
Abstract
A new avenue of mining published genome-wide association studies includes the joint analysis of related traits. The power of this approach depends on the genetic correlation of traits, which reflects the number of pleiotropic loci, i.e. genetic loci influencing multiple traits. Here, we applied new meta-analyses of optic nerve head (ONH) related traits implicated in primary open-angle glaucoma (POAG); intraocular pressure and central corneal thickness using Haplotype reference consortium imputations. We performed a multi-trait analysis of ONH parameters cup area, disc area and vertical cup-disc ratio. We uncover new variants; rs11158547 in PPP1R36-PLEKHG3 and rs1028727 near SERPINE3 at genome-wide significance that replicate in independent Asian cohorts imputed to 1000 Genomes. At this point, validation of these variants in POAG cohorts is hampered by the high degree of heterogeneity. Our results show that multi-trait analysis is a valid approach to identify novel pleiotropic variants for ONH.
Similar content being viewed by others


Introduction
Glaucoma is the most common cause of irreversible blindness in the world1. Primary open angle glaucoma (POAG) is the most prevalent type of glaucoma accounting for 74% of all glaucoma cases2,3. Intraocular pressure (IOP) and the morphology of the optic nerve head (cup area (CA), disc area (DA), and vertical cup–disc ratio (VCDR)) are important features of the glaucomatous process. For each of these traits, twin studies showed a high heritability (h2CA = 0.75, h2DA = 0.72, h2IOP = 0.55, and h2VCDR = 0.48)4. Central corneal thickness (CCT) is also a highly heritable trait (h2CCT = 0.68–0.95)5, which is most likely non-physiologically associated with POAG, but rather biases IOP measurement, the major risk factor of POAG6,7. CA, DA, and VCDR, are significantly correlated both at the genetic level8. The high genetic correlation found between the optic nerve head (ONH) traits (Rg = 0.31–0.83) raises the question whether multi-trait analyses will improve the statistical power of the individual GWAS and will find variants with pleiotropic effects9.
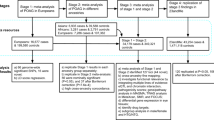
For this study, we generated new data on these 5 quantitative traits by imputing 12 European ancestry cohorts from the International Glaucoma Genetic Consortium (IGGC) (nMAX = 31,269) to haplotype reference consortium (HRC) release 1 imputation panel, which includes over 39 million variants10. A meta-analysis of these 12 European ancestry studies served as a discovery cohort in the analyses. Replication was performed in five Asian ancestry cohorts that were part of the IGGC. The cohorts of Asian descent were imputed to 1000 Genomes as there is little gain in HRC imputation in this ancestry group because there are no additional Asian samples included in HRC (http://www.haplotype-reference-consortium.org/participating-cohorts)11. We evaluated the added value of multi-trait analyses using two programs: CPASSOC and multi-trait analysis of GWAS (MTAG). Both use aggregated GWAS results. Whereas CPASSOS performs a meta-analysis assuming homogeneous and heterogeneous effects across traits by applying a inter-trait correlation matrix, MTAG basically increases the power of a single trait analyses by incorporating the GWAS findings of correlated traits.
By multi-trait analysis we identified two novel loci associated with the ONH at rs11158547 in-between PPP1R36 and PLEKHG3 and at rs1028727 near SERPINE3 in those of European descent. These loci replicated in the Asian replication sample. Findings for these loci were consistent using a distinct multi-trait approach, MTAG, in both the European and Asian cohorts. This study emphasis that multi-trait analysis in GWAS pleiotropic traits is an effective approach to identify variants harboring correlated traits
Results
Replication of previous CA, DA, VCDR, IOP, and CCT GWAS results
As a validation we first confirmed previously identified loci for CA, DA, VCDR, IOP, and CCT by Springelkamp et al.8 (nCA = 22,489; nDA = 22,504; nVCDR = 23,899; nIOP = 37,930) and Iglesias et al.12 (n = 17,803) based on 1000 Genomes imputation. Supplementary Fig. 1 and Supplementary Datas 1 and 2 show the per trait comparison of our meta-analysis of all European ancestry discovery cohorts using the HRC imputation with the results of the meta-analysis by Springelkamp8 and Iglesias12 based on 1000 Genomes. Out of 113 (95%), 107 available variants in HRC replicated at a Bonferroni significance level.
Optic nerve head parameters
In the single trait meta-analyses of ONH traits (CA, DA, and VCDR) in those of European descent (nCA = 24,493, LDSC interceptca = 1.024 (SE = 0.0083); nDA = 24,509, LDSC interceptDA = 1.041 (SE = 0.0071); nVCDR = 25,180, LDSC interceptVCDR = 1.029 (SE = 0.0081) Supplementary Data 3), 59 loci showed genome-wide significant association with at least one of the traits (Fig. 1a–c, Supplementary Data 4). The ONH analyses yielded six loci not previously reported (Table 1, Supplementary Data 5), however, none of these novel variants replicated in the Asian replication sample comprising five Asian studies. As the correlation analysis between the ONH traits showed significant correlations at the genetic and phenotype level (Fig. 2), we applied multi-trait analysis to uncover pleiotropic effects. Using multi-trait approach CPASSOC13, we identified three new loci at p < 5 × 10−8 by CPASSOC’s SHom (KIF6, EPB41L3, PPP1R36-PLEKHG3) (Fig. 1d, Supplementary Data 6). This method assumes that genetic effects are homogenous across traits and cohorts. Two additional new loci were identified by SHet (ZAK, SERPINE3) (Fig. 1e, Supplementary Data 6), which assumes the genetic effects are heterogenous. Locuszoom plots for these novel variants a depicted in Supplementary Fig. 2. Using an alternative approach (MTAG)14, the loci emerged consistently as genome-wide significant: rs9471130 near KIF6 in the DA analysis (p = 2.63 × 10−08), rs11158547 near PPP1R36-PLEKHG3 in the CA analysis (p = 2.13 × 10−08) and rs1028727 near SERPINE3 in the DA analysis (p = 4.50 × 10−09) (Supplementary Data 7). rs11158547 (PPP1R36-PLEKHG3) and rs1028727 (SERPINE3) displayed nominally significant association in the multi-trait analysis (CPASSOC and MTAG) in individuals of Asian ancestry (Supplementary Datas 6 and 7). Both variants were not in LD (r2 < 0.1) with neighboring known variants near SIX6, DDHD1, and DLCK1.

