
Abstract
Although a physiological role for redox signaling is now clearly established, the processes sensitive to redox signaling remains to be identified. Ratiometric probes selective for H2O2 have revealed its complex spatiotemporal dynamics during neural development and adult regeneration and perturbations of H2O2 levels disturb cell plasticity and morphogenesis. Here we ask whether endogenous H2O2 could participate in the patterning of the embryo. We find that perturbations of endogenous H2O2 levels impact on the distribution of the Engrailed homeoprotein, a strong determinant of midbrain patterning. Engrailed 2 is secreted from cells with high H2O2 levels and taken up by cells with low H2O2 levels where it leads to increased H2O2 production, steering the directional spread of the Engrailed gradient. These results illustrate the interplay between protein signaling pathways and metabolic processes during morphogenetic events.
Similar content being viewed by others

Introduction
Reactive oxygen species (ROS), including hydrogen peroxide (H2O2), once only considered as deleterious compounds, have recently raised novel interest due to their action as bona fide-signaling molecules1,2,3. Ratiometric probes selective for H2O2 have revealed its complex spatiotemporal dynamics during neural development and adult regeneration4,5. In addition, modifying H2O2 levels disturbs cell plasticity and morphogenesis6,7. Proteins targeted by H2O2 during development, by mechanisms still largely elusive, belong to many categories and include homeoproteins (HPs)8,9. HPs play important roles in the control of cellular and regional identity during development. Although first characterized as purely cell autonomous transcription factors10, HPs are also transferred between cells via non-conventional secretion and internalization routes, providing them with direct paracrine activity11. Intercellular transfer is a general property of HPs, and its efficiency is context-dependent12,13. Within recipient cells, transferred HPs may act not only as transcription factors but also on processes as diverse as translation, DNA repair, mitochondrial activity, and epigenetic modification14.
We have previously shown that the zebrafish midbrain–hindbrain boundary (MHB) displays high levels of H2O2 that cannot be lowered without affecting tectum topography6, a process that also requires the graded distribution of the Engrailed HPs (EN in amniotes, Eng in fish)15,16,17,18. We thus asked whether H2O2 levels and Engrailed distribution interplay with each other by conducting in parallel a close examination of both parameters at the time of the tectum antero-posterior polarization. Importantly, the Engrailed extracellular gradient is instrumental in establishing tectum polarity in frogs and chicks19, requires Eng2b paracrine activity in the zebrafish20. We combined ex vivo and in vivo approaches to demonstrate an unsuspected role of H2O2 in Engrailed homeoprotein spreading during tectum development. Engrailed is released from cells with high H2O2 levels and transfer to cells with low H2O2 levels in which it stimulates H2O2 production, thereby controlling its own polarized traffic. In addition, we identify cysteine 175 as a key residue in the redox regulation of Engrailed traffic.
Results and discussion
H2O2 levels shape Engrailed 2 distribution in the tectum
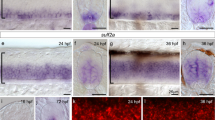
The graded distribution of Eng2 evolves during development, particularly between 24 and 26 h post fertilization (hpf) (Fig. 1a, b and Supplementary Fig. 1). As previously shown in several vertebrates15,16,17,18, Eng2 level is maximum at the MHB where it is known to be synthesized and decreases along the posterior–anterior axis until becoming undetectable at the most anterior part of the tectum. The temporal analysis of Eng2 distribution throughout the tectum (24–28 hpf) showed that, at the MHB, Eng2 level is highest at 24 hpf and decreased at 26 and 28 hpf (Fig. 1b). These modifications in Eng2 distribution were concomitant with remarkable changes in H2O2 levels in the same structure (Fig. 1d, e). In the whole tectum, H2O2 levels increased over time (Fig. 1e), but with a marked gradient from the MHB to the most anterior part of the tectum (Fig. 1d). We noticed that in zebrafish embryos, the clear graded distribution of nuclear Eng2 proteins is accompanied by an extranuclear gradient in the tectum along the anteroposterior axis (Fig. 1a′–a′′′), previously observed in chick and Xenopus embryos19 and shown to correlate with Engrailed intercellular transfer ex vivo21. Using DAPI staining as a marker of the nuclear compartment, nuclear and extranuclear Eng2 signals were quantified separately. The ratio of nuclear to extranuclear Eng2 staining showed that the cellular distribution of Eng2 varied both spatially and temporally along the antero-posterior axis of the tectum from 24 to 28 hpf (Fig. 1c). The nuclear/extranuclear ratio was highest at 26 hpf and decreased at 28 hpf while H2O2 increased between 24 and 28 hpf. We thus concluded that EN2 distribution is regulated, at least partially, by H2O2 levels between 24 and 28 hpf, a temporal window during which both are highly dynamic.

