunopathology. Immunol. Rev. 229, 216–231 (2009).
Mahnke, Y. D., Brodie, T. M., Sallusto, F., Roederer, M. & Lugli, E. The who’s who of T-cell differentiation: human memory T-cell subsets. Eur. J. Immunol. 43, 2797–2809 (2013).
Tesselaar, K. et al. Expression of the murine CD27 ligand CD70 in vitro and in vivo. J. Immunol. 170, 33–40 (2003).
Borst, J., Hendriks, J. & Xiao, Y. CD27 and CD70 in T cell and B cell activation. Curr. Opin. Immunol. 17, 275–281 (2005).
De Milito, A. et al. Mechanisms of hypergammaglobulinemia and impaired antigen-specific humoral immunity in HIV-1 infection. Blood 103, 2180–2186 (2004).
Hishima, T. et al. CD70 expression in thymic carcinoma. Am. J. Surg. Pathol. 24, 742–746 (2000).
Diegmann, J. et al. Identification of CD70 as a diagnostic biomarker for clear cell renal cell carcinoma by gene expression profiling, real-time RT-PCR and immunohistochemistry. Eur. J. Cancer 41, 1794–1801 (2005).
Held-Feindt, J. & Mentlein, R. CD70/CD27 ligand, a member of the TNF family, is expressed in human brain tumors. Int. J. Cancer 98, 352–356 (2002).
Han, B. K. et al. Increased prevalence of activated CD70+CD4+ T cells in the periphery of patients with systemic lupus erythematosus. Lupus 14, 598–606 (2005).
Yin, H. et al. Hypomethylation and overexpression of CD70 (TNFSF7) in CD4+ T cells of patients with primary Sjogren’s syndrome. J. Dermatol Sci. 59, 198–203 (2010).
Park, J. K. et al. CD70-expressing CD4 T cells produce IFN-gamma and IL-17 in rheumatoid arthritis. Rheumatology 53, 1896–1900 (2014).
Manocha, M. et al. Blocking CD27-CD70 costimulatory pathway suppresses experimental colitis. J. Immunol. 183, 270–276 (2009).
Hintzen, R. Q. et al. Engagement of CD27 with its ligand CD70 provides a second signal for T cell activation. J. Immunol. 154, 2612–2623 (1995).
Soares, H. et al. A subset of dendritic cells induces CD4+ T cells to produce IFN-gamma by an IL-12-independent but CD70-dependent mechanism in vivo. J. Exp. Med. 204, 1095–1106 (2007).
Xiao, Y., Peperzak, V., Keller, A. M. & Borst, J. CD27 instructs CD4+ T cells to provide help for the memory CD8+ T cell response after protein immunization. J. Immunol. 181, 1071–1082 (2008).
Hendriks, J. et al. CD27 is required for generation and long-term maintenance of T cell immunity. Nat. Immunol. 1, 433–440 (2000).
Arens, R. et al. Constitutive CD27/CD70 interaction induces expansion of effector-type T cells and results in IFNgamma-mediated B cell depletion. Immunity 15, 801–812 (2001).
Hendriks, J., Xiao, Y. & Borst, J. CD27 promotes survival of activated T cells and complements CD28 in generation and establishment of the effector T cell pool. J. Exp. Med. 198, 1369–1380 (2003).
Yamada, A. et al. CD70 signaling is critical for CD28-independent CD8+ T cell-mediated alloimmune responses in vivo. J. Immunol. 174, 1357–1364 (2005).
Latour, S. & Winter, S. Inherited immunodeficiencies with high predisposition to Epstein-Barr virus-driven lymphoproliferative diseases. Front. Immunol. 9, 1103, https://doi.org/10.3389/fimmu.2018.01103 (2018).
Izawa, K. et al. Inherited CD70 deficiency in humans reveals a critical role for the CD70-CD27 pathway in immunity to Epstein-Barr virus infection. J. Exp. Med. 214, 73–89 (2017).
Alkhairy, O. K. et al. Novel mutations in TNFRSF7/CD27: clinical, immunologic, and genetic characterization of human CD27 deficiency. J. Allergy Clin. Immunol. 136, 703–712 e710 (2015).