Manhattan plot of single trait analysis for cup area (a), disc area (b), and vertical cup–disc ratio (c). Manhattan plot for multi-trait analysis of the optic nerve head (ONH) SHom (d) and SHet (e).

Phenotype (a) and genetic (b) correlations between cup area, disc area, vertical cup–disc ratio, intraocular pressure, and central corneal thickness. a Partial pearson correlation coefficient s between cup area (CA), disc area (DA), vertical cup–disc ratio (VCDR), intraocular pressure (IOP), and central corneal thickness (CCT) adjusted for age and sex in the Rotterdam study I. b Genetic correlation coefficient (Rg) for CA, DA, VCDR, IOP, and CCT calculated by LD score regression; *p < 0.05, **p < 0.0001.
IOP and CCT
Next, we conducted a single trait meta-analysis for IOP and for CCT, the two traits that are not likely physiologically related. For IOP, we meta-analyzed a total of 31,269 participants (LDSC intercept = 1.028; SE = 0.0078, Supplementary Data 4) and identified 9 genome-wide significant regions of which two were novel in the HRC-based imputations and had not been uncovered in the IGGC 1000 Genomes analyses before (Table 1, Supplementary Data 5). The lead single-nucleotide polymorphisms (SNPs) in these genomic regions were a common variant rs9853115 near DGKG on 3q27.3 and a rare variant rs150202082[T] (frequency 0.03) near TCF4 on 18q21.2. rs9853115 failed replication in the Asians (p = 0.9315) and rs150202082 could not be examined since this variant was monoallelic in the Asian individuals. A GWAS by Choquet et al.15 also identified rs9853115 as new variant associated with IOP in multiethnic cohort of predominately (83%) European ancestry. The same study also identified a novel variant near TCF4, rs11659764, approximately 300 kb upstream of rs150202082 which was in relatively weak LD (r2 = 0.4). In a recent study from the UKbiobank by Khawaja et al.16 the same variant near DGKG showed genome-wide significant association with similar effectsize, however, rs150202082 near TCF4 could not be validated in this study.
In the meta-analysis of CCT, a total of 16,204 participants were included (LDSC intercept = 0.989; SE = 0.0082). We identified 31 independent genome-wide significant signals of which three were novel (Table 1 and Supplementary Data 5), including a variant, rs34869, near CDO1. Again, none of the three new variants replicated at a nominal significance level in the Asian samples. Multi-trait analysis by CPASSOC identified four novel variants (Supplementary Datas 8 and 9). In contrast to ONH cross trait analyses, also these could not be replicated in the Asian.
In silico analysis
To investigate the functional and regulatory potential, we annotated the variants in linkage disequilibrium (European LD, r2 ≥ 0.8) with the lead SNPs at the two new and replicated ONH variants, rs11158547 and rs1028727, using a combination of bioinformatics tools (see Method section). A total of 70 variants in LD with the 2 novel variants were queried. None of the examined variants were predicted to damage protein structure by SIFT, Polyphen, or alternative splicing using Ensembl’s Variant Effect Predictor. As all queried variants are noncoding, we reviewed the possible regulatory annotation of these SNPs in experimental epigenetic evidence, including DNAse hypersensitive sites, histone modifications, and transcription factor-binding sites in human cell lines and tissues from the ENCODE17 and ROADMAP EPIGENOMICS18 projects, integrated in Haploreg19. Annotations of chromatin states indicated that the two novel variants were located in, or in LD with, an active chromatin state region from at least one of the tissues investigated (Supplementary Data 10 and Figs. 3 and 4 for chromatin states in brain tissue). Next, we evaluated the overlaps of cis-expression quantitative trait loci (eQTL) in several databases (see Methods). In both novel loci PPP1R36-PLEKHG3 and SERPINE3, variants were found to be eQTL’s and based on Regulome DB-scores both ONH loci contained variants that were likely to alter binding (Supplementary Data 10).

Regional, chromatin state, and eQTL plot for rs11158547 (PPP1R36-PLEKHG3). Panel a shows the regional assocaiations plots with −log10 p value depicted on the y-axis, genes mapped by either position, eQTL or chromatin interaction are depicted in red on the x-axis; panel b shows 15 core chromatin states of varaints plotted in panel a for 13 brain tissues from Roadmap epidenomes described on the y-axis. E054 ganglion eminence derived primary cultured neurospheres, E053 cortex-derived primary cultured neurospheres, E071 brain hippocampus middle, E074 brain substantia nigra, E068 brain anterior caudate, E069 brain cingulate gyrus, E072 brain inferior temporal lobe, E067 brain angular gyrus, E073 brain dorsolateral prefrontal cortex, E070 brain germinal matrix, E082 fetal brain female, E081 fetal brain male, E125 NH-A astrocytes primary cells. Panel c depicts varaints that overlap eQTLs from selected eQTL databases described in the legend of panel (c).