a Immunodetection of Eng2a and Eng2b (green) and DAPI staining (red) in zebrafish embryos (24 hpf) revealed different nuclear/extranuclear distributions along the anteroposterior axis (a’–a”‘: insets of sections at higher magnification; MHB midbrain hindbrain boundary, HB hindbrain). Eng2 staining alone is shown in Supplementary Fig. 1. b Quantification of total Eng2 levels (inferred from Eng immunostaining) along the anteroposterior axis of the tectum at 24, 26, and 28 hpf. All values were normalized to the maximum value for 26 hpf. c Ratio of Eng2 nuclear over extranuclear signals at 24, 26, and 28 hpf. d H2O2 levels in the tecta of zebrafish embryos from 24 to 28 hpf. H2O2 levels were inferred from the YFP500/YFP420 excitation ratio of HyPer7 in time-lapse recordings. Arrowhead: MHB position. The quantification is presented in e. f H2O2 levels in the tecta of control (Ctrl) and two hours Nox-i (100 nM) treated zebrafish 26 hpf embryos. g Quantification of immunodetected Eng2 along the anteroposterior axis of the tectum in control (Ctrl) and Nox-i-treated embryos (26 hpf). h ratio of Eng2 nuclear/extranuclear signals in control (Ctrl) and Nox-i-treated embryos (26 hpf). Double arrows in g and h indicate the domain where nuclear to extranuclear ratios are statistically different (confidence interval varying from 0.0001 to 0.05 from the MHB to more anterior positions). Scale bars, 50 μm. *p-value ≤ 0.05.
We next tested whether altering endogenous H2O2 levels would affect Eng2 distribution. Thanks to the use of the improved ratiometric H2O2 sensor HyPer722, we were able to detect, in vivo, the modulation of H2O2 levels after treatment with a pan-NADPH oxidase inhibitor (Nox-i) (Fig. 1f). The resulting mild decrease in H2O2 levels led to a marked modification of Eng2 distribution, enhancing Eng2 signal at the MHB but raising the gradient slope in the tectum (Fig. 1g). The higher amount of Eng2 at the MHB was not a consequence of a higher transcription rate since a quantitative RT-PCR performed on the two sets of embryos (Nox-i treated and control embryos) revealed no difference in the amount of eng2a and eng2b mRNA (Supplementary Fig. 2). This suggested that lowering endogenous H2O2 levels reduced Eng2 spreading from its area of production, most likely by modifying its intercellular trafficking. To test this hypothesis, we analyzed the nuclear/extranuclear distribution of Eng2 over the tectum in embryos treated or not with Nox-i (Fig. 1h). In Nox-i-treated embryos, the cellular distribution of Eng2 was significantly altered (Fig. 1h), characterized by a specific increase of Eng2 in the nuclear fraction, indicating that the correct propagation of Eng2 is dependent on H2O2 having reached a given threshold. In summary, mild reduction in H2O2 levels induced both a strong modification in the allocation of Eng2 between the nuclear and extranuclear compartments, and a distorted Eng2 distribution over the tectum. These results suggested that Eng2 intercellular transfer is regulated by H2O2. To directly address the involvement of H2O2 in Engrailed intercellular trafficking, we used ex vivo cell culture models which permit an accurate quantification of this process.
Engrailed 2 intercellular transfer is asymmetrically regulated by H2O2
To evaluate the sensitivity of Engrailed trafficking to H2O2 levels, we performed assays in HeLa cells, where this process has been best charaterized13,21,23. H2O2 levels were fine-tuned with two strategies. To enhance H2O2 levels, we added d-Alanine (d-Ala) to cells expressing a membrane-bound form of d-amino-acid oxidase (Lck-DAO) (Supplementary Fig. S3a, b)24. To reduce H2O2 levels, we expressed Catalase (CAT) deprived of its peroxisome targeting sequence (CATΔC) and targeted to the plasma membrane (Lck-CATΔC, Supplementary Fig. S3c, d). We also reduced H2O2 levels by adding purified CAT to cell culture or by treating cells with Nox-i (Supplementary Fig. S3e, f). Under all conditions, H2O2 levels were monitored with HyPer expressed in the cytoplasm25. The two steps of intercellular transfer, secretion, and internalization, were analyzed separately using dedicated assays set up with the Engrailed 2 homeoprotein (EN2).
