
Abstract
Keratoconus is a common ocular disorder that causes progressive corneal thinning and is the leading indication for corneal transplantation. Central corneal thickness (CCT) is a highly heritable characteristic that is associated with keratoconus. In this two-stage genome-wide association study (GWAS) of CCT, we identified a locus for CCT, namely STON2 rs2371597 (P = 2.32 × 10−13), and confirmed a significant association between STON2 rs2371597 and keratoconus development (P = 0.041). Additionally, strong STON2 expression was observed in mouse corneal epithelial basal cells. We also identified SMAD3 rs12913547 as a susceptibility locus for keratoconus development using predictive analysis with IBM’s Watson question answering computer system (P = 0.001). Further GWAS analyses combined with Watson could effectively reveal detailed pathways underlying keratoconus development.
Similar content being viewed by others


Introduction
Keratoconus is a common, bilateral, noninflammatory type of corneal degeneration, affecting one out of every 375 people in the general population1, and is a major indication for corneal transplantation in developed countries2,3,4. Keratoconus is a progressive eye disease characterised by an asymmetrical conical protrusion due to focal thinning of the cornea, which causes variable and severe visual disturbances. Thus, patients with severe keratoconus usually present with thin corneas, although not all patients with thin corneas develop the disease.
Although previous genome-wide association studies (GWASs) were performed with keratoconus patients, no genetic region with a significant genome-wide association has been identified thus far5,6,7. Most of keratoconus-susceptibility genes were identified through a large meta-GWAS of central corneal thickness (CCT), because CCT is closely associated with keratoconus and is highly heritable trait8,9,10,11. For example, Lu et al. conducted a GWAS on CCT (n = 20,020), screened for associations between 26 CCT-associated loci and keratoconus (ncase = 874, ncontrol = 6085), and identified six loci associated with keratoconus susceptible12. When the sample size of patients with disease is limited, this intermediate phenotype strategy is reasonable because it is less affected by multiple testing correction, when compared to a simple GWAS. However, where possible, it is preferable to reasonably narrow down the number of candidate genes that are screened, in order to avoid ‘the curse of multiplicity’. Indeed, although a recent large meta-GWAS of CCT (n = 25,910) identified multiple additional CCT-associated loci, no novel keratoconus-susceptible genes were identified through sequential screening (ncase = 933, ncontrol = 5946)13. In addition, pathway analysis of keratoconus has not been performed because of the lack of reliable GWASs of keratoconus. To reveal the specific role of various genes in keratoconus development, it may be important to identify susceptibility genes with relatively smaller effect size, which cannot be identified through large sample size GWAS studies14.
In this study, we used the latest artificial intelligence technology, IBM’s Watson for Drug Discovery (WDD), to select candidate genes from the results of our newly performed GWAS on CCT. WDD, which has been configured to support life science research, can help in understanding and identifying relationships between molecules/genes, using various types of enormous datasets from structured databases and PubMed database15,16. To identify new keratoconus-susceptibility loci, we performed a traditional two-stage GWAS on CCT using a Japanese community-based cohort, and assessed the association of CCT-susceptibility loci with keratoconus occurrence. Further, we assessed new keratoconus-susceptibility genes derived from the combination of GWAS results and WDD.
Results
Two-stage GWAS of CCT in Japanese patients
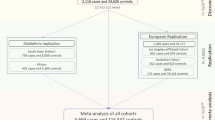
To investigate and identify genetic loci associated with CCT, the average CCT in both eyes was used as the dependent variable for genome-wide quantitative trait locus (QTL) analyses. We included the age, sex, average axial length of both eyes, and the first principal component as covariates. An inflation factor (λGC) of 1.065 indicated good control of the study population substructure (Supplementary Fig. 1). We also found that inclusion of additional PCs minimally altered the findings.
During the first stage with 3584 participants, we identified two single-nucleotide polymorphisms (SNPs), namely STON2 rs2371597 (β = 5.35, P = 1.91 × 10−11) and FNDC3B rs4894538 (β = −4.13, P = 1.90 × 10−8), which exceeded genome-wide significance (Fig. 1 and Table 1). Since FNDC3B rs4894538 was in strong linkage disequilibrium with rs4894535 (R2 = 0.991 in the discovery set), which is an established SNP associated with CCT and keratoconus, we only carried STON2 rs2371597 forward to the replication stage. In the replication stage, we analysed 2942 participants and found that STON2 rs2371597 showed a significant association with CCT (β = 2.95, P = 3.58 × 10−4). Meta-analysis of the discovery and replication sets further confirmed a robust association of STON2 rs2371597 with CCT (βmeta-Japanese = 4.20, Pmeta-Japanese = 2.32 × 10−13).