Regional, chromatin state and eQTL plot for rs1028727 (SERPINE3). Panel a shows the regional assocaiations plots with −log10 p value depicted on the y-axis, genes mapped by either position, eQTL or chromatin interaction are depicted in red on the x-axis; panel b shows 15 core chromatin states of varaints plotted in panel a for 13 brain tissues from Roadmap epidenomes described on the y-axis. E054 ganglion eminence derived primary cultured neurospheres, E053 cortex-derived primary cultured neurospheres, E071 brain hippocampus middle, E074 brain substantia nigra, E068 brain anterior caudate, E069 brain cingulate gyrus, E072 brain inferior temporal lobe, E067 brain angular gyrus, E073 brain dorsolateral prefrontal cortex, E070 brain germinal matrix, E082 fetal brain female, E081 fetal brain male, E125 NH-A astrocytes primary cells. Panel c depicts varaints that overlap eQTLs from selected eQTL databases described in the legend of panel (c).
Gene prioritization, pathway analysis, and gene expression
We explored possible tissue expression and biological functions by pathway analysis for the two novel SNPs. We annotated these SNPs to genes by positional gene mapping, eQTL mapping and chromatin interaction mapping strategies implemented in FUMA20 (see Method section). For, rs11158547 (PPP1R36-PLEKHG3) 9 genes were assigned to this locus and for rs1028727 (SERPINE3) 21 genes were mapped to this locus (Supplementary Data 11). Pathway analysis based on enrichment of gene-set terms (MsigDB21 and Wikipathways22) found 5 and 11 Bonferroni significant gene-sets comprising genes mapped to SERPINE3 locus and PPP1R36-PLEKHG3 locus respectively. These pathways were in particular involved in immune response and cancer development (Supplementary Data 12 highlighted gene-sets).
As expression in eye tissues is not available in GTex23, we assessed the Ocular Tissue Database24. For 26 out of the 30 genes mapped to either rs11158547 or rs1028727 expression data were present in the Ocular Tissue Database (Supplementary Data 11). The highest levels of expression in the optic nerve was found for HSPA2, a gene associated to rs11158547 via lung, tibial nerve and fibroblasts eQTL’s and chromatin interaction mapping (Supplementary Fig. 3).
From endophenotypes to glaucoma
We also investigated the translational potential of these two loci to POAG by carrying out a meta-analysis of three POAG studies, NEIGHBOR, Southampton and UK Biobank Eye and Vision Consortium (Ncase = 9450; Ncontrol = 436,824), from European origin. For rs1028727 a negative effect on the VCDR in the present study predicts a decreased risk of POAG which was seen in NEIGHBOR study and the UKBiobank but not in the Southampton study (Supplementary Data 13). rs11158547 in PPP1R36-PLEKHG3 is predicted to be associated with increased POAG risk based on the positive effect of VCDR. Indeed in all three POAG studies the effect of the SNP is also positive. Pooling the studies based on a fixed effect analysis yields an OR 1.28 (95% CI: 1.15–1.39; p = 4.83 × 10−8) and showed Bonferroni significance (p = 0.025). Given the high degree of heterogeneity of effects at this locus a random effect meta-analysis was carried out which could not confirm this finding in POAG (Supplementary Data 14). Thus the findings were partly but not consistently replicated, awaiting larger and more homogeneous data sets for the final replication.
Discussion
Our results implicate two novel loci, one downstream SERPINE3 and one other in-between PPP1R36 and PLEKHG3, both associated with ONH morphology via CPASSOC multi-trait analysis. SERPINE3 belongs to the clade E family of extracellular serpins. Family members have been described to play a role in other neurodegenerative diseases such as Alzheimer’s disease25. Recent studies in glaucomatous human postmortem samples and in rat models identified oxidative inactivation of serpins (neuroserpin) as a molecular mechanism of increased plasmin activity leading to neurodegeneration in high ocular pressure conditions26. In the trabecular meshwork, serpins (plasminogen activator inhibitor) may mediate the inhibition of matrix metaloprotienase (MMPs) activity induced by transforming growth factor-beta enhancement27,28. Inactivity of MMPs were found to increase aqueous humor outflow resistance leading to rising IOP29. rs11158547 downstream the PLEKHG3 gene, a pleckstrin homology domain containing protein, is also a relatively unknown gene. It contains a guanide nucleotide exchange factor (GEF) domain which is important for Rho-dependent signal transduction30. In mice PLEKHG3 knockout is associated with an abnormal anterior chamber depth of the eye (IMPC release 3.2 http://www.mousephenotype.org/data/experiments?geneAccession=MGI:2388284). The other gene close to this locus, PPP1R36, is less likely a candidate gene for ONH. PPP1R36, has been described in connection with autophagy during spermatogenesis. PPP1R36 encodes a regulatory subunit of protein phosphatase 1 which is involved in multiple cellular functions such as metabolism, immune response, apoptosis, meiosis, mitosis, cytoskeletal reorganization, and synthesis. However, its function to the eye has not been characterized.