To study EN2 secretion, this process was quantified with a new strategy (transRUSH26) adapted from the RUSH system27 (Fig. 2a). Two tags were added to EN2: one (SBP-tag) that hooks the protein at the inner side of the plasma membrane when co-expressed with a membrane-bound Streptavidin hook, and another tag (HiBiT), a small nanoluciferase fragment that allows light production28 upon interaction with its large counterpart fragment (LgBit) addressed to the extracellular side of the plasma membrane. In this system, intracellular-trapped EN2 is released upon biotin addition, and secretion is monitored by light production upon interaction of EN2-HiBiT with LgBiT at the cell surface. Increasing H2O2 levels (via d-Ala addition) stimulated EN2 secretion (Fig. 2b), whereas decreasing them by either expressing CATΔC or treating the cells with purified CAT or Nox-i reduced EN2 secretion (Fig. 2c–e).

a Method for quantification of EN2 secretion synchronized at the plasma membrane via a modified RUSH strategy, the transRUSH method26. EN2 is tagged with SBP (black) that hooks the protein at the inner side of the plasma membrane in presence of a Streptavidin hook (gray) and HiBiT (white bulb) that emits light when combined to LgBit (yellow crescent). b–e Quantification of EN2 secretion from cells expressing Lck-DAO with or without d-Ala b, expressing inactive or active Catalase (CATmut or CAT, respectively) c, treated with extracellular CAT (CAText,) d, or treated with Nox-i e. f Method for quantification of EN2 internalization. g–i Quantification of EN2 internalization in cells expressing Lck-DAO with or without d-Ala g, treated with CAText h or Nox-i i. *p-value ≤ 0.05; **p-value ≤ 0.01; ***p-value ≤ 0.001; and ****p-value ≤ 0.0001.
To study EN2 internalization, purified recombinant HiBiT-tagged EN2 was added to the medium, and its specific delivery into the cytosol was quantified by light production resulting from its interaction with a cytosolic LgBiT (Fig. 2f). Increasing H2O2 levels (via d-Ala addition) reduced EN2 internalization (Fig. 2g), while decreasing them (by treatment with either purified CAT or Nox-i) enhanced EN2 internalization (Fig. 2h, i). These results were confirmed by direct visualization of FITC-tagged EN2 uptake (Supplementary Fig. S4). In summary, redox levels modulate the two steps of EN2 trafficking in an uneven manner: low levels of H2O2 stimulate internalization and reduce secretion, while high levels of H2O2 have the opposite effects. The dual role of H2O2 levels on EN2 trafficking observed in cell culture nicely fits with the in vivo effects of Nox-i treatment presented above (Fig. 1h): the nuclear accumulation in producing cells close to the MHB and the reduction of Eng spreading through the tectum are best explained by an inhibition of Eng2 secretion supporting the view that H2O2 levels directly regulate Eng2 distribution in the zebrafish embryonic tectum.
Cysteine 175 is involved in the redox regulation of Engrailed spreading
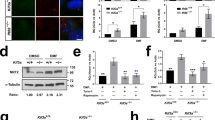
Reversible oxidation of cysteine residues is the main target of ROS action within proteins, affecting their conformation and activity29. Only one cysteine is conserved through evolution in Engrailed proteins. It is located at position 175 in chicken EN2, next to the hexapeptide motif, that is essential both for EN2 transcriptional activity30 and intercellular transfer20 (Fig. 3a). To test whether this conserved cysteine is required for EN2 functions, we first evaluated the DNA-binding and transcriptional activities of a C175S EN2 mutant (EN2C>S) using as negative control EN2W>K, a W169>K,W>172>K double mutant deficient for transcriptional activity and intercellular transfer20,31. In the electrophoretic mobility shift assay (EMSA), EN2C>S bound its target nucleic acid sequence in the presence of its PBX co-factor with the same efficiency as EN2 (Fig. 3b). In co-transfection experiments, EN2C>S stimulated transcription from the MAP1b promoter (a known target of Engrailed32) to the same extent as EN2 (Fig. 3c). The transfer ability of EN2C>S was then analyzed in secretion and internalization assays. As shown in Fig. 3d–g, EN2C>S transfer between cells was strongly impaired at both the secretion and internalization steps. Importantly, EN2C>S secretion became insensitive to H2O2 level modulation (Fig. 3e, f). Reducing EN2 cysteine by DTT decreased its internalization to EN2C>S levels, which was insensitive to DTT treatment (Fig. 3h). Part of this effect might involve the ability of EN2 to dimerize as a covalent EN2 dimer (Supplementary Fig. S5a). Indeed, promoting covalent redox-insensitive EN2 homodimerization using a chemical cross-linker (Supplementary Fig. S5b) stimulated internalization compared to native EN2 (Fig. 3i). In summary, the EN2C>S mutation had no broad effect on the transcriptional activity of EN2, but it lessened EN2 intercellular transfer and abolished its sensitivity to H2O2.

