
Abstract
Deaminase base editing has emerged as a tool to install or correct point mutations in the genomes of living cells in a wide range of organisms. However, the genome-wide off-target effects introduced by base editors in the mammalian genome have been examined in only one study. Here, we have investigated the fidelity of cytosine base editor 4 (BE4) and adenine base editors (ABE) in mouse embryos using unbiased whole-genome sequencing of a family-based trio cohort. The same sgRNA was used for BE4 and ABE. We demonstrate that BE4-edited mice carry an excess of single-nucleotide variants and deletions compared to ABE-edited mice and controls. Therefore, an optimization of cytosine base editors is required to improve its fidelity. While the remarkable fidelity of ABE has implications for a wide range of applications, the occurrence of rare aberrant C-to-T conversions at specific target sites needs to be addressed.
Similar content being viewed by others


Introduction
Deaminase base editing1,2 directly converts target C·G base pairs to T·A by cytosine base editors (CBE), or target A·T base pairs to G·C by adenine base editors (ABE), without inducing double-stranded DNA breaks3. Since the majority of known human pathogenic variants are single-nucleotide alterations2,4, base editing has been heralded as a high-fidelity tool to correct single-nucleotide polymorphisms (SNPs) associated with many human disorders. While exceptional precision is paramount in a quest to correct somatic and in particular germline mutations, recent studies have revealed that CBEs can induce bystander mutations, including deletions, in mouse zygotes5 and plants6. In contrast, ABE displays a greater fidelity5,7, even though unexpected C-to-T conversions have been observed with ABE at some target sites5,8.
Whole-genome sequencing (WGS) of base-edited rice9 and mouse embryos10 revealed that BE3, a commonly used CBE, induces a large number of inadvertent base changes throughout the genome, while ABE displays high fidelity. In a separate study, WGS of BE3-edited sheep did not reveal an obvious increase of off-target mutations11. Since BE3 can introduce unwanted indels1,5 and other undesirable base substitutions in addition to C-to-T conversions5,7,12, the fourth-generation BE4, containing a second uracil glycosylase inhibitor (UGI) domain and optimized linker architectures, appears to have an increased fidelity in vitro13, in mouse zygotes5 and in rabbit embryos14. Off-target effects for BE4 might be expected based on WGS studies that examined off-target mutations introduced by BE3 and ABE9,10. However, as different sgRNAs, editors and computational analytic methods might yield different results9,10, there is a definitive need for additional studies investigating in vivo genomic effects of BE4 in comparison with ABE at a greater depth. As a note of caution, the original WGS study of CRISPR/Cas9-edited mice suggested the presence of extensive off-target mutations15, which was, however, likely the result of an imperfect experimental design as pointed out in editorials16,17,18,19,20. Further WGS investigations by Iyer and colleagues as well as our group21,22 using trio studies demonstrated that CRISPR/Cas9 does not introduce an excess of off-target mutations. Therefore, it is scientifically prudent and warranted to examine critical issues of base editing, such as the extent of off-target mutations, with a larger number of mice and under additional conditions. Having several independent studies should provide confidence to those investigators who actively explore therapeutic use in many laboratories and companies. In this study, we have addressed the question of base editing fidelity and conducted unbiased WGS on a total of 44 BE4- and ABE-edited mice, control mice and their wild-type parents, providing more evidence to support previous data and conclusions9,10.
Results
Targeting mouse embryos with BE4 and ABE
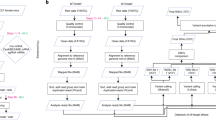
To assess on- and off-target fidelities of the advanced BE4 and ABE in mouse embryos, we conducted a family based trio WGS study (Fig. 1a). Fertilized eggs were injected with BE4 or ABE7.10 mRNA together with a single sgRNA used by both editors, which permitted a direct comparison. Two-cell stage embryos were implanted into surrogate mothers and 13 ABE-edited and nine CBE-edited founder mice were born, together with 13 non-injected controls (Table 1). Tail tissues from 3- to 4-week-old founder mice (Fig. 1b and Supplementary Fig. 1) were screened by Sanger sequencing to identify mutants and targeted deep sequencing was performed to determine haplotypes. ABE introduced A-to-G transitions in the target window and, except one, no bystander or proximal off-target mutations were detected in the 33 edited alleles. In contrast, BE4 induced not only the expected C-to-T transitions but also C-to-G and C-to-A conversions in the target site, frequent proximal off-target mutations, and deletions in four of the nine founders (12 out of the 17 edited alleles) (Fig. 1b and Supplementary Fig. 1). The presence of more than two mutant alleles in some founders is indicative of mosaicism where targeting had also occurred at the two-cell stage, or maybe even later.