The plot shows −log10-transformed P-values for all SNPs adjusted for age, sex, average axial length in both eyes, and the first principal component. The horizontal line represents the genome-wide significance threshold of 5.0 × 10−8, and the lower dashed line represents the threshold of 1.0 × 10−4 to identify candidate genes for Watson Drug Discovery analysis.
The baseline characteristics of the participants and SNPs that showed P-values of <1.0 × 10−4 are shown in the Supplementary Table 1 and Supplementary Data 1.
Replication with patients of other ethnicities
To confirm the association between STON2 rs2371597 and CCT in individuals with other ethnicities, we conducted trans-ethnic replication analyses using three different cohorts comprised of Malay, Chinese, and Indian subjects (N = 2510, 2469, and 2508, respectively) (Table 2). In all cohorts, the rs2371597 C allele was associated with increased CCTs (β = 2.14, P = 0.0406, Malay cohort; β = 2.44, P = 0.021, Chinese cohort; β = 1.56, P = 0.130, Indian cohort), which was in agreement with the data from the Japanese cohort. Thus, meta-analysis of the data from all ethnic groups revealed a strong association between rs2371597 and CCT (Pmeta-all = 2.18 × 10−4). Additionally, publicly available GWAS data enabled us to discover an association of the STON2 rs2371597 C allele with increased CCTs in a Latino population (P = 6.12 × 10−3, Table 2).
Association of previously reported CCT-associated loci with CCT
We also evaluated the association of 93 previously reported CCT-susceptible SNPs from 57 genetic regions in our discovery GWAS, using 3584 samples10,11. Of these SNPs, 53 were included in our discovery dataset, and 30 (56.6%) from 18 genetic regions showed an association with CCT with the same effect directions reported previously (P < 0.05, Supplementary Data 2). After Bonferroni’s correction, 14 of the 53 SNPs (26.4%) showed a significant association with CCT. Additionally, we assessed the associations of SNPs with the lowest P-value in the 48 genetic regions. We found 39 of the top 48 SNPs (81.3%) had a P-value of <0.05, and 16 SNPs (33.3%) showed significant associations with CCT after Bonferroni’s correction (Supplementary Data 2).
Association of STON2 with keratoconus
To evaluate the possible association between STON2 rs2371597 and keratoconus, we conducted a case-controlled study using 179 keratoconus patients, regardless of age, sex, or physical impairment, from the Yokohama City University and 11,084 Japanese healthy controls. This analysis revealed that minor-allele frequency (MAF) at rs2371597 C was significantly high in keratoconus patients than that in controls (MAFkeratocnus = 0.293, MAFcontrols = 0.246, odds ratio [OR] (95% confidence interval [CI]) = 1.27 (1.01–1.60), P = 0.041).
Expression of STON2 in human tissues and in the mouse cornea
A search of a publicly available expression quantitative trait loci analysis (eQTL) database revealed that rs2371597 was significantly associated with STON2 expression (GTEx Portal; https://gtexportal.org/home/). A multi-tissue eQTL plot revealed that the normalised effect size (NES) of rs2371597 on STON2 expression was strongest in the skeletal muscle (NES = −0.189, P = 1.3 × 10−8; Fig. 2, https://www.gtexportal.org/home/snp/rs2371597) in which collagen plays an important role in providing its tensile strength and elasticity. However, data on the association of rs2371597 with gene expression in human corneal tissue were not available.

Multi-tissue eQTL plot generated by performing an eQTL search of a publicly available database (GTEx Portal; https://gtexportal.org/home/). The effect size of rs2371597 on STON2 expression was strongest in the skeletal muscle (NES = −0.189, P = 1.3 × 10−8), followed by the adrenal gland (NES = −0.175, P = 0.03).
Our immunohistochemical study of mouse corneas showed that STON2 was expressed in the corneal epithelial cell layer (Fig. 3). Our results demonstrated that strong STON2 expression mainly occurred in basal cells rather than superficial cells in the corneal epithelium. In the stroma and endothelium layer, only minimal STON2 expression was observed.