a Conserved cysteine residue in Engrailed 2 proteins across species (the hexapeptide is underlined). b Gel-shift assay comparing the DNA-binding properties of EN2, EN2C>S and DNA-binding deficient EN2W>K. c Quantification of EN2C>S and EN2 transcriptional activity on the MAP1b promoter. d–f Quantification of EN2 or EN2C>S secretion via the transRUSH method from control cells d or cells expressing Lck-DAO with or without d-Ala e or inactive or active Catalase (CATmut or CAT, respectively) f. Quantification of EN2 or EN2C>S internalization g after DTT pretreatment h or EN2 dimerization i. *p-value ≤ 0.05; **p-value ≤ 0.01; ***p-value ≤ 0.001; and ****p-value ≤ 0.0001.
Engrailed 2 internalization increases H2O2 levels
Signaling pathways are not just linear chains of events, but use crosstalk, feedback, and reciprocal interactions to control biological processes33. In this context, because Engrailed is known to influence the oxidative metabolism of recipient cells11, we wondered whether its trafficking34 could have an impact on intracellular H2O2 levels. To address this question, EN2 was added to the medium of cells expressing HyPer throughout their cytoplasm. EN2 addition, but not of either mutant proteins deficient for internalization (EN2W>K and EN2C>S), induced a quick increase in H2O2 levels (Fig. 4a, b). To verify that EN2 internalization was responsible for this increase, this step was specifically blocked by preincubation with the 4G11 monoclonal antibody19. Under these conditions, EN2 addition no longer enhanced intracellular H2O2 levels (Fig. 4c, d). EN2 import is thus necessary and sufficient to modify cytoplasmic H2O2 levels in recipient cells ex vivo. We then asked whether Eng intercellular transfer also modulates H2O2 levels in vivo. Extracellular anti-En single-chain antibody 4F11scFv, which blocks Engrailed transfer35, was expressed in the Eng domains using eng2a mini-enhancer36 in zebrafish embryos expressing HyPer7. This led to a strong reduction in H2O2 levels in the tecta of embryos expressing the blocking antibody, but not of embryos expressing a fluorescent protein as a control (Fig. 4e, f).

a, b H2O2 imaging and quantification in cells treated with extracellular EN2, EN2W>K, or EN2C>S. H2O2 levels were inferred as in Fig. 1. c, d H2O2 imaging and quantification of cells treated with extracellular EN2 and the 4G11 antibody directed against Engrailed. e, f Representative images e and quantification f of H2O2 levels (inferred as in Fig. 1) in the tecta of zebrafish embryos (26 hpf) expressing scFv4F11 or mCherry as a control. g Synthetic model of H2O2–Engrailed interaction during tectum development (H2O2: red; Engrailed: green). In the tectum, H2O2 levels increase over time and are gradualy distributed from the MHB to the anterior part of the embryo (A: anterior, P: posterior). Engrailed is released from cells with high H2O2 levels and enters cells with lower H2O2 levels, where it stimulates the production of H2O2. This results in the establishment of a polarized gradient of Engrailed. *p-value ≤ 0.05; **p-value ≤ 0.01; ***p-value ≤ 0.001; and ****p-value ≤ 0.0001.
In summary, Engrailed internalization enhanced intracellular levels of H2O2, modifying the properties of the recipient cells.
Conclusion
Together, these results demonstrate that Engrailed is transferred from cell to cell in the zebrafish tectum in an H2O2-dependent manner, and itself acts rapidly to establish an H2O2 gradient in vivo. We propose that the global wave of H2O2 that takes place during development (Fig. 1d, e) sets the spatio-temporal window for heightened Engrailed transfer. Engrailed internalization increases H2O2 levels in recipient cells, making them more competent to secrete Engrailed and reducing Engrailed uptake once they have reached an H2O2 threshold. Both events propagate the Engrailed signal forward (posterior to anterior), which leads to a directional spread of the Engrailed gradient.
As a consequence, the extent and intensity of the morphogenetic action of Engrailed mediated via its non-cell autonomous diffusion37 is shaped by its reciprocal interactions with the main redox signaling molecule (Fig. 4g). We still do not know if this finding can be generalized to other HPs endowed with paracrine activity13, but if so, may give new insights into how tissue morphogenesis and cell metabolism influence each other. Given the role of HPs expression in evolution, it is tempting to consider how such a mechanism may have contributed to link the second rise in atmospheric oxygen to the concomitant increase in metazoan complexity38.