a Experimental design of the family based trio study. b Alignment of sequences from founder mice. 33 mutant alleles edited by ABE, and 17 alleles edited by BE4 were analyzed. The sgRNA sequence is underlined and the PAM sites shown in green. The target nucleotides and edited nucleotide are shown in bold black and blue, respectively. Unintended nucleotide substitutions are shown in red. Deletions are marked with a deletion symbol.
Off-target analysis by WGS
Unbiased WGS was performed on the 22 edited mice, 13 controls and nine parents at an average depth of 60X (Table 1 and Supplementary Data 1). As non-injected or Cas9-treated embryos don’t display significant level of SNVs10, we used non-injected mice as controls. The WGS data were analyzed using GATK with Joint Genotyping and subsequent filtering to identify single-nucleotide variants (SNVs) and simple indels for each individual mouse (Fig. 2a). Lumpy with SvTyper was used to identify complex and large indels. To explicitly identify de novo mutations located outside the sgRNA, the SNVs and indels present in the parents were subtracted from those identified in the progeny (Supplementary Data 2 and 3). Non-edited control mice had accumulated an average of 132 de novo SNVs (Fig. 2b and Supplementary Data 2). On average 119 de novo SNVs were detected in ABE-edited mice, comparable to that in controls. In contrast, BE4-edited mice carried on average 221 de novo SNVs, a significant increase (Mann–Whitney-U-test: p = 0.002), especially C-to-T variants (Fig. 2b, Supplementary Fig. 2 Fig and Supplementary Data 2).

a Workflow of whole-genome sequencing analysis for SNVs and indels. b, c Numbers of de novo SNVs (b) and indels (c) identified in mutants (13 ABE-edited mice, n = 13; BE4-edited mice, n = 9) and controls (non-injected control mice, n = 13). Boxplot: center line, mean; box limits, plus and minus standard deviation; whiskers, minimum and maximum. Wilcoxon rank-sum test and Mann–Whitney-U-Test for the pairwise comparison were used to evaluate the statistical significance of differences between groups.
About 2% of off-target SNVs coincided with predicted off-target sites (see M&M for details) suggesting that the majority of mutations were not dependent on the sgRNA and by predicted off-target sites (Supplementary Data 4). The increased off-target editing observed in BE4 but not ABE implies that these mutations were the result of cytosine deaminase AID/APOBEC1 which can induce SNVs in the absence of sgRNAs1. C-to-T conversions (plus some C-to-A and C-to-G) are overrepresented in de novo SNVs observed in BE4-edited mice, consistent with enzymatic activity of BE4. Since four out of the nine BE4-edited mice carried additional deletions proximal to the target region, we analyzed globally their indel frequencies compared with controls (Fig. 2c and Supplementary Data 3). The numbers of indels in the BE4- and ABE-edited group showed no differences from the control group (Fig. 2c), also not regarding their characteristics (Supplementary Data 5).
