
Abstract
Billions of people have obesity-related metabolic syndromes such as diabetes and hyperlipidemia. Promoting the browning of white adipose tissue has been suggested as a potential strategy, but a drug still needs to be identified. Here, genetic deletion of activating transcription factor 3 (ATF3−/−) in mice under a high-fat diet (HFD) resulted in obesity and insulin resistance, which was abrogated by virus-mediated ATF3 restoration. ST32da, a synthetic ATF3 inducer isolated from Salvia miltiorrhiza, promoted ATF3 expression to downregulate adipokine genes and induce adipocyte browning by suppressing the carbohydrate-responsive element-binding protein–stearoyl-CoA desaturase-1 axis. Furthermore, ST32da increased white adipose tissue browning and reduced lipogenesis in HFD-induced obese mice. The anti-obesity efficacy of oral ST32da administration was similar to that of the clinical drug orlistat. Our study identified the ATF3 inducer ST32da as a promising therapeutic drug for treating diet-induced obesity and related metabolic disorders.
Similar content being viewed by others


Introduction
The prevalence of obesity, a risk factor for type 2 diabetes, hyperlipidemia and non-alchoholic fatty liver disease, has reached epidemic proportions worldwide. Mammals have two different types of adipose tissue: white adipose tissue (WAT) and brown adipose tissue (BAT). WAT primarily stores lipids, and BAT confers anti-obesity effects by metabolizing lipids through uncoupling protein 1 (UCP1)-mediated uncoupled respiration1,2. In small mammals, BAT plays a key role in the thermogenic response and regulation of energy balance3. Moreover, BAT activation promotes energy expenditure, reduces adiposity, and protects against diet-induced obesity4,5. Recent studies have shown the existence of another type of adipocyte: beige or “brite” (brown in white) cells2,6,7. In addition, morphological and histological data have shown adipocytes with an intermediate phenotype, which may due to white-to-beige adipocyte transdifferentiation8,9. In this regard, strategies to combat obesity have shifted from reducing fat accumulation to promoting energy expenditure by activation of BAT and browning of WAT10,11,12,13.
Two transcription-factor families are key determinants of terminal adipocyte differentiation: CCAAT/enhancer-binding proteins C/EBPα, C/EBPβ, and C/EBPδ, and peroxisome proliferator-activated receptor γ (PPARγ). Previous studies have shown that activating transcription factor 3 (ATF3) can inhibit the expression of C/EBPα, which in turn can inhibit adipocyte differentiation14. ATF3 levels are high in the WAT of obese mice and negatively regulate adiponectin gene expression15. In addition, ATF3 is involved in adipocyte hypoxia-mediated mitochondrial dysfunction in obesity16. Although the biological functions of ATF3 have been widely studied, the specific role of ATF3 in obesity regulation and energy metabolism remains to be fully explored.
In the present study, deletion of ATF3 in mice aggravated high-fat diet (HFD)-induced obesity and metabolic dysfunction. Furthermore, ATF3 overexpression inhibited adipogenic/lipogenic gene expression and upregulated lipolytic and browning-related gene expression, which was due to suppressing the gene expression of carbohydrate-responsive element-binding protein (ChREBP) and stearoyl-CoA desaturase-1 (Scd1). We used ATF3-specific promoter-screening approaches to explore ATF3 inducers from a modified Chinese herb single-compound library and uncovered a single compound, ST32da, a synthetic compound related to neo-tanshinlactone and isolated from Salvia miltiorrhiza, that showed good anti-obesity effects in mice with HFD-induced obesity. We confirmed that ATF3 is an important metabolic regulator. The ATF3-inducer ST32da may have therapeutic benefits in individuals with obesity and metabolic dysfunction.
Results
Obese patients had reduced ATF3 levels in different organs
Prior gene polymorphism study indicated that ATF3 is associated with human obesity17. Furthermore, after analysis the relationship between ATF3 and obesity in human GEO DataSet Browser (https://www.ncbi.nlm.nih.gov/sites/GDSbrowser/), we characterize that the gene expression of ATF3 was lower in human liver (Fig. 1a)18, adipose tissue (Fig. 1b)19 and muscle (Fig. 1c)20 specimens of obese than in the lean ones, but the ATF3 expression did not differ in the blood monocytes from normal weight, mildly obese and morbidly obese subjects (Fig. 1d)21.