Salzer, E. et al. Combined immunodeficiency with life-threatening EBV-associated lymphoproliferative disorder in patients lacking functional CD27. Haematologica 98, 473–478 (2013).
van Montfrans, J. M. et al. CD27 deficiency is associated with combined immunodeficiency and persistent symptomatic EBV viremia. J. Allergy Clin. Immunol. 129, 787–793 (2012).
Abolhassani, H. et al. Combined immunodeficiency and Epstein-Barr virus-induced B cell malignancy in humans with inherited CD70 deficiency. J. Exp. Med. 214, 91–106 (2017).
Duggleby, R. C., Shaw, T. N., Jarvis, L. B., Kaur, G. & Gaston, J. S. CD27 expression discriminates between regulatory and non-regulatory cells after expansion of human peripheral blood CD4+ CD25+ cells. Immunology 121, 129–139 (2007).
Coenen, J. J., Koenen, H. J., van Rijssen, E., Hilbrands, L. B. & Joosten, I. Rapamycin, and not cyclosporin A, preserves the highly suppressive CD27+ subset of human CD4+CD25+ regulatory T cells. Blood 107, 1018–1023 (2006).
Ruprecht, C. R. et al. Coexpression of CD25 and CD27 identifies FoxP3+ regulatory T cells in inflamed synovia. J. Exp. Med. 201, 1793–1803 (2005).
Nadig, S. N. et al. In vivo prevention of transplant arteriosclerosis by ex vivo-expanded human regulatory T cells. Nat. Med. 16, 809–813 (2010).
Dhainaut, M. et al. Thymus-derived regulatory T cells restrain pro-inflammatory Th1 responses by downregulating CD70 on dendritic cells. EMBO J. 34, 1336–1348 (2015).
Bhairavabhotla, R. et al. Transcriptome profiling of human FoxP3+ regulatory T cells. Hum. Immunol. 77, 201–213 (2016).
De Simone, M. et al. Transcriptional landscape of human tissue lymphocytes unveils uniqueness of tumor-infiltrating T regulatory cells. Immunity 45, 1135–1147 (2016).
Ramakrishna, V. et al. Characterization of the human T cell response to in vitro CD27 costimulation with varlilumab. J. Immunother. Cancer 3, 37 (2015).
Ferraro, A. et al. Interindividual variation in human T regulatory cells. Proc. Natl Acad. Sci. USA 111, E1111–E1120 (2014).
Miyara, M. et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity 30, 899–911 (2009).
He, L. Z. et al. Agonist anti-human CD27 monoclonal antibody induces T cell activation and tumor immunity in human CD27-transgenic mice. J. Immunol. 191, 4174–4183 (2013).
Trzupek, D. et al. Discovery of CD80 and CD86 as recent activation markers on regulatory T cells by protein-RNA single-cell analysis. Genome Medicine 12, 55 (2020).
Singh, S. P., Zhang, H. H., Foley, J. F., Hedrick, M. N. & Farber, J. M. Human T cells that are able to produce IL-17 express the chemokine receptor CCR6. J. Immunol. 180, 214–221 (2008).
Bailey-Bucktrout, S. L. et al. Self-antigen-driven activation induces instability of regulatory T cells during an inflammatory autoimmune response. Immunity 39, 949–962 (2013).
Vukmanovic-Stejic, M. et al. The kinetics of CD4+Foxp3+ T cell accumulation during a human cutaneous antigen-specific memory response in vivo. J. Clin. Invest. 118, 3639–3650 (2008).
Arroyo Hornero, R. et al. CD45RA distinguishes CD4+CD25+CD127−/low TSDR demethylated regulatory T cell subpopulations with differential stability and susceptibility to tacrolimus-mediated inhibition of suppression. Transplantation 101, 302–309 (2017).
Fraser, H. et al. A rapamycin-based GMP-compatible process for the isolation and expansion of regulatory T cells for clinical trials. Mol. Ther. Methods Clin. Dev. 8, 198–209 (2018).
Hoffmann, P., Eder, R., Kunz-Schughart, L. A., Andreesen, R. & Edinger, M. Large-scale in vitro expansion of polyclonal human CD4(+)CD25high regulatory T cells. Blood 104, 895–903 (2004).