Discussion
Our results confirm and extend previous work that BE3 but not ABE increases off-target SNVs in mouse embryos10 and rice9. Based on WGS data sets of nine BE4-edited and 13 ABE-edited mice (a total of 50 mutant alleles), we observed a significant mutation rate with the improved BE4, but not ABE, in mouse embryos. While base-edited mouse embryos10 acquire off-targets independent of sgRNAs, in base-edited rice off-target mutations can coincide with predicted off-target sites9 suggesting some sgRNA dependence. Here we used the same sgRNA for both BE4 and ABE to eliminate latent sequence-specificity targeting as explanations for off-target mutations in BE4. At this point we cannot assert that off-target effects are due to sgRNA-independence as the TadA enzyme may be much slower to perform its chemistry than APOBEC, resulting in little or no ABE editing at weak Cas9 off-target binding sites. Although different experimental design and analysis methods might lead to different outcomes9,10, we demonstrated an approximately two-fold increase of de novo off-target mutations in BE4-edited one-cell embryos, which favorably compares to the more than 20-fold increase observed in BE3-edited mouse two-cell embryos10. However, given the variation between individual mice, our study and that in rice9 show a statistically significant difference in off-target SNVs. BE4, an advanced version of BE3, contains an additional UGI (uracil DNA glycosylase inhibitor) and a longer linker, as a means to enhance its specificity13. Further studies will be needed to validate the higher genome-wide fidelity of BE4. Notably, BE4 caused inadvertent proximal off-target mutations and deletions in four of the nine founders, which has implications in its use as therapeutic agent. These adverse mutations are likely independent of the sgRNA used as none of the 13 embryos edited with ABE and the same sgRNA displayed proximal off-target mutations and deletions. In addition, although different sgRNAs could influence rates of bystander mutations, previous studies9,10 show that de novo mutations are induced independent of sgRNAs, and are rather the result of different type of base editors5. Since only a few SNVs (<0.01%) coincided with potential off-target sites that had been identified in silico, they are likely the result sgRNA-independent edits. However, it is not clear whether BE3-induced proximal off-target mutations were detected in the two WGS studies9,10.
In summary, our study emphasizes the high fidelity of ABE as compared to BE4, which also induces increased unwanted base substitutions and deletions in close proximity to the designed target site. Such unwanted mutations are of particular concern when correcting disease-associated SNPs in proteins, as they could adversely alter non-targeted amino acids. Our study also emphasizes the need to monitor off-target mutations in clonal populations, as the analysis of large pools of cells with variable editing, as commonly conducted in in vitro experiments23,24,25, results in population averaging. Based on our study and previous experiments5,9,10, ABE appears to be the current choice for base editing because of its fidelity at target sites and throughout the genome. However, caution about the fidelity of deaminase base editors comes from recent studies that demonstrated extensive off-target RNA editing26,27 as well as illicit C-to-T conversions introduced by ABE at the target window5,8.
Methods
Mice
All animals were housed and handled according to the guidelines of the Animal Care and Use Committee (ACUC) of the NIH (https://oacu.oir.nih.gov) and all animal experiments were approved by the ACUC of National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK, MD) and performed under the NIDDK animal protocol K089-LGP-17. Base-edited founder mice were generated using C57BL/6N mice (Charles River Laboratories, MD) by the Transgenic Core of the National Heart, Lung, and Blood Institute (NHLBI, MD).
CRISPR reagents and microinjection of mouse zygotes
We targeted enhancer 1 within the Wap super-enhancer28 (Wap Gene ID: 22373). The Wap-E1 sgRNA (GGCACAGTATGGGCCCTTCT)28, which contains two cytidines and two adenines near the editing window, was designed and synthesized using ThermoFiser’s sgRNA in vitro transcription service. The pCMV-BE4 plasmid (from David Liu’s laboratory) and pCMV-ABE7.10 plasmid (Addgene plasmid #102919) were linearized and then their mRNAs were synthesized in vitro using the mMESSAGE mMACHINE T7 kit (ThermoFisher Scientific). Mouse zygotes were produced by in vitro fertilization (IVF) using eggs collected from eight superovulated C57BL/6N female mice and sperm collected from one C57BL/6N male (Charles River Laboratories). The ABE and BE4 mRNAs (50 ng/ul) were separately microinjected with the sgRNA (20 ng/ul) into the cytoplasm of the IVF zygotes. After culturing overnight in M16 medium, those embryos reached 2-cell stage of development were implanted into oviducts of pseudopregnant foster mothers (Swiss Webster, NY). Mice born to the foster mothers were genotyped and subsequently analyzed by WGS.
Genotyping
Genomic DNA was isolated from the tip of tails of three to four-week-old founder mice using Wizard Genomic DNA purification Kit (Promega), amplified by PCR, and followed by Sanger sequencing (Quintarabio, CA). Mutations were identified by PCR amplifying a 599 bp fragment encompassing the target sequence, followed by Sanger sequencing. Library preparation and WGS was conducted by the Broad Institute (Cambridge, MA) using Illumina HiSeq X, at a coverage of 60X using 150 bp paired-end reads (Supplementary Data 1).