Analysis of ATF3 expression level among liver, adipose tissue, muscle and blood monocytes from lean, obese and morbidly obese patients by NCBI GEO DataSets. a–d ATF3 expression level in different organs. a Liver. b Adipose tissue. c Muscle. d Blood monocytes. For a, Lean (n = 13), Obese (n = 26). For b, Lean (n = 5), Obese (n = 18). For c, ControlLean (n = 8), Obese (n = 8), m-Obese (n = 7). For d, Lean (n = 4), Obese (n = 7), m-Obese (n = 7). Data are mean ± SEM. and *p < 0.05 compared to control group
ATF3 −/− mice conferred HFD-induced metabolic dyshomeostasis
To examine whether ATF3 modulates energy metabolism and body-weight changes in mice, we analyzed the metabolic syndrome and its related traits in ATF3-null (ATF3−/−) mice. ATF3−/− mice, with a normal diet or HFD, showed higher body weight than their wild-type littermates (Fig. 2a). HFD-fed ATF3−/− mice showed increased body-fat percentage, serum triglyceride (TG) levels (Fig. 2b, c), glucose intolerance and insulin resistance (Fig. 2d, e), WAT and BAT depot weights (Fig. 2f), BAT whitening (Fig. 2g, Supplementary Fig. 1a), white-adipocyte cell size, diameter and perigonadal fat-pad weight (Fig. 2g, h), and liver lipid deposition (Fig. 2i, Supplementary Fig. 1b). In addition, we found that chronic HFD (12 weeks) in mice conferred ATF3 overexpression in WAT and BAT, found only in wild-type but not ATF3−/− mice (Fig. 3a, Supplementary Fig. 1c), implying WAT and BAT act as a stress response organ to chronic HFD. Because obesity is closely associated with a state of chronic, low-grade inflammation in adipose tissues22, the expression of intercellular adhesion molecule 1 (ICAM-1) but not adiponectin was higher in WAT of ATF3−/− than wild-type mice (Fig. 3b, Supplementary Fig. 1d). Adipokines secreted from adipose tissue may have systemic effects and shift to pro-inflammation as adipose tissue expands during the development of obesity23. Accordingly, protein array and ELISA analysis of adipokines revealed higher serum levels of pro-inflammatory adipokines except for adiponectin from ATF3−/− than wild-type mice (Fig. 3c, d, Supplementary Fig. 1e). Further evidence of systemic metabolic involvement, including levels of tumor necrosis factor-α (TNFα), interleukin 6 (IL-6), and inducible nitric oxide synthase (iNOS), were also higher in liver tissue of ATF3−/− than wild-type mice by real-time PCR analysis (Fig. 3e). These results from immunofluorescence, protein array and real-time PCR analyses all showed that ATF3 may inhibit the secretion of inflammatory hormones in adipose tissue.

Loss of ATF3 in mice aggravated high fat diet (HFD)-induced obesity and metabolic dysfunction. ATF3 gene-deleted mice (ATF3−/−) and their wild-type littermates (WT) fed a normal diet (ND) or a HFD for 22 or 14 weeks, respectively. Measurement was performed after 12 weeks of HFD feeding for both groups. a Body weight of wild-type (WT) and ATF3−/− mice fed a normal diet (ND) or HFD and body image. b Body composition of wild-type and ATF3−/− mice. Proportion of body fat and lean mass as a percentage of their respective body weights. c Serum triglycerides (TG) level. d Glucose tolerance test. e Insulin tolerance test. f White adipose tissue (WAT) and brown adipose tissue (BAT) fat-pad weights. g Representative H&E staining of inguinal WAT (iWAT), epididymal WAT (eWAT), and BAT. h Adipocyte diameter (µm), size (μm2), number per area (mm2), and perigonadal fat pad weight per body weight (g). i Oil-red O staining in liver. For a, n = 3 per group (ND); wild-type, HFD (n = 4), ATF3−/−, HFD (n = 9). For b, n = 12 per group. For c, n = 5 per group. For d, wild-type (n = 4), ATF3−/− (n = 5). For e, n = 3 per group. For f, wild-type (n = 3), ATF3−/− (n = 3 in 6-week HFD; n = 7 in 12-week HFD; n = 6 in 16-week HFD). For g–i, n = 3 per group, except wild-type (n = 8), ATF3−/− (n = 9) in % of perigonadal fat pads weight/body weight. Scale bar for images g: 200 µm; i: 100 µm. Data are mean ± SEM. and *p < 0.05 compared to wild-type

Loss of ATF3 aggravated the expression of inflammation-related genes in HFD-induced obese mice. a ATF3 protein level in iWAT and BAT of wild-type and ATF3−/− mice after HFD feeding for 12 weeks. b Representative immunofluorescence images of adiponectin (red IF) and ICAM-1 (green IF) in wild-type and ATF3−/− mice. Yellow scale bar indicated the size of adipocyte tissues. c Serum protein levels of adipokine and inflammation-related genes in wild-type and ATF3−/− mice after HFD feeding for 8 weeks by adipokine assays; Gel-Pro Analyzer software was used for densitometry of blots. d Serum protein level of adiponectin, ICAM-1 and resistin by ELISA assays in wild-type and ATF3−/−mice after HFD feeding for 8 weeks. e Quantified real-time PCR analysis of mRNA levels of iNOS, IL-6, and TNFα in livers of wild-type and ATF3−/− mice. For a–c, n = 3 per group. For d, wild-type (n = 4 in adiponectin; n = 5 in ICAM1; n = 8 in resistin), ATF3−/− (n = 7 in adiponectin; n = 5 in ICAM1; n = 7 in resistin). For e, n = 6 per group. Scale bar for image b: 50 µm. Data are mean ± SEM; *p < 0.05 compared to wild-type
ATF3 ameliorated HFD-induced metabolic dyshomeostasis
As a complementary approach to our knockout mouse studies, we performed phenotype-rescue studies in which ATF3−/− mice received adeno-associated virus (AAV)-mediated gene transfer of ATF3 (AAV8-ATF3)24,25. We found first that ATF3 expression in adipocytes was restored by AAV8-ATF3 injection (Supplementary Fig. 2, Supplementary Fig. 1f). Next, 12 weeks after intravenously injecting HFD-fed ATF3−/− mice with AAV8-ATF3, body weight, serum TG levels, glucose intolerance, and insulin resistance were lower than in ATF3−/− mice injected with AAV8-GFP (Fig. 4a–d). In addition, peri-gonadal fat-pad weight per body weight, adipocyte diameter, and adipocyte size were lower with AAV8-ATF3 than AAV8-GFP injection (Fig. 4e, f). These results suggest that ATF3 is a key regulator in HFD-induced obesity and related forms of metabolic dyshomeostasis.