Methods
DNA constructs, recombinant proteins, and biochemical assays
Details of the DNA constructs are given in Supplementary Table S1. His6-tagged recombinant proteins were produced in BL21 (DE3) grown in MagicMedia (Invitrogen) 24 h, 28 °C and purified on HisTrap columns (GE Healthcare) by Imidazole gradient elution on AKTA Prime according to manufacturer instructions. Following tag removal by incubation with PreScisson protease (6 h, 4 °C), the protein was purified on Heparin column (GE Healthcare), eluted by NaCl gradient and dialyzed for 2 days (20 mM phosphate buffer, 100 mM NaCl, pH 7.5). For protein FITC labeling, 100 μM of dialyzed purified protein was incubated with a two-fold molar excess of fluorescein isothiocyanate in carbonate buffer (50 mM pH 9.5, 100 mM NaCl) overnight at 4 °C and free FITC was removed by dialysis (48 h, 4 °C). The efficacy of FITC incorporation was determined by SDS–PAGE and spectral analysis. The molecular FITC:protein ratio was confined to a range between 1.5 and 2 for all proteins. Gel-shift assays were performed as in ref. 39. Redox-insensitive En2 covalent dimerization was obtained with the homobifunctional crosslinker 1,8-bis(maleimido)diethylene glycol (Thermo #22336), according to manufacturer instructions.
Fish care and manipulation
Fish husbandry: Zebrafish were maintained and staged as previously described40. Experiments were performed using the standard AB wild-type strain. The embryos were incubated at 28 °C. Developmental stages were determined and indicated as hours postfertilization (hpf). The animal facility obtained permission from the French Ministry of Agriculture for all the experiments described in this manuscript (agreement no. C 75-05-12).
Nucleic acid injection: Plasmids (2 ng/µl) were injected into the one-cell stage embryos with 20 ng/μl transposase mRNA to induce DNA recombination into the genome. Transgenic embryos were screened for fluorescent protein expression prior to the analysis. mRNA synthesis was performed using the mMESSAGE mMACHINE transcription kit from Thermofisher, Inc. Equivalent volumes of 75 ng/µl mRNA were injected into one-cell stage embryos to induce ubiquitous expression. EN2-ERT2 fusions were activated at 90% epiboly by adding cyclofen as described in ref. 20.
Pharmacological treatments: To decrease H2O2 levels, embryos were incubated in VAS-2870 (Nox-i) (100 nM) from Enzo Life Sciences (#BML-El395-0010, Enzo Life Sciences, Inc.; Farmingdale, NY, USA) or an equivalent amount of DMSO as a control for the duration of the time-lapse analysis.
Whole-mount immunostaining: Embryos were fixed with paraformaldehyde (4%, 2 h, room temperature) in PBS. The embryos were permeabilized with cold acetone (−20 °C, 10 min), washed in Triton X-100 (0.8% in PBS, room temperature), and saturated with PBS containing 10% sheep serum, 1% DMSO (Sigma), and 0.8% Triton X-100 (1 h, room temperature) before incubation with the primary antibody (4D9 anti-Engrailed antibody was deposited to DSHB by Goodman C, and used at a 1:500 dilution, overnight, 4 °C) and secondary antibody (Alexa Fluor-488 goat anti-mouse IgG, diluted 1:1500, with DAPI diluted 1:500, overnight, 4 °C).
RNA extraction and quantitative PCR: Total mRNA was extracted form 30 embryos per sample using Monarch total RNA miniprep kit according to the manufacturer’s protocol (NEB). 500 ng of total mRNA was reverse-transcribed by superscript II reverse transcriptase using oligo(dT) primers (Invitrogen). Quantitative PCR was performed using LightCycler® 480 detection system from Roche, Taqman gene expression master mix and Taqman probes from Applied Biosystem (rpl13: Dr03119261_m1; en2a: Dr03079901_m1; en2b: Dr03118700_m1). eng2a and eng2b gene expressions were determined using ΔΔCt method and normalized to rpl13 levels. Each sample was tested in triplicate. The run protocols were performed according to manufacturer’s recommendations.
Embryo imaging: The larvae were anesthetized in tricaine solution and embedded in low-melting agarose (0.5%). Imaging was performed with a CSU-W1 Yokogawa spinning disk coupled to a Zeiss Axio Observer Z1 inverted microscope equipped with a sCMOS Hamamatsu camera and a ×25 (Zeiss 0.8 Imm DIC WD: 0.57 mm) oil objective. DPSS 100 mW 405 nm and 150 mW 491 nm lasers and a 525/50 bandpass emission filter were used.