PCR Primers
Wap-S1_F1: GTTGGAACCCATCACAGACAAAGG
Wap-S1_R1: TGTAGAAACAGAGCAGAGAGGTGG
GATK analysis
WGS (60X) was performed on 44 mice, nine parents (one male and eight females), and their progeny, including 22 founder mice carrying base substitutions at target sites induced by ABE or BE4 using one guide RNA and 13 non-injected control ones. The analysis was performed accordingly to the GATK best practices guidelines29,30,31 for germline mutations (version 3.8-0). Quality control and alignment was done by BBmap32 (version 37.36) and BWA MEM33 (version 0.7.15), respectively, using the reference genome mm10.
For runtime optimization, the aligned BAM files were split up to a chromosome level (for runtime optimization) and reads aligned to different chromosomes were filtered using SAMtools34 (version 1.5), followed by Picard tools35 (version 2.9.2) to mark duplicates. The GATK analysis workflow was applied as follows: base recalibration—GATK BaseRecalibrator, AnalyzeCovariates, and PrintReads—using the databases of known polymorphic sites, dbSNP142 and MGPv5 (provided by the high-performance computing team of the NIH (Biowulf)); variant calling—GATK HaplotypeCaller—with the genotyping mode discovery, the ERC parameter for creating gvcf and a minimum phred-scaled confidence threshold of 30. The final step included merging the VCF files of each chromosome (GenomeAnalysisTK, GATK).
GATK SNV analysis
Joint genotyping was applied on all 44 samples together and hard filters were applied: QD < 2.0 || FS > 60.0 || MQ < 40.0 || MQRankSum < −12.5 || ReadPosRankSum < −8.0 || SOR > 3. The resulting SNVs were additionally filtered by removing those overlapping with repetitive elements36 (UCSC’s masked repeats plus simple repeats; http://hgdownload.soe.ucsc.edu/goldenPath/mm10/database) and black regions (ENCODE37; http://mitra.stanford.edu/kundaje/akundaje/release/blacklists/mm10-mouse/). On an individual level, only SNVs with a genotype of 0/1 or 1/1 were kept. Further filtering steps comprised the removal of SNVs with a read depth smaller than 10, an excessive read depth38 (d + 3√d, where d is the average read depth), an allele frequency less than 10% using a variety of tools (BEDtools, version 2.26.0; BEDOPS, version 2.4.3; VCFtools, version 0.1.17)39,40,41. All SNVs within ±5 bp of an indel border were removed as likely false-positives.
Simple GATK indel analysis
Indels identified by GATK where extracted after joint genotyping and subsequently hard filters were applied according to the GATK recommendations: QD < 2.0 || FS > 200.0 || ReadPosRankSum < −20.0 || SOR > 10.0. Indels overlapping with repetitive elements36 (UCSC’s masked repeats plus simple repeats; http://hgdownload.soe.ucsc.edu/goldenPath/mm10/database) or black regions (ENCODE37; http://mitra.stanford.edu/kundaje/akundaje/release/blacklists/mm10-mouse/) were removed. The individual samples were filtered keeping only indels with the genotypes of 0/1 and 1/1, removing those with a read depth smaller than 10 as well as sites with an excessive number of reads38 (d + 3√d, d = average read depth). Last, all simple indels that overlap with complex indels identified using LUMPY (version 0.2.13) were excluded. For all those steps a variety of tools39,40,41 was applied.
Complex indel analysis using Lumpy
The analysis of complex indels was done on the same samples using Lumpy42 according to the guidelines. Mapping was done using BWA MEM33, with the parameters –excludeDups –addMateTags –maxSplitCount 2 –minNonOverlap 20 (reference genome mm10), followed by Lumpy42 using the discordant and split reads as input and genotypes were identified using SVTyper43. The subsequent filtering steps comprised the selection of indels with a genotype of 0/1 and 1/1 and the removal of indels with a quality smaller than 100 and an excessive read coverage (d + 3√d38, where d is the average read depth) or a SU value (Number of pieces of evidence supporting the variant across all samples) smaller than 5. Indels overlapping with repetitive elements36 or black regions37 were excluded.