Mouse corneal sections were immunostained with an antibody against STON2 (green). The nuclei are counterstained with 40,6-diamidino-2-phenylindole (blue) in the merged image. The upper portion of the panel shows the corneal epithelium.
Predictive analysis using the WDD system
WDD predictive analysis scores and visualisation of the relationships between ‘teaching genes’ and candidate genes (based on searching millions of Medline abstracts in PubMed [accessed on March 1st, 2018], as described in the Methods section). The similarity score (i.e. the degree of similarity to ‘teaching genes’) of each candidate gene is shown in Supplementary Table 2. The tree plot and scatter plot results are shown in Figs. 4 and 5. Three major streams were observed, and STON2 was part of the FOXO1–SMAD3 stream (Fig. 4).

Nine teacher genes (green triangles) and 42 candidate genes (red circles) are shown, based on predictive analysis with WDD. The relationships between the genes are visualised.

A scatter plot of genes was generated based on their distances in the literature. Nine teacher genes (green triangles) and 42 candidate genes (red circles) are shown.
We focused on candidate genes within the FOXO1–SMAD–STON2 stream. Specifically, we selected seven keratoconus-susceptibility genes (CDH13, SMAD3, ADAM12, CSMD1, NRXN1, CPLX2, and WWOX) for further analysis. Among these candidate genes, SMAD3 showed the highest similarity score when compared to the ‘teaching genes’ (Supplementary Table 3). Since the SMAD3 rs11333560 genotype was not available in the Tohoku Medical Megabank Project, we instead employed a proxy SNP (rs12913547 in SMAD3). The SMAD3 rs12913547 T allele was significantly associated with an increased risk for keratoconus development after Bonferroni’s correction (OR (95% CI) = 1.44 (1.16–1.80), P = 0.001, Table 3). CPLX2 rs4242187 tended to show an association with keratoconus, but the association was not statistically significant (OR (95% CI) = 0.70 (0.46–1.05), P = 0.082).
Discussion
In this study, we identified two keratoconus-susceptibility genes by integrating conventional GWAS of CCT and artificial intelligence, using the WDD platform. We found that the STON2 rs2371597 allele was significantly associated with CCT and keratoconus, as well as significantly altered STON2 expression in human tissues. WDD analysis assisted in identifying another keratoconus-susceptibility SNP, i.e. SMAD3 rs12913547. Considering that recent association analyses of keratoconus failed to identify novel keratoconus-susceptibility genes despite their large sample sizes5,6,13, the current approach of combining GWAS and artificial intelligence data appears to have been quite efficient.
We first conducted a conventional GWAS of CCT with trans-ethnic replication. This analysis enabled us (1) to confirm STON2 as a susceptibility gene for CCT and (2) to show that our Japanese population shared most of underlying genetic effects not only with other Asian populations, but also with European and Latino populations12,17. ZNF469 rs9938149, the most established CCT-susceptibility SNP, was replicated in our GWAS (n = 3584, β = −3.35, P = 7.45 × 10−3). The relatively high P-value was due to its small MAF in Japanese subjects, compared to European ethnic groups (MAFJapanese = 0.034, MAFEuropean = 0.349; data from the 1000 Genomes database).
We also identified STON2 rs2371597 (located at chromosome14q31) as a keratoconus-susceptibility SNP. STON2 was previously identified as an endocytic adaptor dedicated to the retrieval of surface-stranded synaptic vesicle proteins18. A previous genetic study reported that STON2 SNPs were associated with CCT, but not with keratoconus development in Caucasians. Therefore, as the prevalence of keratoconus is known to be higher in the Asian population compared to Caucasians, we speculate that the genetic and pathological background of keratoconus differs between ethnicities. Moreover, as it is possible that certain keratoconus-susceptibility genes identified in Asians are not associated with keratoconus development in Caucasians, further studies are required to compare the effect size of keratoconus-susceptibility genes on keratoconus development across various ethnicities, which will serve to reveal ethnical differences.
The eQTL data revealed that the effect size of rs2371597 on STON2 expression was strongest in skeletal muscle, which is rich in collagen, a protein that reportedly plays a key role in keratoconus pathogenesis. Although no previous reports have investigated the expression of STON2 in human corneal tissue, we speculate that STON2 may play an important role in keratoconus development by interacting with extracellular matrix remodelling. Our immunohistochemical staining results for STON2 are compatible with a previous histopathological study of keratoconus, which showed that thinning of the corneal stroma, breaks in Bowman’s layer, and degeneration of the corneal epithelium were the characteristics of corneas in patients with keratoconus2,19. STON2 might be associated with the vulnerability to physical damage or immunological changes. Although biological studies examining the role of STON2 in human corneal tissue are required to prove our hypothesis, pathways involving STON2 may serve as targets for treating keratoconus by controlling basal cell degeneration.