A general insight from this study was that the strength of genetic correlation is an important condition for investigating the pleiotropic effect of genetic variants on traits. The high genetic and phenotypic correlation observed between the ONH traits enabled multi-trait analyses and yielded plausible and replicable results. We validated these novel variants identified by CPASSOC with another multi-trait analysis method MTAG, which also uses summary statistics as input. The main difference between both methods is that CPASSOC test whether a SNP is not associated with any of the traits under the null hypothesis. MTAG, on the contrary produces trait-specific effect estimates for each SNP. The variants discovered in the European CPASSOC analysis that replicated in the Asian CPASSOC analysis also replicated in the MTAG analysis. Sensitivity analysis excluding the Rotterdam studies showed a high correlation of the Shom and Shet statistic with the SHom/SHet statistic from the full analysis including the Rotterdam studies (r = 0.71, p < 2.2 × 10−16; r = 0.68, p < 2.2 × 10−16, respectively). The correlation in MTAG for CA, DA, and VCDR was considerably low yet very significant (rCA = 0. 31, p < 2.2 × 10−16; rDA = 0.56, p < 2.2 × 10−16, rvcdr = 0.17).
In contrast, the multi-trait analysis between IOP and CCT could not uncover robust new variants. A reason for this observation might be the moderate magnitude of the genetic correlation between IOP and CCT. Also, clinical research has shown that this relation is largely driven by measurement errors in Goldmann applanation tonometry, rather than a pathophysiological process31,32.
A potential limitation of this study is the application of a different imputation panel for the discovery and replication phase. The European studies were all imputed to the HRC panel which has a beneficial imputation quality compared to 1000 Genomes. By contract the Asian replications set, was imputed to 1000 Genomes since a recent publication showed that HRC imputations perform less adequate in Asians11. Variants associated with cup area and other endophenotypes at genome-wide significance in the European single trait analysis could not be replicated in the Asian replication sample. The use of different imputation panels may be a source bias hampering replication. A theoretical shortcoming, is that the various studies used different methods and equipment to assess ONH parameters among studies. This has most likely reduced the power of the study and has generated most probably false negative rather than false-positive results. To prevent false-positive findings using novel methods, we aimed to replicate the findings of the primary CPASSOC analyses by another analyses using MTAG. The variants discovered in the CPASSOC analysis could also be replicated in MTAG. As both methods are mathematically distinct we concluded that our results are rather robust and independent of the statistical approach. This underscores the strength of the association as it is consistently found by two independent approaches.
We have no doubt that association of the variants to ONH are of interest to the biology community. To evaluate the implications of our findings in the context of glaucoma we studied three independent POAG studies. Up until now we are not able to link our finding to POAG. Although fixed effect meta-analysis showed Bonferroni significance (p = 0.025) for rs11158547 in PPP1R36-PLEKHG3, random effect meta-analysis that takes into account the heterogeneity could not confirm this finding in POAG. The source of the high variability in estimates is unknown and may involve clinical variability and ethnic differences. It is important to realize that the identification of POAG genes is far from complete and work in progress.
In conclusion, we conducted single and multi-trait meta-analysis of five endophenotypes of glaucoma based on HRC imputations in European ancestral populations. The HRC single trait analyses in those of European descent did not yield new loci that could be replicated in Asians. We identified two novel loci for ONH in-between PPP1R36-PLEKHG3 at chromosome 14q23.3 and near SERPINE3 at chromosome 13q14.3 by multi-trait analysis in those of European descent that could be replicated in Asians using CPASSOC. Findings for these loci were consistent using MTAG. The present study underscores that multi-trait analysis in GWAS of true pleiotropic traits in relatively small sample sizes is a powerful approach to identify variants harboring correlated traits. Although these novel loci could not be directly associated with POAG it is likely that the genes in these regions mediate the glaucomatous process by their effect on the optic nerve morphology. For instance the PLEKHG3 gene identified in this study is involved in the Rho signaling cascade, this pathway is known to play a crucial role in POAG pathophysiology and is currently targeted for new therapies for POAG33. Our bioinformatic analyses suggests that both the PPP1R36-PLEKHG3 and SERPINE3 variants are eQTL’s opening avenues to counteract the problem by RNA interference. Further research including exome sequencing and functional studies are needed to further define these genes in the mechanism of POAG.
Methods
Study design
We performed a meta-analysis of European origin GWAS’s imputed to HRC reference panel release 1. We analyzed five outcomes: CA, DA, VCDR, IOP, and CCT. The CA phenotype was adjusted for DA in all analyses since these phenotypes are clearly correlated (Pearson correlation coefficient 0.6). Subsequently, we performed multi-trait analysis for CA, DA, VCDR, IOP, and CCT. Replication was carried out for the single trait as well as the multi-trait analysis in a meta-analysis of 5 Asian cohorts imputed to 1000 genomes. We also tested significance of lead SNPs in three independent POAG cohorts.
Study samples, phenotyping, and genotyping
All studies included in this meta-analysis are part of the International Glaucoma Genetics Consortium (IGGC). A description of the details of all cohorts participating in this study can be found in Supplementary Note and Supplementary Tables 1–6. The mean IOP, VCDR, CCT, CA, and DA of both eyes was used for the analyses. In case of missing or unreliable data for one eye, the measurement of the other eye was used instead. For subjects who received IOP-lowering medication, the measured IOP was multiplied by a factor of 1.3. The total number of individuals in the meta-analysis was 24,493 for CA, 24,509 for DA, 25,180 for VCDR, 31,269 for IOP, and 16,204 for CCT. All studies were performed with the approval of the local institutional review board (Supplementary Note) and written informed consent was obtained from all participants in accordance with the Declaration of Helsinki.