Adeno-associated virus 8 (AAV8)-mediated expression of ATF3 reversed metabolic dysfunction in ATF3−/− mice. Analysis of mice fed an HFD for 12 weeks: untreated wild-type mice, AAV8-GFP–treated ATF3−/− mice (AAV8-GFP- ATF3−/−), or AAV8-ATF3-treated ATF3−/− mice (AAV8-ATF3-ATF3−/−). a Body weights. b Serum TG level. c Glucose tolerance test. d Insulin tolerance test. e Representative H&E staining of epididymal WAT. f Adipocyte diameter (μm), size (μm2), number per area (mm2), and perigonadal fat pad weight per body weight (g). For a, n = 3 per group. For b, n = 5 per group. For c–e, n = 3 per group. For f, n = 3 per group, except wild-type (n = 5), AAV8-GFP-ATF3−/− (n = 5), AAV8-ATF3-ATF3−/− (n = 5) in % of perigonadal fat pads weight/body weight. Scale bar for image e: 50 µm. Data are mean ± SEM; #p < 0.05 compared to wild-type. *p < 0.05 AAV8-GFP-ATF3−/− vs. AAV8-ATF3-ATF3−/−
ATF3 −/− mice impaired WAT/BAT balance and thermoregulation
In mice, induction of heat production in BAT helps prevent fluctuations in body temperature upon cold exposure. We checked whether ATF3−/− mice fed with a HFD affected their WAT/BAT depot distribution, energy expenditure, and thermoregulation, thus resulting in metabolic dysregulation. ATF3−/− mice showed increased weight in inguinal WAT (iWAT), mesenteric WAT (mWAT), and retroperitoneal WAT (rWAT) white fat depots, with no difference in BAT or epididymal WAT (eWAT) (Fig. 5a). In addition, the expression of adipogenesis and lipogenesis biomarkers, including C/EBPα, c/EBPβ, PPARγ1, PPARγ2, fatty acid binding protein 4 (FABP4), resistin and ChREBP was elevated in iWAT of ATF3−/− mice (Fig. 5b). The expression of some adipogenesis and lipogenesis biomarkers, including adiponectin, perilipin 1/2, ACC1/2, FAS, and SCD1, was not altered (Fig. 5b). In contrast, the expression of selected markers of WAT browning and BAT activation such as UCP1, CIDEA, and CD137 in iWAT (Fig. 5c) and UCP1, PGC1α, Prdm16, Zic1, and Elovl3 in BAT (Fig. 5d) was lower in ATF3−/− than wild-type mice. Furthermore, the protein levels of ChREBP, SCD1 and adiponectin in iWAT did not differ between ATF3−/− and wild-type mice (Fig. 5e, Supplementary Fig. 1g). Conversely, UCP1 protein was lower in iWAT (Fig. 5e, Supplementary Fig. 1g) but not BAT (Fig. 5f, Supplementary Fig. 1h) in ATF3−/− than wild-type mice. As an alternative approach to ATF3−/− mice, restoration of ATF3 could inhibit both ChREBP and SCD1 protein levels (Supplementary Fig. 3, Supplementary Fig. 1i). These results suggest that the loss of ATF3 in vivo affected the WAT/BAT balance and normal BAT activation. Moreover, whitening of BAT was greater for ATF3−/− than wild-type mice (Fig. 2g), which was consistent with the downregulation of mRNA levels of BAT/beige markers in BAT of ATF3−/− compared to wild-type mice. These results indicate that ATF3 may play an important role in promoting the white fat phenotype to brown/beige fat phenotype. Interestingly, positive correlations between ATF3 and HSL and CIDEA were observed in patients by using GEO and the GDS3679 dataset (Supplementary Fig. 4)19. The dysregulated balance of WAT/BAT seen in ATF3−/− mice was also associated with impaired thermoregulation under acute cold stress (Fig. 6a). We found that, even when the body weight differences between ATF3−/− and wild-type mice was not obvious 9 weeks after HFD feeding (Fig. 6b), the total body oxygen consumption and heat production were lower and respiratory exchange ratio was higher in ATF3−/− than wild-type mice (Fig. 6c–e). These data implied a decrease in the use of fat (versus carbohydrate) for heat production and confirm that ATF3 ablation in mice modulated adipose tissue and mitochondria adaptive gene programming, thereby affecting the WAT/BAT balance and impairing energy expenditure and thermoregulation.