Through WDD, we identified SMAD3 as an additional keratoconus-susceptibility gene (OR = 1.44 (1.16–1.80), P = 0.001), and CPLX2 as a future candidate gene for keratoconus-susceptibility (OR = 0.70 (0.46–1.05), P = 0.082). SMAD3 is known to play a significant role in the transforming growth factor-β (TGF-β) signalling pathway, which may modulate extracellular matrix (ECM) alterations in keratoconus20. Data from a previous study showed TGF-β/SMAD3 signalling promoted gluconeogenesis through an interaction with FOXO1, which is an established keratoconus-susceptibility gene21. Further studies of the roles of SMAD3, FOXO1, and TGF-β signalling in keratoconus development are needed. In addition, CPLX2 is another interesting target because (1) both CPLX2 and STON2 are reported to be associated with schizophrenia18,22, and (2) comorbidity between schizophrenia and keratoconus was also demonstrated previously23,24,25,26. We speculate that schizophrenia and keratoconus employ shared underlying pathophysiological processes, which would be another pathway apart from gluconeogenesis. We expect that the relationships between these two diseases will be studied more in the future.
We expected that CCT-reducing alleles would be associated with an increased risk of developing keratoconus, given the known association between thin corneas and the disease. Indeed, most CCT-reducing alleles have been associated with an increased risk of keratoconus, and this was true for SMAD3 rs12913547 in the current study. However, with STON2, a CCT-increasing allele led to increased keratoconus risk. Although this finding was unexpected, such complex association patterns have often been observed in previous studies. One example is that a body fat-decreasing allele near IRS1 was also associated with adverse lipid profiles, and increased risks for coronary artery disease and type-2 diabetes27,28,29. Another example is that the most famous CCT-reducing SNP, rs9938149, located between BANP and ZNF469, is known to decrease keratoconus risk12. We anticipate that further research on ZNF469 and STON2 will reveal pleiotropic effects for these genes in terms of corneal thickness and the risk for keratoconus development.
We applied a approach, i.e. WDD, to screen for keratoconus-susceptibility SNPs from thousands of potential CCT-susceptibility SNPs. While we focused on seven genes out of 52 candidate genes (based on the predictive analysis results of WDD), SNPs in only two genes showed a significant or marginal association with keratoconus. Since the similarity score was calculated based on Medline abstract searches in PubMed, genes that have been more extensively studied tend to have higher scores. We focused on genes located between ‘teacher genes’ and STON2 in this study to identify genes that were not sufficiently evaluated in previous studies. Although CPLX2 (ranked number 16 by Watson) showed a marginally significant association with keratoconus, only SMAD3 (with the highest similarity score) showed a robust association with keratoconus. However, other genes with a relatively high similarity score, such as CDH13 and MEIS1, were not associated with keratoconus. Interestingly, CPLX2 and SMAD3 were located next to teaching genes, whereas CDH13 and MEIS1 were not. We speculate that the rank of the similarity score may be an important index; however, the approach used to rank scores needs to be improved. To identify disease-susceptible genes with Watson, we propose that focusing on genes located between and/or next to teaching genes may be important. Further studies will be required to establish a method for combining GWAS with Watson.
In summary, we identified two keratoconus-susceptibility loci, STON2 and SMAD3, by integrating conventional GWAS and artificial intelligence, using WDD. Cognitive-computing technology combined with GWAS can assist in identifying hidden relationships among disease-susceptibility genes and potential susceptibility genes, enabling more efficient interpretation of GWAS results. We believe that the current approach can be generalised for application to numerous other diseases. Since samples from patients with a disease are more difficult to obtain than samples from healthy individuals, which can be collected through cohort studies, the current approach will prove particularly helpful in facilitating the exploration of disease-susceptible genes.
Methods
Participants in the two-staged GWAS of CCT
For the two-staged GWAS, we analysed data from healthy Japanese volunteers enrolled in the Nagahama Prospective Cohort for Comprehensive Human Bioscience (the Nagahama Study). The initial follow-up data were collected from 9850 participants between July 2013 and February 2017; meanwhile data collected from 8289 participants (34–80 years of age) between July 2013 and February 2016 were used for the CCT measurements.