Quantification and statistical analyses: Total Engrailed was quantified by measuring the mean fluorescence along an 80 pixel-width line starting from the MHB border on Z-projections based on MHB-crossing slices. Nuclear and extranuclear Engrailed levels were analyzed by using the DAPI staining as a mask. Nuclear over extranuclear ratio was then calculated on the raw fluorescence values. Ordinary one-way ANOVA followed by Tukey’s multiple comparison test was performed to evaluate the significant differences between the conditions along the tectum. H2O2 levels were quantified by measuring the mean ratio value of HyPer7 per time point and normalized by the mean ratio value determined before treatment. A t-test was then performed to statistically determine the differences between the conditions over time.
Cell culture, ex vivo H2O2 manipulation, and transcriptional activity analysis
Cell culture experiments were performed with HeLa cells grown in DMEM supplemented with 10% fetal bovine serum. Transient transfection was performed with Lipofectamine 2000 (Life Technologies) according to the manufacturer’s instructions. The cells were cultured for an additional 24 h before being processed for analysis. HeLa cells constitutively expressed a tetracycline repressor (HeLa-Flp-In/T-Rex, Life Technologies), and doxycycline was used to induce protein expression when tetracycline-sensitive expression plasmids were transfected. Stable HeLa cells in which protein expression was controlled by doxycycline were prepared using the HeLa Flp-In cell line, which was kindly provided by Stephen Taylor41). A list of the stable cell lines used in this study is given in Supplementary Table S3.
Pharmacological treatments: To decrease H2O2 levels, cells were either treated with extracellular CAT (Sigma-Aldrich #C1345, 4 U/mL) or pretreated for 1 h with VAS-2870 (Nox-i) (10 μM) (#BML-El395-0010, Enzo Life Sciences, Inc.; Farmingdale, NY, USA) or an equivalent amount of DMSO as a control. To increase H2O2 levels, cells expressing d-amino acid oxidase (DAO) were treated with 10 mM d-alanine (Sigma-Aldrich #A7377) before the internalization or secretion assays were performed. Blocking anti-Engrailed monoclonal antibody 4G11 was deposited to the DSHB by Jessel TM and Brenner-Morton S.
Quantitative secretion assay: Cells (13,000 per well) stably expressing doubly tagged EN2 (SBP-EN2-HiBiT) were plated on 96-well plates (Greiner Bio-one) coated with polyornithine (10 μg/mL) and induced for constitutive protein expression with doxycycline. After 10 h, the cells were transfected with bidirectional expression plasmids of the transRUSH series making use of the sCMV enhancer, all expressing transmembrane fusions of LgBiT (outside) and core streptavidin (inside) downstream the CMV minimal promoter, and expressing in the opposite direction—downstream the sCMV promoter—either one of Lck-tagged DAO, active CAT devoid of a peroxisome signal, or its inactive counterpart. After 24 h, media were removed, and cells were incubated with fresh medium at 37 °C. Secretion was induced with biotin (100 μM final), and luciferase activity was measured 1 h later with a 96-well plate luminometer (Tristar, Berthold) as described in the HiBit assay kit (Promega). The cells were then lysed to measure intracellular protein expression. Normalization with biotin-untreated wells enabled us to calculate the secretion index and report the secretion efficiency.
Qualitative internalization assay: Cells (30,000 per well) were plated on μ-slide six-well plates (Ibidi). After 24 h, the medium was removed, and cells were incubated with a fluorescent protein (1 µM) diluted in DMEM without serum for 30 min at 37 °C before visualization on a CSU-W1 Yokogawa spinning disk coupled to a Zeiss Axio Observer Z1 inverted microscope equipped with a sCMOS Hamamatsu camera with a ×63/1.4 oil WD: 0.17 mm objective. Cells were analyzed following addition of Trypan blue (0.1% final concentration), an efficient quencher of all extracellular fluorescence (and that of permeabilized cells), to visualize intracellular staining.
Quantitative internalization assay: Cells (90,000 per well) stably expressing LgBiT were plated in 24-well culture dishes. After 24 h, the purified protein fused to HiBiT was added to the medium for 30 min at 37 °C before incubating the cells with trypsin and removing them. After centrifugation, cells were resuspended in PBS and 2% FBS, and the luciferase activity of internalized protein was measured with a 96-well plate luminometer (Tristar, Berthold) with a HiBiT assay kit (Promega).
H2O2 imaging with the HyPer probe: HyPer fluorescence was excited with 501/16 and 420/40 bandpass excitation filters, and the corresponding YFP emission was measured using a 530/35 bandpass emission filter. Spinning-disk images were acquired using a ×63 objective (×63/1.4 oil WD: 0.17 mm) on a Spinning-Disk CSU-W1 (Yokogawa) equipped sCMOS Hamamatsu 2048×2048 camera. To calculate the HyPer ratio, images were treated as previously described42.