ATF3−/− mice showed dysregulated WAT/BAT balance. Analysis of ATF3−/− and wild-type mice after 12 weeks of HFD feeding. a Weights of brown adipose tissue (BAT) and white adipose tissue (WAT) in individual depots including inguinal WAT (iWAT), epididymal WAT (eWAT), mesenteric WAT (mWAT), and retroperitoneal WAT (rWAT) fat pads. b Analysis of gene expression of adipogenic, lipogenic, and lipolytic genes in iWAT. c Analysis of gene expression of brown (BAT), beige (Bei), mitochondria (Mito), and β-oxidation (β-oxi) markers in iWAT. d Analysis of expression of brown/mitochondria/β-oxidation markers in BAT. e Protein levels of ChREBP, SCD1, UCP1 and adiponectin in iWAT. f Protein level of UCP1 in BAT. For a, wild-type (n = 8), ATF3−/− (n = 9). For b, n = 3 per group. For c, n = 4 per group. For d, n = 6 per group. For e, f, n = 3 per group. Data are mean ± SEM; *p < 0.05 compared to wild-type

ATF3−/− mice showed impaired energy metabolism and thermoregulation. a Body and rectal temperature during acute cold exposure. b Body weight after 9 weeks of HFD feeding. c Measurement of oxygen consumption levels, d respiratory exchange ratio (RER), and e energy expenditure in ATF3−/− and wild-type mice after 9 weeks of HFD feeding. For a, wild-type (n = 6), ATF3−/− (n = 5). For b, wild-type (n = 7), ATF3−/− (n = 9). For c–e, wild-type (n = 4), ATF3−/− (n = 5). Data are mean ± SEM; *p < 0.05 compared to wild-type
ATF3 regulated adipocyte browning via ChREBP-SCD1 pathway
To clarify the molecular mechanism underlying the regulation of lipogenesis/lipolysis and energy expenditure in white adipocytes by ATF3, we stably overexpressed ATF3 in 3T3-Ll cells. Overexpression of ATF3 decreased (>80%) oil droplet deposition in 3T3-Ll cells after 8 days of differentiation (Supplementary Fig. 5), so normal adipogenesis was suppressed. Further examination of markers related to adipogenesis and lipogenesis, including PPARγ, c/EBPα, ACC1/2, ChREBP, and SCD1, showed reduced levels in ATF3-overexpressing cells26 (Fig. 7a, b). By contrast, the expression of genes involved in BAT/beige fat programs and β-oxidation, such as UCP1, PGC1α, Cpt1α and Mcad, was upregulated in ATF3-overexpressing cells (Fig. 7c, d). These data were consistent with our in vivo results that expression of adipogenesis and lipogenesis biomarkers was oppositely elevated in iWAT of ATF3−/− mice (Fig. 5b).