All participants underwent ophthalmic examinations, including objective determinations of the corneal thicknesses (TX-20P; Canon, Tokyo, Japan) and axial lengths (IOLMaster; Carl Zeiss Meditec, Inc., Dublin, CA, USA). The study participants comprised individuals with available DNA samples; CCTs for both eyes; and information regarding the patient’s age, sex, and axial length of both eyes. Of the participants, 84 who lacked CCT data for both eyes, 617 with a history of intraocular surgery, and 480 who lacked axial length data for both eyes were excluded. Of the remaining participants, samples from those who underwent genome-wide SNP genotyping were used for the discovery GWAS, and remaining subset of samples was used for the replication stage.
The Kyoto University Graduate School and Faculty of Medicine Ethics Committee and the Nagahama Municipal Review Board of Personal Information Protection approved the study protocol and the procedures used to obtain informed consent (Kyoto University Graduate School and Faculty of Medicine Ethics Committee approval number; G0278). All study procedures adhered to the tenets of the Declaration of Helsinki. All participants were fully informed of the purpose and procedures of the study, and written consent was obtained from each participant. Patient records and information were anonymised prior to analysis.
Genome-wide SNP genotyping
Genomic DNA was extracted from peripheral blood samples according to standard laboratory procedures. Genome-wide SNP genotyping was performed on samples from 5299 participants. A series of BeadChip DNA arrays, namely the HumanHap610 Quad (1818 samples), HumanOmni2.5-4 (1606 samples), HumanOmni2.5-8 (375 samples), HumanOmni2.5 s (671 samples), CoreExome24 (1,722 samples), and HumanExome (671 samples; Illumina, San Diego, CA, USA) arrays, were used for the analysis. Some samples were repeatedly genotyped using multiple kinds of DNA arrays.
Quality-control filters were used to remove poorly performing samples and SNP markers. SNPs with a call rate of <99%, an MAF of <1%, and a significant deviation from the Hardy–Weinberg equilibrium (P < 1.0 × 10−6) were excluded from further statistical analysis. Samples with a call rate of <95% were excluded from the analysis. Among the remaining participants, 636 were estimated to have a first- or second-degree kinship within this population (pi-hat > 0.35, PLINK ver. 1.90 [https://www.cog-genomics.org/plink/1.9/]) and were excluded from the analysis.
Genotype imputation was performed using MACH software (http://www.sph.umich.edu/csg/abecasis/MACH/tour/imputation.html), with a 1000 Genomes dataset (phase 3, v5 release) serving as a reference panel. Imputed SNPs for which R2 < 0.5 were excluded from the association analysis. Finally, 4,710,779 SNPs from 3584 individuals were used for the discovery stage analysis.
Replication genotyping for the GWAS of CCT
To conduct a replication study for confirming genetic associations with CCT, genotypes of the remaining samples from the Nagahama Study (n = 3422) were determined using commercially available TaqMan SNP assays and the ABI PRISM 7700 system (Applied Biosystems, Foster City, CA, USA). Fourteen patients without CCT data in both eyes, six patients lacking axial length data for both eyes, and 460 patients with history of intraocular surgery were excluded. Finally, 2942 samples were used for the replication stage analysis.
Trans-ethnic replication
All patients enrolled in the study provided informed consent, and ethical approval was granted by Singapore Malay Eye Study (SiMES) group, the SingHealth Centralized Institutional Review Board (SCES), and the Singapore Indian Eye Study (SINDI) group. For the trans-ethnic replication stage, we included three Asian cohorts, namely the SiMES, SCES, and SINDI cohorts, which consisted of Malay (n = 2510), Chinese (n = 2469), and Indian (n = 2508) subjects, respectively. We also included publicly available data on a Latino population (n = 3584). Detailed information and the sample-collection methods for these cohorts can be found in the Supplementary Table 4 and Supplementary Note and in a previous publication17.