Genotyping was performed using commercially available Affymetrix or Illumina genotyping arrays (Supplementary Table 7). Quality control was executed independently for each study. To facilitate meta-analysis, each cohort performed genotype imputation using either the Sanger imputation service (https://imputation.sanger.ac.uk) or the Michigan imputation server (https://imputationserver.sph.umich.edu) with reference to the HRC panel, version 1 or 1.134.
Association analysis in discovery cohorts
Within each discovery cohort, each genotyped or imputed variant was tested for association with each of the traits, assuming an additive genetic model. The measurements were adjusted for sex, age, and five principal components in all cohorts and if necessary also for cohort-specific covariates (Supplementary Table 8). Family-based studies were adjusted for family structure. Given the clear correlation of CA with DA (Pearson’s correlation r = 0.59 in Rotterdam Study I), the CA GWAS was adjusted for DA in all discovery cohorts prior to meta-analysis. Linear regression was employed for studies with unrelated individuals, and linear mixed effects models were used to account for family structure in the family-based studies.
Centralized quality control
Before meta-analysis, a centralized quality control procedure implemented in EasyQC was applied to individual study association summary statistics to identify outlying studies35. We included variants with imputation quality ≥ 0.3 (e.g., Minimac R2) and expected minor allele count >6. Additional checks for quality control were applied on the already filtered datasets including review of P–Z-plots, allele frequency plots and calculation of genomic inflation factor λ.
Meta-analysis of discovery cohorts
The association results of all studies were combined in a fixed effect inverse variance meta-analysis in METAL36, since there was no sample overlap or cryptic relatedness as checked by LD score regression (see methods genetic overlap). This tool also applies genomic control by correcting the test statistics to account for small amounts of population stratification or unaccounted relatedness. We also assessed heterogeneity by calculating I2 values and Cochrans Q-test for heterogeneity as implemented in METAL. After meta-analyses of all available variants, we excluded the variants that were not present in at least three studies. This resulted in 11,830,838 variants for CA, 11,764,957 for DA, 11,901,698 variants for VCDR, 12,426,120 for IOP, and 9,249,813 variants for CCT. The remaining variants per trait were used to create Manhattan plots and QQ-plots, see Supplementary Figs. 4 and 5. The meta-analysis resulted in 1918 SNPs with a p value less than 5 × 10−8 for CA, 2029 for DA, 2473 for VCDR, 156 for IOP, and 1288 for CCT. Re-running the meta-analysis excluding TEST-BATS study to show that the significantly younger mean age in this study did not distorted our findings showed nearly perfect correlation between effect estimates from the full analysis and the effect estimates from in the analysis excluding TEST-BATS (CA r = 0.99; DA r = 0.99; VCDR = 0.99; IOP = 0.99). Furthermore, the mean differences between effect estimates found in the full analysis and the effect estimates found in the analysis excluding TEST-BATS were zero (CA mean difference = 0, SD = 0.00; DA mean difference = 0, SD = 0.00; VCDR mean difference = 0, SD = 0.01; IOP mean difference = 0, SD = 0.06), this also suggests that the younger age in the TEST-BATS study has not biased the results.
Selection of independent variants
We examined whether multiple independent variants at a given locus influenced a trait and if they were independent of previous findings, we used the genome-wide complex trait analysis software (GCTA)37. This tool performs a stepwise selection procedure to select multiple associated SNPs by a conditional and joint (–CoJo) analysis approach using summary-level statistics from a meta-analysis and LD corrections between SNPs. The three Rotterdam Study cohorts (N = 5815), imputed with the HRC reference panel version 1, were used as the reference to calculate the LD, because it represents the largest discovery studies. LD was calculated between pairwise SNPs, but any SNP further than 10 Mb apart were assumed to not be in LD. All autosomal chromosomes were analyzed, with MAF restricted to ≥0.01 estimated from the three Rotterdam Study cohorts. The independent variants were annotated by Haploreg19, see Supplementary Table 9.
Identification of potential novel variants
Previously, Springelkamp et al.8,38,39, Iglesias et al.12, Hysi et al.40, and Lu et al.41 identified various loci associated with CA, CCT, DA, IOP, and VCDR by GWAS with the HapMap and 1000 Genomes as a reference panel for imputations. To identify new variants, we investigated if any of the independent variants were within 1 Mb of a known loci identified for the same trait by Springelkamp et al.8,38,39, Iglesias et al.12, Hysi et al.40, and Lu et al.41. We created locuszoom plots and forest plots of all potential novel variants, see Supplementary Figs. 5 and 6. Variants showing significant association with a trait and are within 1 Mb of a previous identified locus were annotated to the known variant.