ATF3-overexpressing 3T3-L1 adipocytes showed suppression of lipogenesis/adipogenesis and activation of mitochondrial, brown or beige fat programs. a, b Real-time PCR analysis of mRNA levels of adipogenic, lipogenic, and lipolytic genes; c, d BAT, beige (Bei), mitochondria (Mito), and β-oxidation (β-oxi) genes after 2 and 8 days of differentiation in ATF3-overexpressing 3T3-L1 cells, normalized to GAPDH and relative to pcDNA control. e, f The expression level of ChREBPand FABP4 in iWAT of wild-type and ATF3−/− mice after 6, 8, and 16 weeks of HFD feeding. f The expression level of FABP4 in iWAT of wild-type and ATF3−/− mice after 6, 8, and 16 weeks of HFD feeding. g FABP4 promoter activity measured with or without overexpression of ATF3 in 3T3-L1 pre-adipocytes. h Overexpression of ATF3 repressed the ChREBP promoter activity of the p (−2980)/Luc reporter but not other reporters in 3T3-L1 pre-adipocytes. i The sequence of 3 potential binding sites for ATF3 in ChREBP promoter, including region #1 (–2810/–2803), region #2 (−2790/−2783) and region #3 (−2721/−2714) of the ChREBP locus. j Chromatin immunoprecipitation (ChIP) experiments with ATF3-specific antibody and primers to amplify region #1, region #2 and region #3 of the ChREBP locus, which contains one predicted ATF/CRE binding site in 3T3-L1 preadipocytes. k Real-time PCR analysis of gene levels of brown (BAT), mitochondrial (Mi), beige (Bei), and β-oxidation (β-oxi) genes in ATF3-overexpressing 3T3-L1 pre-adipocyte stable clone with or without Scd1 transfection. For a, b, n = 4 per group. For c, d, pc-DNA (n = 4), pc-DNA-ATF3 (n = 6). For e, n = 4 per group. For f, n = 6 per group. For g, h, n = 5 per group. For j, n = 3 per group. For k, control (n = 3), ATF3 (n = 4), ATF3 + SCD1 (n = 4). Data are mean ± SEM; *p < 0.05 compared to control. #p < 0.05 ATF3 vs. ATF3 + SCD1
ChREBP is a glucose-responsive transcription factor that drives de novo lipogenesis and increases adiposity and potentially worsens insulin resistance27,28. Therefore, we checked whether ChREBP is involved in the ATF3-dependent lipogenesis/lipolysis balance. The expressions of ChREBP (Fig. 7e) and FABP4 (Fig. 7f) were increased in ATF3−/− mice after a HFD for 6–16 weeks. To examine whether ATF3 could regulate ChREBP or FABP4 directly, we analyzed the proximal promoter regions of ChREBP and FABP4. Sequential 5′-deleted luciferase reporter constructs with different lengths of ChREBP and FABP4 promoter regions were created and expressed with and without ATF3 in 3T3-L1 pre-adipocytes. We found that FABP4 promoter activity was not repressed by ATF3 (Fig. 7g). Only the −2980 construct of ChREBP promoter was repressed by ATF3 (Fig. 7h), which suggested that the ChREBP promoter (from −2980 to −2700) is involved in the ATF3-dependent regulation of ChREBP. Furthermore, we identified three potential ATF3-binding sites (Fig. 7i). To confirm this finding, we used chromatin immunoprecipitation assay to examine whether ATF3 could bind to its potential binding sites upstream of the ChREBP promoter. ATF3 bound to site 1 but not sites 2 and 3 (Fig. 7j).
ChREBP can promote lipogenesis by directly regulating SCD129, and mice with Scd1 deletion show increased white adipocyte browning30. To check whether ATF3 activates white adipocyte browning by suppressing ChREBP–SCD1 signaling, we overexpressed SCD1 in ATF3-overexpressing 3T3-L1 cells and examined the expression of BAT/beige markers. SCD1 overexpression reduced the upregulation of BAT/beige markers, including UCP1, Zic1, CIDEA, and Tbx1, in ATF3-overexpressing 3T3-L1 cells (Fig. 7k). Thus, ATF3 can suppress adipocyte adipogenesis and lipogenesis while activating white adipocyte transdifferentiation by inhibiting ChREBP and SCD1.
Identification of the small-molecule ATF3-inducer ST32da
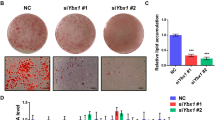
Overexpression of ATF3 decreased (>80%) oil droplet deposition, reduced the expression of adipogenic/lipogenic markers, and increased that of lipolytic markers in 3T3-Ll cells (Fig. 7a, b and Supplementary Fig. 5). These findings suggested that upregulation of ATF3 could be effective in treating or preventing obesity. We subcloned the ATF3 promoter into the pGL4.17-Luc luciferase reporter vector and selected stable clones overexpressing pGL4.17-ATF3 in 3T3-L1 pre-adipocytes (Fig. 8a, b). We then screened more than 400 small molecules from modified Chinese herbs single compound library at the National Research Institute of Chinese Medicine, Ministry of Health and Welfare and uncovered a single compound by using this stable clone with pGL4-ATF3 overexpression. tBHQ, a known ATF3 inducer, was a positive control. We found 19 compounds that increased ATF3 promoter activity and 44 that decreased it as compared with the tBHQ-treated positive control (Supplementary Table 1). Compound ST32da showed the highest luciferase activity and was chosen for further analysis. The chemical structure and purity of ST32da were characterized by mass spectrophotometry (Supplementary Fig. 6). Because we found that ATF3 inhibited ChREBP promoter activity (Fig. 7h, j), we attempted to clarify whether ST32da would produce similar effects. ST32da dose-dependently reduced ChREBP promoter activity within the p (−2980)/Luc reporter (Fig. 8c). Furthermore, ST32da dose-dependently reduced lipid accumulation in 3T3-L1 adipocytes (Fig. 8d). ST32da-treated 3T3-L1 cells showed increased ATF3 expression and high levels of lipolytic markers such as ATGL and MGL but downregulated expression of adipogenesis- and lipogenesis-related genes (Fig. 8e). In addition, ST32da-treated cells showed increased expression of BAT, beige, β-oxidation, and mitochondrial gene markers (Fig. 8f). Therefore, the effects of ST32da were similar to those of ATF3 overexpression in vitro.