Statistics and reproducibility
Genome-wide linear-regression analysis was conducted for the average CCT in both eyes in the discovery set of the Nagahama Cohort (N = 3584). This regression framework enabled us to adjust for covariates, such as age, sex, the average axial length in both eyes, and the first principal component. In the replication analysis with data from the SiMES (N = 2510), SCES (N = 2469), and SINDI (N = 2508) cohorts, we performed association testing under an additive model to examine the effect of the risk allele, while adjusting for the age, sex, and at least the first five principle components. Experiment-wide significance in the discovery stage was set at P < 5.0 × 10−8. All meta-analyses were performed using an inverse variance weighted fixed effect model. Thereafter, differences were considered statistically significant at P < 0.05. Deviations in genotype distributions from the Hardy–Weinberg equilibrium were assessed by performing Chi-squared tests. These statistical analyses were performed using R software (R Foundation for Statistical Computing, Vienna, Austria; available at http://www. rproject.org/, version 3.5.2) and PLINK software, version 1.90 (https://www.cog-genomics.org/plink/1.9/).
Association of identified keratoconus-susceptibility gene with keratoconus
To investigate the association between identified gene and keratoconus, we performed a case-control analysis using data from 179 patients with keratoconus from Yokohama City University and pooled the data of 9589 Japanese healthy control subjects from Yokohama City University, Nagahama Cohort and Tohoku Medical Megabank Project30,31 A detailed description of these cohorts and the genotyping methods used are provided in the Supplementary Note. Briefly, both cases and controls were recruited from the Yokohama City University, and genotyped using the same genotyping platform, OmniExpress, and imputed using the same pipelines. Control samples from the Nagahama Cohort were genotyped as described above. Control samples from the Tohoku Medical Megabank Project were derived from a publicly available database. Differences in the genotype distributions of these cohorts were tested using the Cochran–Armitage test. P < 0.05 was set as the threshold of statistical significance.
Immunohistochemical staining
To confirm the expression of identified gene, we conducted immunohistochemical staining with the corneas of normal adult C57BL/6 mice. After each mouse was sacrificed, the eyes were enucleated and fixed with 4% paraformaldehyde in 0.1 M PB for 20 min. Expression in the cornea was examined in vertical sections. For this purpose, the eyes were cryoprotected in a sucrose gradient (10 and 30% w/v sucrose in 0.1 M PB), and 13-μm cryostat sections were cut. The sections were blocked for 1 h in a solution containing 10% normal goat serum (Millipore), 0.3% Triton X-100 (Bio-Rad, Hercules, CA, USA), and 0.1 M PB. The sections were incubated overnight at 4 °C with a primary rabbit anti-human STON2 antibody (catalogue number NBP1-90658; Novus Biologicals, LLC), diluted 1:10 in blocking solution. Subsequently, the sections were incubated for 1 h with a secondary Alexa Fluor 488-conjugated goat anti-rabit IgG antibody (catalogue number A-11005; Invitrogen, Eugene, OR, USA). All incubation steps were performed at 4 °C. All images were obtained using a Keyence BZ-9000 microscope.
Predictive pathway analysis using the WDD system
WDD is one of the most famous cognitive-computing systems. The details of the WDD predictive analysis algorithm are described elsewhere15; thus, the system is introduced briefly below. WDD is configured to support life sciences research and extracts text features from medical literature to identify new connections between entities of interest, such as genes and diseases. From these documents, Watson can create semantic connections between a known set of genes associated with keratoconus and candidate genes. WDD can also rank the candidate genes by their degrees of similarity to the known set, using a graph-diffusion algorithm.
To detect additional keratoconus-susceptibility loci and infer pathways associated with keratoconus, we performed WDD predictive analysis. Eight previously reported keratoconus-susceptibility genes (FNDC3B, COL5A1, FOXO1, MPDZ, NF1B, RXRA, BANP, and ZNF469) and the newly identified keratoconus-susceptibility gene was input as a ‘teaching gene’12,32,33.
The candidate genes were selected as follows. First, from 1215 SNPs with a P-value of <1.0 × 10−4 in the discovery GWAS (Supplementary Data 1), we selected 745 SNPs that were located within a gene. These SNPs were distributed within 53 genomic regions. Second, genes that had already been input as ‘teaching genes’ and genes that had never been mentioned previously in the literature were automatically excluded, resulting in the exclusion of three and eight genes as candidates, respectively. Finally, 42 genes were selected as candidate genes. This process is summarised in Supplementary Table 2.