Transcriptional activity: MAP1B promoter activation by EN2 or EN2C>S was quantitated in co-transfection experiments as previously described32 except that the reporter construct expressed Nanoluciferase instead of Luciferase.
Statistics and reproducibility: Data were analyzed using GraphPad Prism 6 and expressed as the mean ± standard error of the mean (SEM). Statistical significance was calculated using a two-sided paired Student’s t-test. For multiple conditions, ordinary one-way ANOVA followed by Tukey’s multiple comparison test was performed to evaluate the significant differences. Sample sizes and number of replicates are given in Supplementary Table 4. For each experiment, at least two independent experiment with similar results were performed.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Source data are provided with this paper. The DNA constructs and cell lines and transgenic fish are available upon request. All other data underlying the findings of the study are available from the corresponding author upon reasonable request.
References
Holmstrom, K. M. & Finkel, T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 15, 411–421 (2014).
Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: oxidative eustress. Redox Biol. 11, 613–619 (2017).
Sies, H. & Jones, D. P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. https://doi.org/10.1038/s41580-020-0230-3 (2020).
Rampon, C., Volovitch, M., Joliot, A. & Vriz, S. Hydrogen peroxide and redox regulation of developments. Antioxidants 7, https://doi.org/10.3390/antiox7110159 (2018).
Vriz, S. Redox signalling in development and regeneration. Semin. Cell Dev. Biol. 80, 1–2 (2018).
Gauron, C. et al. Hydrogen peroxide (H2O2) controls axon pathfinding during zebrafish development. Dev. Biol. 414, 133–141 (2016).
Albadri, S. et al. Redox signaling via lipid peroxidation regulates retinal progenitor cell differentiation. Dev. Cell 50, 73–89 e76 (2019).
Covarrubias, L., Hernandez-Garcia, D., Schnabel, D., Salas-Vidal, E. & Castro-Obregon, S. Function of reactive oxygen species during animal development: passive or active? Dev. Biol. 320, 1–11 (2008).
Marinho, H. S., Real, C., Cyrne, L., Soares, H. & Antunes, F. Hydrogen peroxide sensing, signaling and regulation of transcription factors. Redox Biol. 2, 535–562 (2014).
Krumlauf, R. Hox genes in vertebrate development. Cell 78, 191–201 (1994).
Spatazza, J. et al. Homeoprotein signaling in development, health, and disease: a shaking of dogmas offers challenges and promises from bench to bed. Pharm. Rev. 65, 90–104 (2013).
Bernard, C. & Prochiantz, A. Otx2–PNN interaction to regulate cortical plasticity. Neural Plast. 2016, 7931693 (2016).
Lee, E. J. et al. Global analysis of intercellular homeodomain protein transfer. Cell Rep. 28, 712–722 e713 (2019).
Di Nardo, A. A., Fuchs, J., Joshi, R. L., Moya, K. L. & Prochiantz, A. The physiology of homeoprotein transduction. Physiol. Rev. 98, 1943–1982 (2018).
Retaux, S. & Harris, W. A. Engrailed and retinotectal topography. Trends Neurosci. 19, 542–546 (1996).
Nakamura, H. Regionalization of the optic tectum: combinations of gene expression that define the tectum. Trends Neurosci. 24, 32–39 (2001).
Omi, M. & Nakamura, H. Engrailed and tectum development. Dev. Growth Differ. 57, 135–145 (2015).
Scholpp, S. & Brand, M. Morpholino-induced knockdown of zebrafish Engrailed genes eng2 and eng3 reveals redundant and unique functions in midbrain–hindbrain boundary development. Genesis 30, 129–133 (2001).
Wizenmann, A. et al. Extracellular Engrailed participates in the topographic guidance of retinal axons in vivo. Neuron 64, 355–366 (2009).
Rampon, C. et al. Control of brain patterning by Engrailed paracrine transfer: a new function of the Pbx interaction domain. Development 142, 1840–1849 (2015).
Maizel, A., Bensaude, O., Prochiantz, A. & Joliot, A. A short region of its homeodomain is necessary for Engrailed nuclear export and secretion. Development 126, 3183–3190 (1999).
Pak, V. V. et al. Ultrasensitive genetically encoded indicator for intracellular hydrogen peroxide identifies novel roles for cellular oxidants in cell migration and mitochondrial function. Cell Metab. 32, 642–653 (2020).