Multi-trait analysis
For multi-trait genome-wide association analysis we applied the CPASSOC package developed by Zhu et al.13. We used CPASSOC for two analyses to combine the association results from CA, DA, VCDR, and from IOP and CCT. CPASSOC generates two statistics, SHom and SHet. SHom is similar to the fixed effect meta-analysis method but accounts for the correlation of summary statistics because of the correlated traits. SHom uses the sample size of a trait as a weight instead of variance, so that it is possible to combine traits with different measurement scales. SHet is an extension of SHom, but power can be improved when the genetic effect sizes are different for different traits. To compute statistics SHom and SHet, a correlation matrix is required to account for the correlation among traits or induced by overlapped or related samples from different cohorts. We followed the approach previously described by Park et al.42, to calculate this correlation matrix. Briefly, we used all independent SNPs (r2 < 0.2) present in datasets that were not associated with any of the traits (−1.96 > Z score < 1.96), and took the Pearson’s correlation of their Z-scores13. For both tests QQ-plots were created (Supplementary Fig. 8). Novel loci identified by CPASSOC (p < 5 × 10−8) that were not implicated in the single-trait analysis were validated using a second multi-trait method, MTAG. Similarly, MTAG also utilizes summary statistics as input, but performs LD score regression to estimate the genotypic and phenotypic variance-covariance matrices. In contrast to CPASSOC, MTAG performs association tests for each individual trait by boosting the power of a signal and providing an estimation of the underlying association via the multi-trait variance-covariance structure. We applied MTAG to SNPs MAF > 0.01 for combing the analysis of CA, DA, and VCDR, and the analysis of IOP and CCT. For the European sample we used the 1000Genomes European pre-calculated LD scores and for the Asians the 1000Genomes East-Asian pre-calculated LD scores (https://data.broadinstitute.org/alkesgroup/LDSCORE/). We then validated each of the genome-wide significant signals identified by CPASSOC in the MTAG results.
Replication in Asian cohorts imputed to 1000Genomes
All independent SNPs identified with p < 5 × 10−8 in the discovery stage (single and multi-trait analysis) were carried forward for replication in Asians. For single-trait analyses, we validated these signals in fixed effect meta-analyses previously reported by Springelkamp et al. (CA, DA, VCDR, and IOP) and Iglesias et al. (CCT). Similar as in the discovery stage, we also performed a multi-trait CPASSOC and MTAG analysis of CA, DA, VCDR, and IOP, CCT in the Asians using the 1000Genomes summary statistics. Association replication was sought at nominal (p < 0.05) levels. A brief description of the cohorts participating in this study can be found in the Supplementary Note. Descriptive statistics, phenotyping methods, genotype, and 1000 Genomes phase I version 3 (March 2012) imputation quality and control has been described previously in Springelkamp et al.8 and Iglesias et al.10.
Validation in POAG case–control studies
To evaluate whether SNPs identified in the European HRC discovery stage (p < 5 × 10−8) that replicated at nominal significance (p < 0.05) in Asians 1000Genomes have a shared component with primary open-angle glaucoma we validated these SNPs in three POAG case–control studies from NEIGBOHR/MEEI, Southampton and UK Biobank Eye and Vision Consortium. Phenotyping and genotyping methods are provided in Supplementary Note 1.3 and Supplementary Table 9. For the queried SNPs summary statistics from NEIGBOHR/MEEI and Southampton were combined in a fixed-effect and random-effect meta-analysis as implemented in Metasoft43. Statistical significant level was corrected for the number of queried SNPs by the Bonferroni method.
The genetic overlap between CA, DA, VCDR, IOP, and CCT
To further investigate the genetic overlap among CA, DA, VCDR, CCT, and IOP we used the LD Score regression implemented in LDSC44 to examine the pattern of genetic correlations. The LD score for each SNP measures the amount of pairwise LD(r2) with other SNPs within 1-cM (centimorgan) windows based linkage disequilibrium. Bivariate LD score regression can estimate the extent to which two phenotypes share genetic variance.
Summary statistics of the five meta-analysis were formatted to LDSC input files, we followed quality control as implemented by the LDSC software package (https://github.com/bulik/ldsc). We used pre-calculated LD scores provided by the developers for each SNP using individuals of European ancestry from the 1000 Genomes project that are suitable for LD score analysis in European populations. SNP heritability estimates for all traits and genetic correlations were then calculated between the traits, see Supplementary Data 3 and Fig. 2.
Bioinformatical annotation
Using the software HaploReg (version 4.1)19 and RegulomeDB v1.145, we annotated the potential regulatory functions of the replicated GWAS SNPs and their proxies (r2 > 0.8, 1000 genomes CEU) based on epigenetic signatures. We examined whether these variants (GWAS SNPs and variants in LD with the GWAS SNPs) overlapped with regulatory elements including DNAse hypersensitive sites, histone modifications, and transcription factor-binding sites in human cell lines and tissues from the ENCODE Project and the Epigenetic Roadmap Project. We then used the RegulomeDB score to assess their potential functional consequence, as described previously46.
Pathway analysis
We applied FUMA, which uses a three way gene-mapping strategy, to assign genome-wide significant SNPs to genes of interest. For positional mapping, SNPs in LD with the independent SNPs were mapped to genes using a window of 10 kb. eQTL mapping was performed by mapping SNPs to genes up to 1 Mb (cis-eQTL). eQTLs from all tissues available in GTEx v623, Blood eQTL browser47, BIOS eQTL browser48, and BRAINEAC49 were selected for the mapping. Chromatin interaction was based on GSE87112 (Hi-C) database as implemented in FUMA. We explored possible biological functions by pathway analysis for all variants that reached genome wide significance in the discovery stage and were nominal significant in the Asian replication set. These 55 associated variants (ONH = 32, IOP = 3, CCT = 20) were assigned to genes by FUMA mapping strategies. Prioritized genes for ONH traits were highly overlapping and were combined to form a set of 295 unique genes for further functional annotation in FUMA. For IOP and CCT 11 and 116 genes were prioritized respectively. We further investigated the FUMA-mapped genes for enrichment using hypergeometric enrichment tests on pre-defined gene-sets derived from MsigDB and WikiPathways. p Values were corrected based on Bonferroni method for the number of tested gene-sets.