Identification of ATF3 inducers and their functional assays. a Construction map of ATF3 promoter in pGL4.17 plasmid containing luciferase cassette. b Luciferase activity of stable clones of 3T3-L1 pre-adipocytes expressing pGL4.17-ATF3, with tBHQ as a positive control. c Luciferase activity measured in 3T3-L1 pre-adipocytes transfected with pGL4.17-ChREBP (p (−2980)/Luc reporter), then treated with ST32da or ST32db or ST32c. d Oil-red O staining in differentiated 3T3-L1 adipocytes with and without the ATF3 inducer ST32da (10 and 50 µM) for 8 days. e Real-time PCR analysis of mRNA levels of adipogenic, lipogenic, and lipolytic genes; f BAT, beige (Bei), mitochondria (Mito) and β-oxidation (β-oxi) genes with 2 and 8 days of ST32da treatment during 3T3-L1 differentiation normalized to GAPDH and relative to control. For b–d, n = 3 per group. For e, n = 4 per group. For f, n = 6 per group. Data are mean ± SEM; *p < 0.05 compared to control. #p < 0.05 compared to pGL4.17-ATF3
ST32da ameliorated HFD-induced metabolic dyshomeostasis
Having identified ST32da as the most effective ATF3 inducer, we then tested whether ST32da could influence the lipogenesis/lipolysis balance or WAT browning in vivo. HFD-fed obese mice received a daily intraperitoneal (i.p.) administration of ST32da, and food intake and body weight were measured weekly. The mice received ST32da at two different dosages (1 and 2 mg kg−1 per day), only higher dose (2 mg kg−1 per day) leading to a reduced body weight as compared with non-treated HFD-fed mice but with no significant difference in food intake (Fig. 9a).

ATF3 inducer, ST32da, protects against HFD-induced obesity and metabolic dysfunction by promoting browning in vivo. Analysis of wild-type mice fed a HFD for 12 weeks with or without i.p. ST32da 1 or 2 mg kg−1 per day. a Body weight and food intake. b Change in adipose tissue depot weight in BAT and WAT. c H&E staining of inguinal WAT, epididymal WAT, and BAT fat depots. d Glucose tolerance test (GTT). e Insulin tolerance test (ITT). f Real-time PCR analysis of mRNA levels of ATF3, c-Jun, PGC-1α and UCP1. g Real-time PCR analysis of mRNA levels of brown (BAT) and beige (Bei), mitochondria (Mito), and β-oxidation (β-oxi) genes in iWAT; h adipogenic, lipogenic, and lipolytic genes in iWAT; and i brown fat programs in BAT. For a, HFD (n = 10), HFD + 1 mg kg−1 per day ST32da (n = 8), HFD + 2 mg kg−1 per day ST32da (n = 8). For b, HFD (n = 9), HFD + 1 mg kg−1 per day ST32da (n = 7), HFD + 2 mg kg−1 per day ST32da (n = 6). For c, n = 3 per group. For d, e, HFD (n = 4), HFD + 1 mg kg−1 per day ST32da (n = 5), HFD + 2 mg kg−1 per day ST32da (n = 4). For f, g, n = 6 per group. For h, HFD (n = 6), HFD + 1 mg kg−1 per day ST32da (n = 5), HFD + 2 mg kg−1 per day ST32da (n = 6). For i, n = 6 per group. Data are mean ± SEM; *p < 0.05 compared to HFD group
Because ATF3−/− mice showed increased WAT (Fig. 2f), we then examined whether the role of ST32da in mitigating the weight gain was mediated by reducing WAT weight. The weight of iWAT, eWAT, mWAT, rWAT, and BAT was dose-dependently reduced in ST32da-treated mice after 12 weeks of treatment (Fig. 9b). Different adipose depots, including iWAT, eWAT, and BAT, showed decreases in adipocyte size and diameter after ST32da treatment (Fig. 9c). In addition, ST32da enhanced the glucose tolerance and insulin sensitivity of HFD-fed obese mice (Fig. 9d, e) while decreasing serum TG and creatinine levels (Supplementary Fig. 7a). However, serum BUN levels, liver function and weight, and cardiac/renal histology remained unchanged (Supplementary Fig. 7a–d).