Tassetto, M., Maizel, A., Osorio, J. & Joliot, A. Plant and animal homeodomains use convergent mechanisms for intercellular transfer. EMBO Rep. 6, 885–890 (2005).
Matlashov, M. E., Belousov, V. V. & Enikolopov, G. How much H(2)O(2) is produced by recombinant d-amino acid oxidase in mammalian cells? Antioxid. Redox Signal 20, 1039–1044 (2014).
Belousov, V. V. et al. Genetically encoded fluorescent indicator for intracellular hydrogen peroxide. Nat. Methods 3, 281–286 (2006).
Amblard, I. et al. Bidirectional transfer of Engrailed homeoprotein across the plasma membrane requires PIP2. J. Cell Sci. https://doi.org/10.1242/jcs.244327 (2020).
Boncompain, G. et al. Synchronization of secretory protein traffic in populations of cells. Nat. Methods 9, 493–498 (2012).
Dixon, A. S. et al. NanoLuc complementation reporter optimized for accurate measurement of protein interactions in cells. ACS Chem. Biol. 11, 400–408 (2016).
Leichert, L. I. & Dick, T. P. Incidence and physiological relevance of protein thiol switches. Biol. Chem. 396, 389–399 (2015).
Moens, C. B. & Selleri, L. Hox cofactors in vertebrate development. Dev. Biol. 291, 193–206 (2006).
Erickson, T., Scholpp, S., Brand, M., Moens, C. B. & Waskiewicz, A. J. Pbx proteins cooperate with Engrailed to pattern the midbrain–hindbrain and diencephalic–mesencephalic boundaries. Dev. Biol. 301, 504–517 (2007).
Montesinos, M. L. et al. The neuronal microtubule-associated protein 1B is under homeoprotein transcriptional control. J. Neurosci. 21, 3350–3359 (2001).
Brandman, O. & Meyer, T. Feedback loops shape cellular signals in space and time. Science 322, 390–395 (2008).
Sagan, S. et al. Homeoproteins and homeoprotein-derived peptides: going in and out. Curr. Pharm. Des. 19, 2851–2862 (2013).
Layalle, S. et al. Engrailed homeoprotein acts as a signaling molecule in the developing fly. Development 138, 2315–2323 (2011).
Maurya, A. K. et al. Integration of Hedgehog and BMP signalling by the engrailed2a gene in the zebrafish myotome. Development 138, 755–765 (2011).
Quininao, C., Prochiantz, A. & Touboul, J. Local homeoprotein diffusion can stabilize boundaries generated by graded positional cues. Development 142, 1860–1868 (2015).
Thannickal, V. J. Oxygen in the evolution of complex life and the price we pay. Am. J. Respir. Cell Mol. Biol. 40, 507–510 (2009).
Joliot, A., Pernelle, C., Deagostini-Bazin, H. & Prochiantz, A. Antennapedia homeobox peptide regulates neural morphogenesis. Proc. Natl Acad. Sci. USA 88, 1864–1868 (1991).
Westerfield, M. The Zebrafish Book: a Guide for the Laboratory Use of Zebrafish (Brachydanio rerio) (University of Oregon Press, Eugene, OR, 1995).
Tighe, A., Staples, O. & Taylor, S. Mps1 kinase activity restrains anaphase during an unperturbed mitosis and targets Mad2 to kinetochores. J. Cell Biol. 181, 893–901 (2008).
Mishina, N. M. et al. Visualization of intracellular hydrogen peroxide with HyPer, a genetically encoded fluorescent probe. Methods Enzymol. 526, 45–59 (2013).
Acknowledgements
We thank Francesca Meda for preliminary experiments in H2O2 level manipulation in cell culture, and Filippo Del Bene for fruitful comments on the manuscript. We also thank anomymous reviewers for insightful suggestions. This work was funded by CNRS, INSERM, Collège de France, and Université de Paris and grant #075-15-2019-1789 from the Ministry of Science and Higher Education of the Russian Federation.
Author information
Authors and Affiliations
Contributions
S.V. and A.J. conceived the project and designed the experiments. I.Q., M.T., I.A., A.J. and M.V. prepared the D.N.A. constructs used in this study. I.A., M.T. and C.R. performed the experiments. V.B. and V.V.P. provided the HyPer7 sensor and useful advice. I.A., M.T., C.R., M.V., A.J., and S.V. analyzed the experimental data. A.P., M.V., A.J. and S.V. wrote the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Amblard, I., Thauvin, M., Rampon, C. et al. H2O2 and Engrailed 2 paracrine activity synergize to shape the zebrafish optic tectum. Commun Biol 3, 536 (2020). https://doi.org/10.1038/s42003-020-01268-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-020-01268-7
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