Statistics and reproducibility
Software used for the data analysis of this study: METAL (https://genome.sph.umich.edu/wiki/METAL), EasyQC (www.genepi-regensburg.de/easyqc), GCTA (http://cnsgenomics.com/software/gcta/), FUMA (https://fuma.ctglab.nl/), LDSC (https://github.com/bulik/ldsc), CPASSOC (http://hal.case.edu/zhu-web/), and MTAG (https://github.com/omeed-maghzian/mtag). In the single trait analyses and the multi-trait analyses variants surpassing a p value less than 5 × 10−8 were considered genome-wide significant and followed up for replication in the Asian replication sample. Replication was defined as variants surpassing nominal significance level p value < 0.05. Variants were only considered novel when located >1 Mb away from a previously reported variant.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The genome-wide summary statistics that support the findings of this study will be made available via the NHGRI-EBI GWAS Catalog website (https://www.ebi.ac.uk/gwas/downloads/summarystatistics) upon publication.
Code availability
No previously unreported custom computer code or mathematical algorithm was used to generate results central to the conclusions.
References
Tham, Y. C. et al. Global prevalence of glaucoma and projections of glaucoma burden through 2040: a systematic review and meta-analysis. Ophthalmology 121, 2081–2090 (2014).
Kapetanakis, V. V. et al. Global variations and time trends in the prevalence of primary open angle glaucoma (POAG): a systematic review and meta-analysis. Br. J. Ophthalmol. 100, 86–93 (2016).
Quigley, H. A. & Broman, A. T. The number of people with glaucoma worldwide in 2010 and 2020. Br. J. Ophthalmol. 90, 262–267 (2006).
Sanfilippo, P. G., Hewitt, A. W., Hammond, C. J. & Mackey, D. A. The heritability of ocular traits. Surv. Ophthalmol. 55, 561–583 (2010).
Dimasi, D. P., Burdon, K. P. & Craig, J. E. The genetics of central corneal thickness. Br. J. Ophthalmol. 94, 971–976 (2010).
Gordon, M. O. et al. The Ocular Hypertension Treatment Study: baseline factors that predict the onset of primary open-angle glaucoma. Arch. Ophthalmol. 120, 714–720 (2002). discussion 829-30.
Landers, J. A. et al. Heritability of central corneal thickness in nuclear families. Invest Ophthalmol. Vis. Sci. 50, 4087–4090 (2009).
Springelkamp, H. et al. New insights into the genetics of primary open-angle glaucoma based on meta-analyses of intraocular pressure and optic disc characteristics. Hum. Mol. Genet. 26, 438–453 (2017).
Gharahkhani, P. et al. Analysis combining correlated glaucoma traits identifies five new risk loci for open-angle glaucoma. Sci. Rep. 8, 3124 (2018).
Iglesias, A. et al. Haplotype Reference Consortium Panel: Practical implications of imputations with large reference panels. Hum. Mutat. 38, 1025–1032 (2017).
Lin, Y. et al. Genotype imputation for Han Chinese population using Haplotype Reference Consortium as reference. Hum. Genet. 137, 431–436 (2018).
Iglesias, A. I. et al. Cross-ancestry genome-wide association analysis of corneal thickness strengthens link between complex and Mendelian eye diseases. Nat. Commun. 9, 1864 (2018).
Zhu, X. et al. Meta-analysis of correlated traits via summary statistics from GWASs with an application in hypertension. Am. J. Hum. Genet. 96, 21–36 (2015).
Turley, P. et al. MTAG: Multi-trait analysis of GWAS. Nat. Genet. 50, 229–237 (2018).
Choquet, H. et al. A large multi-ethnic genome-wide association study identifies novel genetic loci for intraocular pressure. Nat. Commun. 8, 2108 (2017).
Khawaja, A. P. et al. Genome-wide analyses identify 68 new loci associated with intraocular pressure and improve risk prediction for primary open-angle glaucoma. Nat. Genet. 50, 778–782 (2018).
Consortium, E. P. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012).
Roadmap Epigenomics, C. et al. Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015).
Ward, L. D. & Kellis, M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 40, D930–D934 (2012).
Watanabe, K., Taskesen, E., van Bochoven, A. & Posthuma, D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 8, 1826 (2017).
Liberzon, A. et al. Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740 (2011).
Kutmon, M. et al. WikiPathways: capturing the full diversity of pathway knowledge. Nucleic Acids Res. 44, D488–D494 (2016).
Consortium, G. T. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348, 648–660 (2015).
Wagner, A. H. et al. Exon-level expression profiling of ocular tissues. Exp. Eye Res. 111, 105–111 (2013).
Fabbro, S. & Seeds, N. W. Plasminogen activator activity is inhibited while neuroserpin is up-regulated in the Alzheimer disease brain. J. Neurochem. 109, 303–315 (2009).
Gupta, V. et al. Glaucoma is associated with plasmin proteolytic activation mediated through oxidative inactivation of neuroserpin. Sci. Rep. 7, 8412 (2017).
Fuchshofer, R., Welge-Lussen, U. & Lutjen-Drecoll, E. The effect of TGF-beta2 on human trabecular meshwork extracellular proteolytic system. Exp. Eye Res. 77, 757–765 (2003).
Han, H., Kampik, D., Grehn, F. & Schlunck, G. TGF-beta2-induced invadosomes in human trabecular meshwork cells. PLoS One 8, e70595 (2013).