Hoffmann, P. et al. Loss of FOXP3 expression in natural human CD4+CD25+ regulatory T cells upon repetitive in vitro stimulation. Eur. J. Immunol. 39, 1088–1097 (2009).
Procaccini, C. et al. The proteomic landscape of human ex vivo regulatory and conventional T cells reveals specific metabolic requirements. Immunity 44, 712 (2016).
Cuadrado, E. et al. Proteomic analyses of human regulatory T cells reveal adaptations in signaling pathways that protect cellular identity. Immunity 48, 1046–1059 e1046 (2018).
Joly, A. L. et al. Alternative splicing of FOXP3 controls regulatory T cell effector functions and is associated with human atherosclerotic plaque stability. Circ. Res. 122, 1385–1394 (2018).
Matter, M., Odermatt, B., Yagita, H., Nuoffer, J. M. & Ochsenbein, A. F. Elimination of chronic viral infection by blocking CD27 signaling. J. Exp. Med. 203, 2145–2155 (2006).
van Gisbergen, K. P. et al. Protective CD8 T cell memory is impaired during chronic CD70-driven costimulation. J. Immunol. 182, 5352–5362 (2009).
O’Neill, R. E. et al. T cell-derived CD70 delivers an immune checkpoint function in inflammatory T cell responses. J. Immunol. 199, 3700–3710 (2017).
Ahrends, T. et al.c CD4(+) T cell help confers a cytotoxic T cell effector program including coinhibitory receptor downregulation and Increased tissue invasiveness. Immunity 47, 848–861 (2017).
Matter, M. S., Claus, C. & Ochsenbein, A. F. CD4+ T cell help improves CD8+ T cell memory by retained CD27 expression. Eur. J. Immunol. 38, 1847–1856, https://doi.org/10.1002/eji.200737824 (2008).
Feau, S. et al. The CD4(+) T-cell help signal is transmitted from APC to CD8(+) T-cells via CD27-CD70 interactions. Nat. Commun. 3, 948 (2012).
Croft, M. The role of TNF superfamily members in T-cell function and diseases. Nat. Rev. Immunol. 9, 271–285 (2009).
Brahmer, J. R. et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 366, 2455–2465 (2012).
Esensten, J. H., Helou, Y. A., Chopra, G., Weiss, A. & Bluestone, J. A. CD28 costimulation: from mechanism to therapy. Immunity 44, 973–988 (2016).
Vonderheide, R. H. & Glennie, M. J. Agonistic CD40 antibodies and cancer therapy. Clin. Cancer Res. 19, 1035–1043 (2013).
Yonezawa, A., Dutt, S., Chester, C., Kim, J. & Kohrt, H. E. Boosting cancer immunotherapy with anti-CD137 antibody therapy. Clin. Cancer Res. 21, 3113–3120 (2015).
Curti, B. D. et al. OX40 is a potent immune-stimulating target in late-stage cancer patients. Cancer Res. 73, 7189–7198 (2013).
Burris, H. A. et al. Safety and activity of varlilumab, a novel and first-in-class agonist anti-CD27 antibody, in patients with advanced solid tumors. J. Clin. Oncol. 35, 2028–2036 (2017).
Hester, J., Schiopu, A., Nadig, S. N. & Wood, K. J. Low-dose rapamycin treatment increases the ability of human regulatory T cells to inhibit transplant arteriosclerosis in vivo. Am. J. Transpl. 12, 2008–2016 (2012).
Boiers, C. et al. Lymphomyeloid contribution of an immune-restricted progenitor emerging prior to definitive hematopoietic stem cells. Cell Stem Cell 13, 535–548 (2013).
Stuart, T. et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902 (2019).
Finak, G. et al. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 16, 278 (2015).
Georgiadis, C. et al. Long terminal repeat CRISPR-CAR-coupled “Universal” T cells mediate potent anti-leukemic effects. Mol. Ther. 26, 1215–1227 (2018).
Demaison, C. et al. High-level transduction and gene expression in hematopoietic repopulating cells using a human immunodeficiency [correction of imunodeficiency] virus type 1-based lentiviral vector containing an internal spleen focus forming virus promoter. Hum. Gene Ther. 13, 803–813 (2002).