Overexpression of ATF3 induced transdifferentiation of white adipocytes to beige/brown adipocytes in vitro (Fig. 7c, d). Similarly, ST32da treatment increased BAT/beige-related gene expression and decreased adipogenesis/lipogenesis gene expression in the iWAT of HFD-fed mice (Fig. 9f–h). Furthermore, the expression of BAT- and beige-related genes was enhanced by ST32da in BAT (Fig. 9i). To further support our hypothesis that ST32da exerts its beneficial effect through ATF3 signaling, we treated ATF3−/− mice with ST32da. ST32da treatment did not reduce the body weight of HFD-fed ATF3−/− mice (Supplementary Fig. 8a). Food intake was not affected in ST32da-treated ATF3−/− mice (Supplementary Fig. 8b). The beneficial effect of ST32da in ameliorating the metabolic disorder and in reducing the depot weight of iWAT, eWAT, mWAT, rWAT, and BAT was lost in these HFD-fed ATF3−/− mice (Supplementary Fig. 8c–e). In addition, the inhibitory effect of ST32da on SCD1 and ChREBP expression also vanished in these HFD-fed ATF3−/− mice (Supplementary Fig. 8f), and ST32da treatment appeared to have no effect on liver weight, adipocyte size and number in iWAT (Supplementary Fig. 8g, h) and serum adiponectin levels (Supplementary Fig. 9). Therefore, ST32da may exert its beneficial effects specifically via ATF3 activation and effectively protect against obesity in addition to lowering metabolic dysregulation in HFD-fed obese mice.
Oral ST32da administration conferred anti-obesity effects similar to those of orlistat, an FDA-approved oral anti-obesity drug. In general, oral drug therapy is more convenient than intravenous injection in terms of treating obesity and obesity-related chronic disease such as type 2 diabetes. Therefore, we checked whether oral administration of ST32da to HFD-fed obese mice was beneficial and compared the results with those of orlistat (a US FDA approved anti-obesity drug). ST32da and orlistat were administered orally at 50 mg kg−1 to HFD-fed mice three times per week for 12 weeks. Similar to our results with i.p. ST32da treatment, oral ST32da treatment ameliorated HFD-induced obesity without affecting food intake (Fig. 10a). The decrease in WAT depot weight in ST32da-treated mice was similar to the effect of orlistat on iWAT and better than the effect on eWAT, mWAT and rWAT (Fig. 10b). Along with fat mass reduction, size of adipocytes was decreased in both WAT and BAT (Fig. 10c, Supplementary Fig. 1j). Oral ST32da decreased TG level and liver weight but did not change the biochemical profiles of renal function and serum glucose concentration as compared with orlistat and HFD only (Fig. 10d, e). Oral ST32da also lowered glutamic oxaloacetic transaminase (GOT) and glutamate pyruvate transaminase (GPT) levels as compared with orlistat and HFD only (Fig. 10f).

Oral administration of ATF3 inducer, ST32da, is effective in preventing HFD-induced obesity. Analysis of wild-type mice fed a HFD for 16 weeks, either without or treated with oral orlistat (50 mg kg−1, three times per week) or oral ST32da (50 mg kg−1, three times per week). a Body weights and food intake. b Variation of adipose tissue depot weight in BAT and WAT. c H&E staining of inguinal WAT, epididymal WAT, and BAT fat depots. d Serum parameters. e Liver weight. f Liver function. g Real-time PCR analysis of mRNA levels of ATF3, c-Jun, PGC-1α and UCP1 in iWAT; h brown (BAT), beige (Bei), mitochondria (Mito), and β-oxidation (β-oxi) genes in iWAT; i adipogenic, lipogenic, and lipolytic genes in iWAT; j brown/mitochondria/β-oxidation markers in BAT. For a, ND (n = 2), HFD (n = 7), HFD + 50 mg kg−1 Orlistat (n = 7), HFD + 50 mg kg−1 ST32da (n = 7). For b, ND (n = 2), HFD (n = 5), HFD + 50 mg kg−1 Orlistat (n = 6), HFD + 50 mg kg−1 ST32da (n = 8). For c, n = 3 per group. For d, HFD (n = 8), HFD + 50 mg kg−1 Orlistat (n = 7), HFD + 50 mg kg−1 ST32da (n = 6). For e, ND (n = 2), HFD (n = 5), HFD + 50 mg kg−1 Orlistat (n = 6), HFD + 50 mg kg−1 ST32da (n = 8). For f, HFD (n = 8), HFD + 50 mg kg−1 Orlistat (n = 8), HFD + 50 mg kg−1 ST32da (n = 6). For g, n = 6 per group. For h, n = 4 per group. For i, HFD (n = 4), HFD + 50 mg kg−1 Orlistat (n = 4), HFD + 50 mg kg−1 ST32da (n = 5). For j, HFD (n = 6), HFD + 50 mg kg−1 Orlistat (n = 5), HFD + 50 mg kg−1 ST32da (n = 4). Data are mean ± SEM; *p < 0.05 compared to HFD; #p < 0.05 HFD + Orlistat vs. HFD + ST32da. GOT, glutamic oxaloacetic transaminase; GPT, glutamate pyruvate transaminase
Similar to i.p. administration of ST32da, oral ST32da activated browning-related gene expression in both iWAT (Fig. 10g, h) and BAT (Fig. 10j), both better than oral orlistat administration and suppressed adipogenesis/lipogenesis-related gene expression in iWAT (Fig. 10i), These data confirm that oral ST32da treatment could reduce body weight and WAT fat mass to a degree similar to that with orlistat.