Statistics and reproducibility
All statistical analyses for 13 non-injected control, 13 ABE-edited and 9 BE4-edited mice were performed with R package 3.3.3 (http://www.R-project.org/). Kruskal-Wallis test was applied using kruskal.test and pairwise comparison was done with a Wilcoxon Rank Sum wilcox.test in R. All values represent means ± S.D.
Targeted deep sequencing
Target sites were amplified from mouse genomic DNA using Phusion polymerase (Thermo Fisher Scientific) and PCR products were prepared as libraries for next-generation sequencing. Pooled PCR amplicons were conducted paired-end sequencing using an Illumina MiSeq (Illumina).
PCR primers
Wap-S1_F2: AAGACAGGAGGTTTTGAGCAAGGC
Wap-S1_R2: CACCAGTGAAGACAAAGGAGTATGG
Off-target analysis
Off-target sites were predicted using CRISPOR http://crispor.tefor.net/44. The resulting off-target sites were filtered using the same criteria as for SNVs and indels, to consider only those areas of the genome which do not coincide with black regions37 (ENCODE37; http://mitra.stanford.edu/kundaje/akundaje/release/blacklists/mm10-mouse) or repetitive elements36 (UCSC’s masked repeats plus simple repeats; http://hgdownload.soe.ucsc.edu/goldenPath/mm10/database). Mutations, which were present in the population and not only in base-edited mice, but identified at predicted off-target sites, were not considered as a consequence of base editing.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The data are available at SRA under project number PRJNA555149.
References
Komor, A. C., Kim, Y. B., Packer, M. S., Zuris, J. A. & Liu, D. R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424 (2016).
Gaudelli, N. M. et al. Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature 551, 464–471 (2017).
Komor, A. C., Badran, A. H. & Liu, D. R. Editing the genome without double-stranded DNA Breaks. ACS Chem. Biol. 13, 383–388 (2018).
Landrum, M. J. et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 44, D862–D868 (2016).
Lee, H. K. et al. Targeting fidelity of adenine and cytosine base editors in mouse embryos. Nat. Commun. 9, 4804 (2018).
Shimatani, Z. et al. Targeted base editing in rice and tomato using a CRISPR-Cas9 cytidine deaminase fusion. Nat. Biotechnol. 35, 441–443 (2017).
Liu, Z. et al. Efficient generation of mouse models of human diseases via ABE- and BE-mediated base editing. Nat. Commun. 9, 2338 (2018).
Kim, H. S., Jeong, Y. K., Hur, J. K., Kim, J. S. & Bae, S. Adenine base editors catalyze cytosine conversions in human cells. Nat. Biotechnol. https://doi.org/10.1038/s41587-019-0254-4 (2019).
Jin, S. et al. Cytosine, but not adenine, base editors induce genome-wide off-target mutations in rice. Science 364, 292–295 (2019).
Zuo, E. et al. Cytosine base editor generates substantial off-target single-nucleotide variants in mouse embryos. Science 364, 289–292 (2019).
Zhou, S. et al. Programmable base editing of the sheep genome revealed no genome-wide off-target mutations. Front Genet 10, 215 (2019).
Kim, K. et al. Highly efficient RNA-guided base editing in mouse embryos. Nat. Biotechnol. 35, 435–437 (2017).
Komor, A. C. et al. Improved base excision repair inhibition and bacteriophage Mu Gam protein yields C:G-to-T:A base editors with higher efficiency and product purity. Sci. Adv. 3, eaao4774 (2017).
Liu, Z. et al. Highly efficient RNA-guided base editing in rabbit. Nat. Commun. 9, 2717 (2018).
Schaefer, K. A. et al. Unexpected mutations after CRISPR-Cas9 editing in vivo. Nat. Methods 14, 547–548 (2017).
Nutter, L. M. J. et al. Response to “Unexpected mutations after CRISPR-Cas9 editing in vivo”. Nat. Methods 15, 235–236 (2018).
Wilson, C. J. et al. Response to “unexpected mutations after CRISPR-Cas9 editing in vivo”. Nat. Methods 15, 236–237 (2018).
Lescarbeau, R. M., Murray, B., Barnes, T. M. & Bermingham, N. Response to "Unexpected mutations after CRISPR-Cas9 editing in vivo". Nat. Methods 15, 237 (2018).
Lareau, C. A. et al. Response to “Unexpected mutations after CRISPR-Cas9 editing in vivo”. Nat. Methods 15, 238–239 (2018).