Based on the predictive analysis results of WDD, we selected possible keratoconus-susceptibility genes with respect to identified gene and assessed the associations between those genes and keratoconus. For association studies with keratoconus, we principally selected the top SNP within the genes. If genotype data of the SNP of interest was not available in any of the control cohorts, we employed proxy SNPs instead. An association study was performed using data from 179 patients with keratoconus and pooled data from 6651 healthy control subjects. The Cochran–Armitage test and Bonferroni’s correction were applied for statistical assessments. The nominal P-value threshold was set to 0.05.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The complete GWAS summary data can be visualised at figshare (https://figshare.com/articles/dataset/CCT/12592529). The datasets generated during the current study are also available from the corresponding author on reasonable request.
Code availability
IBM’s Watson for Drug Discovery was used to select candidate genes in the present study (accessed on March 1st, 2018).
References
Godefrooij, D. A. et al. Age-specific incidence and prevalence of keratoconus: a nationwide registration study. Am. J. Ophthalmol. 175, 169–172 (2017).
Rabinowitz, Y. S. Keratoconus. Surv. Ophthalmol. 42, 297–319 (1998).
Romero-Jiménez, M., Santodomingo-Rubido, J. & Wolffsohn, J. S. Keratoconus: a review. Contact Lens Anterior Eye. 33, 157–166 (2010).
Godefrooij, D. A., Gans, R., Imhof, S. M. & Wisse, R. P. L. Nationwide reduction in the number of corneal transplantations for keratoconus following the implementation of cross-linking. Acta Ophthalmol. 94, 675–678 (2016).
Li, X. et al. Genetic association of COL5A1 variants in keratoconus patients suggests a complex connection between corneal thinning and keratoconus. Invest. Ophthalmol. Vis. Sci. 54, 2696–2704 (2013).
Li, X. et al. A genome-wide association study identifies a potential novel gene locus for keratoconus, one of the commonest causes for corneal transplantation in developed countries. Hum. Mol. Genet. 21, 421–429 (2012).
Hardcastle, A. J. et al. Genome-wide association study on a large multi-ethnic sample identifies new genetic loci that predispose to keratoconus. Invest. Ophth. Vis. Sci. 60, 4249 (2019). https://iovs.arvojournals.org/article.aspx?articleid=2743874.
Dimasi, D. P., Burdon, K. P. & Craig, J. E. The genetics of central corneal thickness. Br. J. Ophthalmol. 94, 971–976 (2010).
Landers, J. A. et al. Heritability of central corneal thickness in nuclear families. Invest. Ophthalmol. Vis. Sci. 50, 4087–4090 (2009).
Zheng, Y. et al. Heritability of central corneal thickness in Chinese: the Guangzhou twin eye study. Invest. Ophthalmol. Vis. Sci. 49, 4303–4307 (2008).
Toh, T. et al. Central corneal thickness is highly heritable: the twin eye studies. Invest. Ophthalmol. Vis. Sci. 46, 3718–3722 (2005).
Lu, Y. et al. Genome-wide association analyses identify multiple loci associated with central corneal thickness and keratoconus. Nat. Genet. 45, 155–163 (2013).
Iglesias, A. I. et al. Cross-ancestry genome-wide association analysis of corneal thickness strengthens link between complex and Mendelian eye diseases. Nat. Commun. 9, 1864 (2018).
Valgaeren, H., Koppen, C. & Van Camp, G. A new perspective on the genetics of keratoconus: why have we not been more successful? Ophthalmic Genet. 3, 158–174 (2018).
Chen, Y., Elenee Argentinis, J. D. & Weber, G. IBM Watson: how cognitive computing can be applied to big data challenges in life sciences research. Clin. Ther. 38, 688–701 (2016).
High, R. The era of cognitive systems: an inside look at IBM Watson and how it works. Int. Bus. Mach. Corp. 1, 1–14 (2012).
Gao, X. et al. Genome-wide association study identifies WNT7B as a novel locus for central corneal thickness in Latinos. Hum. Mol. Genet. 25, 5035–5045 (2016).
Luan, Z. et al. Positive association of the human STON2 gene with schizophrenia. Neuroreport 22, 288–293 (2011).
Wisse, R. P. L. et al. Cytokine expression in keratoconus and its corneal microenvironment: a systematic review. Ocul. Surf. 13, 272–283 (2015).
Nakao, A. et al. Identification of Smad7, a TGFβ-inducible antagonist of TGF-β signalling. Nature 389, 631–635 (1997).
Yadav, H., Devalaraja, S., Chung, S. T. & Rane, S. G. TGF-β1/Smad3 pathway targets PP2A-AMPK-FoxO1 signaling to regulate hepatic gluconeogenesis. J. Biol. Chem. 292, 3420–3432 (2017).