De Groef, L., Van Hove, I., Dekeyster, E., Stalmans, I. & Moons, L. MMPs in the trabecular meshwork: promising targets for future glaucoma therapies? Invest. Ophthalmol. Vis. Sci. 54, 7756–7763 (2013).
Nguyen, T. T. et al. PLEKHG3 enhances polarized cell migration by activating actin filaments at the cell front. Proc. Natl Acad. Sci. USA 113, 10091–10096 (2016).
Dueker, D. K. et al. Corneal thickness measurement in the management of primary open-angle glaucoma: a report by the American Academy of Ophthalmology. Ophthalmology 114, 1779–1787 (2007).
Kohlhaas, M. et al. Effect of central corneal thickness, corneal curvature, and axial length on applanation tonometry. Arch. Ophthalmol. 124, 471–476 (2006).
Rao, P. V., Pattabiraman, P. P. & Kopczynski, C. Role of the Rho GTPase/Rho kinase signaling pathway in pathogenesis and treatment of glaucoma: Bench to bedside research. Exp. Eye Res. 158, 23–32 (2017).
McCarthy, S. et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat. Genet. 48, 1279–1283 (2016).
Winkler, T. W. et al. Quality control and conduct of genome-wide association meta-analyses. Nat. Protoc. 9, 1192–1212 (2014).
Willer, C. J., Li, Y. & Abecasis, G. R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010).
Yang, J., Lee, S. H., Goddard, M. E. & Visscher, P. M. GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82 (2011).
Springelkamp, H. et al. Meta-analysis of Genome-Wide Association Studies identifies novel loci associated with optic disc morphology. Genet. Epidemiol. 39, 207–216 (2015).
Springelkamp, H. et al. Meta-analysis of genome-wide association studies identifies novel loci that influence cupping and the glaucomatous process. Nat. Commun. 5, 4883 (2014).
Hysi, P. G. et al. Genome-wide analysis of multi-ancestry cohorts identifies new loci influencing intraocular pressure and susceptibility to glaucoma. Nat. Genet. 46, 1126–1130 (2014).
Lu, Y. et al. Genome-wide association analyses identify multiple loci associated with central corneal thickness and keratoconus. Nat. Genet. 45, 155–163 (2013).
Park, H., Li, X., Song, Y. E., He, K. Y. & Zhu, X. Multivariate analysis of anthropometric traits using summary statistics of genome-wide association studies from GIANT Consortium. PLoS One 11, e0163912 (2016).
Han, B. & Eskin, E. Random-effects model aimed at discovering associations in meta-analysis of genome-wide association studies. Am. J. Hum. Genet. 88, 586–598 (2011).
Bulik-Sullivan, B. K. et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47, 291–295 (2015).
Boyle, A. P. et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 22, 1790–1797 (2012).
Schaub, M. A., Boyle, A. P., Kundaje, A., Batzoglou, S. & Snyder, M. Linking disease associations with regulatory information in the human genome. Genome Res. 22, 1748–1759 (2012).
Westra, H. J. et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat. Genet. 45, 1238–1243 (2013).
Zhernakova, D. V. et al. Identification of context-dependent expression quantitative trait loci in whole blood. Nat. Genet. 49, 139–145 (2017).
Ramasamy, A. et al. Genetic variability in the regulation of gene expression in ten regions of the human brain. Nat. Neurosci. 17, 1418–1428 (2014).
Acknowledgements
Full acknowledgements are provided in the Supplementary Information.
Author information
Authors and Affiliations
Consortia
Contributions
P.W.M.B. and C.M.D. conceived and designed the study. E.M.L., S.M., P.G., P.G.H., C.J.H., V.V. and T.S.B. contributed to the research design and supervised analyses. P.W.M.B., E.M.L., A.I.I., P.G., V.V., A.P.K., M.S., R.H., A.J.C., R.P.I., T.S.B., C.C.K. and P.G.H. contributed to the data analysis and interpretation. P.G., V.V., A.K., R.H., A.G., S.N., J.F.W., C.H., O.P., T.A., N.A., A.J.L., J.L.W., C.C., C.J.H., A.A.H.J.T., S.M., C.C.W.K. and C.M.D. were involved in the collection of clinical and genetic data and/or supervision of the individual cohorts. All authors provided intellectual input and assisted with the drafting and revising of the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bonnemaijer, P.W.M., Leeuwen, E.M.v., Iglesias, A.I. et al. Multi-trait genome-wide association study identifies new loci associated with optic disc parameters. Commun Biol 2, 435 (2019). https://doi.org/10.1038/s42003-019-0634-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-019-0634-9
This article is cited by
-
A multi-ancestry GWAS of Fuchs corneal dystrophy highlights the contributions of laminins, collagen, and endothelial cell regulation
Communications Biology (2024)
-
Comparison of two multi-trait association testing methods and sequence-based fine mapping of six additive QTL in Swiss Large White pigs
BMC Genomics (2023)
-
Large-scale multitrait genome-wide association analyses identify hundreds of glaucoma risk loci
Nature Genetics (2023)
-
Elevated dimethylarginine, ATP, cytokines, metabolic remodeling involving tryptophan metabolism and potential microglial inflammation characterize primary open angle glaucoma
Scientific Reports (2021)
-
A Comparison of Genomic Advances in Exfoliation Syndrome and Primary Open-Angle Glaucoma
Current Ophthalmology Reports (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