Kim, S. T. et al. Response to “Unexpected mutations after CRISPR-Cas9 editing in vivo”. Nat. Methods 15, 239–240 (2018).
Willi, M., Smith, H. E., Wang, C., Liu, C. & Hennighausen, L. Mutation frequency is not increased in CRISPR-Cas9-edited mice. Nat. Methods 15, 756–758 (2018).
Iyer, V. et al. No unexpected CRISPR-Cas9 off-target activity revealed by trio sequencing of gene-edited mice. PLoS Genet. 14, e1007503 (2018).
Kim, D. et al. Genome-wide target specificities of CRISPR RNA-guided programmable deaminases. Nat. Biotechnol. 35, 475–480 (2017).
Kim, D., Kim, D. E., Lee, G., Cho, S. I. & Kim, J. S. Genome-wide target specificity of CRISPR RNA-guided adenine base editors. Nat. Biotechnol. 37, 430–435 (2019).
Yang, L. et al. Engineering and optimising deaminase fusions for genome editing. Nat. Commun. 7, 13330 (2016).
Grunewald, J. et al. Transcriptome-wide off-target RNA editing induced by CRISPR-guided DNA base editors. Nature 569, 433–437 (2019).
Zhou, C. et al. Off-target RNA mutation induced by DNA base editing and its elimination by mutagenesis. Nature. https://doi.org/10.1038/s41586-019-1314-0 (2019).
Shin, H. Y. et al. Hierarchy within the mammary STAT5-driven Wap super-enhancer. Nat. Genet. 48, 904–911 (2016).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
DePristo, M. A. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498 (2011).
Van der Auwera, G. A. et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinforma. 43, 11.10.1–11.10.33 (2013).
Bushnell, B. BBMap Short-read Aligner, and Other Bioinformatics Tools, http://sourceforge.net/projects/bbmap/ (2016).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Broad Institute. Picard, http://broadinstitute.github.io/picard/ (2016).
Casper, J. et al. The UCSC Genome Browser database: 2018 update. Nucleic Acids Res. 46, D762–D769 (2018).
Consortium, E. P. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012).
Li, H. Toward better understanding of artifacts in variant calling from high-coverage samples. Bioinformatics 30, 2843–2851 (2014).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Neph, S. et al. BEDOPS: high-performance genomic feature operations. Bioinformatics 28, 1919–1920 (2012).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011).
Layer, R. M., Chiang, C., Quinlan, A. R. & Hall, I. M. LUMPY: a probabilistic framework for structural variant discovery. Genome Biol. 15, R84 (2014).
Chiang, C. et al. SpeedSeq: ultra-fast personal genome analysis and interpretation. Nat. Methods 12, 966–968 (2015).
Haeussler, M. et al. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 17, 148 (2016).
Acknowledgements
This work utilized the computational resources of the NIH HPC Biowulf cluster (http://hpc.nih.gov). This work was supported by the Intramural Research Programs (IRPs) of National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) and National Heart, Lung, and Blood Institute (NHLBI).
Author information
Authors and Affiliations
Contributions
All authors designed the study. C.L. generated mutant mice. H.K.L. identified and characterized mutant mice and performed experiments and data analysis. H.E.S. and M.W. performed computational analysis. H.K.L., M.W., and L.H. supervised the study and wrote the manuscript and all authors approved the final version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lee, H.K., Smith, H.E., Liu, C. et al. Cytosine base editor 4 but not adenine base editor generates off-target mutations in mouse embryos. Commun Biol 3, 19 (2020). https://doi.org/10.1038/s42003-019-0745-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-019-0745-3
This article is cited by
-
Cell-specific and shared regulatory elements control a multigene locus active in mammary and salivary glands
Nature Communications (2023)
-
A large-scale genome and transcriptome sequencing analysis reveals the mutation landscapes induced by high-activity adenine base editors in plants
Genome Biology (2022)
-
Off-target effects of base editors: what we know and how we can reduce it
Current Genetics (2022)
-
An unbiased method for evaluating the genome-wide specificity of base editors in rice
Nature Protocols (2021)
-
Novel CRISPR/Cas applications in plants: from prime editing to chromosome engineering
Transgenic Research (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