Acknowledgements
We would like to thank Kate Milward for technical advice and Tibor Keller (Celldex Therapeutics Inc.) for providing anti-CD27 antibodies and expert guidance. This work was supported in part by the Restore Research Trust, Kidney Research UK (grant SF1/2014), and the EU Framework Programe H2020 ReSHAPE Study (Grant Agreement 825392). J.H. is Kidney Research UK Senior Fellow and a recipient of ESOT Junior Basic Science Research grant, F.I. is a Wellcome Trust CRCD Fellow. This work has also been supported by positive discussion through A FACTT network (Cost Action BM1305, supported by the EU Framework Programme Horizon 2020). WQ/CG are supported by National Institute of Health Research (RP-2014-05-007), Children with cancer and the Great Ormond Street Biomedical Research Centre. Part of this work was supported by the JDRF UK Centre for Diabetes—Genes, Autoimmunity and Prevention (D-GAP; 4-2007-1003) in collaboration with M. Peakman and T. Tree at Kings College London, a strategic award to the Diabetes and Inflammation Laboratory from the JDRF (4-SRA-2017-473-A-N) and the Wellcome (107212/A/15/Z), and a grant from the JDRF (1-SRA-2019-657-A-N). We acknowledge the participation of all NIHR Cambridge BioResource volunteers, and thank the NIHR Cambridge BioResource centre and staff for their contribution. We thank C. Guy from the Department of Paediatrics, University of Cambridge for D-GAP sample recruitment.
Author information
Authors and Affiliations
Contributions
R.A.H.—conceived and designed the experiments, performed the experiments, analysed the data, wrote the manuscript, reviewed the manuscript. C.G. provided help, advice and materials for CRISPR/Cas9 experiments and performed some of CRISPR/Cas9 experiments, reviewed the manuscript. P.H. performed RNA sequencing and RNA-seq analysis, reviewed the manuscript. D.T. conceived, performed and analysed experiments investigating combined protein and single cell sequencing of cells from human donors, reviewed the manuscript. J.A.T. conceived, performed, and analysed experiments investigating combined protein and single cell sequencing of cells from human donors, reviewed the manuscript. R.C.F. conceived, performed, and analysed experiments investigating combined protein and single cell sequencing of cells from human donors, reviewed the manuscript. L.Z.H. provided guidance on experimental design using anti-CD27 antibodies, provided crucial materials, reviewed the manuscript. W.Q. provided help, advice and materials for CRISPR/Cas9 experiments, reviewed the manuscript. K.J.W. conceived and designed the experiments, wrote the manuscript, reviewed the manuscript. F.I. conceived and designed the experiments, wrote the manuscript, reviewed the manuscript. J.H. conceived and designed the experiments, analysed the data, wrote the manuscript, reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare the following sole competing financial interest: L.Z.H. is an employee and shareholder of Celldex Therapeutics, Inc. The other authors declare no competing financial or non-financial interests. Unrelated to this work, F.I. is currently Scientific Director of the Restore Research Trust but was not in this role when this study was carried out and receives no personal remuneration, J.H. and F.I. receive research funding from the Oxford-Celgene Fellowship Scheme (now Oxford-BMS), and W.Q. receives research funding from Servier and holds stock in Autolus and Orchard Therapeutics.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Arroyo Hornero, R., Georgiadis, C., Hua, P. et al. CD70 expression determines the therapeutic efficacy of expanded human regulatory T cells. Commun Biol 3, 375 (2020). https://doi.org/10.1038/s42003-020-1097-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-020-1097-8
This article is cited by
-
Mapping the functional interactions at the tumor-immune checkpoint interface
Communications Biology (2023)
-
Nasopharyngeal carcinoma cells promote regulatory T cell development and suppressive activity via CD70-CD27 interaction
Nature Communications (2023)
-
The CD70-CD27 axis in oncology: the new kids on the block
Journal of Experimental & Clinical Cancer Research (2022)
-
Ex vivo-expanded human CD19+TIM-1+ regulatory B cells suppress immune responses in vivo and are dependent upon the TIM-1/STAT3 axis
Nature Communications (2022)
-
CD27 expression on Treg cells limits immune responses against tumors
Journal of Molecular Medicine (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