Lee, H. J. et al. Association study of polymorphisms in synaptic vesicle-associated genes, SYN2 and CPLX2, with schizophrenia. Behav. Brain Funct. 1, 15 (2015).
Rudisch, B., D’Orio, B. & Compton, M. T. Keratoconus and psychosis. Am. J. Psychiatry 160, 1011 (2003).
Gorskova, E. N., Sevost’ianov, E. N. & Baturin, N. A. [Results of psychological testing of patients with keratoconus]. Eur. Pmc. 114, 44–45 (1998).
Mannis, M. J. et al. Personality trends in keratoconus: an analysis. Arch. Ophthalmol. 105, 798–800 (1987).
Schürhoff, F., Leboyer, M. & Szöke, A. Comorbidity between schizophrenia and keratoconus. Psychiatry Res. 247, 315–316 (2017).
Kilpeläinen, T. O. et al. Genetic variation near IRS1 associates with reduced adiposity and an impaired metabolic profile. Nat. Genet. 43, 753–760 (1987).
Samani, N. J. et al. Genomewide association analysis of coronary artery disease. N. Engl. J. Med. 357, 443–453 (2007).
Rung, J. Genetic variant near IRS1 is associated with type 2 diabetes, insulin resistance and hyperinsulinemia. Nat. Genet. 41, 1110–1115 (2009).
Mikami, T. et al. IL1B promoter polymorphisms are associated with keratoconus in a Japanese population. Mol. Vis. 19, 845–851 (2013).
Kuriyama, S. et al. The Tohoku Medical Megabank Project: design and mission. J. Epidemiol. 26, 493–511 (2016).
Edwards, M., McGhee, C. N. & Dean, S. The genetics of keratoconus. Clin. Exp. Ophthalmol. 29, 345–351 (2001).
Gordon-Shaag, A., Millodot, M., Shneor, E. & Liu, Y. The genetic and environmental factors for keratoconus. Biomed. Res. Int. 2015, 795738 (2015).
Acknowledgements
This study was supported by a University Grant from Kyoto University; a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science & Technology in Japan; the Center of Innovation Program; the Global University Project from Japan Science and Technology Agency; the Practical Research Project for Rare/Intractable Diseases; Comprehensive Research on Aging and Health Science Research Grants for Dementia R&D from the Japan Agency for Medical Research and Development (AMED); a research grant from the Takeda Science Foundation; and SNEC HREF funds. We thank Ms. Hatsue Hamanaka and Ms. Miki Kokubo for assistance with genotyping. We are extremely grateful to the Nagahama City Office and the nonprofit organisation, Zeroji Club for their help in conducting the Nagahama Study. We also acknowledge the following sources of funding support for analysing the SiMES, SCES, and SINDI cohorts: the National Medical Research Council, Singapore (grant numbers NMRC/TCR/002-SERI/2008 (R626/47/2008TCR), CSA R613/34/2008, NMRC 0796/2003, and STaR/0003/2008), the National Research Foundation of Singapore, the Biomedical Research Council, Singapore (grant numbers BMRC 09/1/35/19/616, 08/1/35/19/550, and 10/1/35/19/675), and the Genome Institute of Singapore (grant number GIS/12-AR2105). The Singapore Tissue Network and the Genome Institute of Singapore (Agency for Science, Technology and Research, Singapore) provided services.
Author information
Authors and Affiliations
Consortia
Contributions
Y.H. and M.M. designed the study. M.M., S.M.S., and K.Y. supervised the design of the study. Y.H., M.M., A.M., S.I., M.Y., C.C.K., S.M.S., and K.Y. analysed the data. M.M., R.Y., and C.Y.C. supervised the analysis. Y.H., M.M., Y.T., R.Y., F.M., and the Nagahama Study Group managed the cohort data. Y.H., M.M., A.M., S.I., N.U.A., E.N., Y.M., H.N., C.C.K., S.S.M., R.Y., C.C., N.M., A.T., and K.Y. gathered and analysed the clinical data. Y.H. and M.M. drafted the paper. All authors critically revised and gave final approval to this manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hosoda, Y., Miyake, M., Meguro, A. et al. Keratoconus-susceptibility gene identification by corneal thickness genome-wide association study and artificial intelligence IBM Watson. Commun Biol 3, 410 (2020). https://doi.org/10.1038/s42003-020-01137-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-020-01137-3
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