Discussion
Previous studies of ATF3 mainly emphasized the diabetic syndrome and glucose homeostasis. In type 2 diabetes mellitus, ATF3 deficiency reduced serum insulin levels via reduced β-cell function31, and adenovirus-mediated ATF3 overexpression increased glucagon mRNA levels in αTC-1.6 cells32. Our results were consistent with the role of ATF3 in maintaining serum glucose homeostasis shown in these reports (Figs. 2d, e, 4c, d). In liver, ATF3 also mediates the inhibitory effects of ethanol on hepatic gluconeogenesis33. However, ATF3 regulation of the change in WAT and BAT in HFD-induced obesity and related metabolic disorders has not been investigated before. Here, we demonstrated that loss of ATF3 in vivo aggravated HFD-induced obesity and metabolic dysfunction in mice, with increased TG level, insulin resistance, and hepatic steatosis, along with loss of normal thermoregulation under cold stress and decreased energy expenditure. We then showed that inducing ATF3 has substantial metabolic benefits, and an ATF3 inducer, ST32da, could inhibit adipocyte lipogenesis/adipogenesis, enhance UCP1 expression, and promote white adipocyte browning in vitro, in addition to inhibiting obesity and restoring normal insulin sensitivity in HFD-fed obese mice. ATF3 plays a key role in metabolic regulation by directly suppressing the lipogenic gene ChREBP and enhancing adipocyte browning by inhibiting the ChREBP–SCD1 axis. Our findings confirm ATF3 inducers as promising drug candidates in treating and preventing obesity and metabolic dysfunction.
Increased visceral fat is well associated with increased metabolic disorder and morbidity and mortality from coronary heart disease, cancer, and diabetes34. Moreover, accumulation of visceral WAT owing to obesity may result in a stressed and dysfunctional state that leads to the release of pro-inflammatory factors such as IL-6 and TNFα, resulting in major complications such as metabolic syndrome, hepatic steatosis, neointima formation, and atherosclerosis35,36. Previous studies identified the anti-inflammatory, anti-apoptotic, and protective effects of ATF3 in various tissues under stress, including the brain, kidneys25, heart24, lungs37, and blood vessels38,39. ATF3 may have beneficial properties in terms of inhibiting obesity-induced cytokines in vivo by targeting muscle, adipocytes, or other organs. However, ATF3 was not found in skeletal muscle of HFD-fed obese animals31, and macrophage-specific ATF3-overexpressing transgenic mice did not show suppressed HFD-induced obesity40. Therefore, we hypothesized that ATF3 may regulate metabolic homeostasis in vivo by targeting adipocytes. Our data support this hypothesis that ATF3 regulates expression adipogenesis/lipogenesis genes in the models of ATF3-overexpressing pre-adipocytes and ATF3–/– mice (Figs. 5 and 7, Supplementary Fig. 5). In addition, an ATF3 inducer, ST32da, suppressed HFD-induced obesity in mice (Figs. 9 and 10). Jang et al. also indicated that ATF3 interacts with PPARγ and represses PPARγ-mediated transactivation in white adipocytes41. Our data also showed that ATF3 can promote white-to-brown adipocyte transdifferentiation to increase energy expenditure and reduce WAT depots, thus mitigating obesity.
The ATF3−/− mice fed a HFD in our study showed obesity with increased WAT weight in both subcutaneous (iWAT) and visceral depots (mWAT and rWAT) (Fig. 5a) and increased expression of many adipogenic/lipogenic genes in iWAT (Fig. 5b) In addition, the expression of browning and mitochondrial genes was reduced in the iWAT and BAT of obese ATF3−/− mice (Fig. 5c, d), likely because of the reduced transdifferentiation of WAT to BAT; therefore, ATF3 −/− mice showed a loss of normal thermoregulation with low body temperature under acute cold stress (Fig. 6a). In humans, the amount of BAT is also inversely proportional to body mass index and age42 and is barely detectable in obese patients as compared with normal-weight individuals of the same age43. Therefore, activating the limited BAT reserves that already exist in obese patients may be of limited therapeutic value. We speculated that ATF3 can induce WAT-to-BAT transdifferentiation and further increase BAT reserves. Our in vitro studies showed that ATF3 can induce UCP1, an adipocyte-browning indicator, and increase brown/beige-related gene expression during the early stage of 3T3-L1 cell differentiation (day 2), while repressing adipogenic/lipogenic gene expression during the late stage (day 8) (Fig. 7a–d), thereby reducing oil droplet formation (Supplementary Fig. 5b). Efficiently promoting white-to-brown transdifferentiation of adipocytes—a currently neglected topic in anti-obesity drug research—should be a primary area of future investigation.
Our in vitro data showed ATF3-overexpressing 3T3-L1 adipocytes with inhibited adipogenic/lipogenic gene expression in general (except C/EBPβ), whereas the expression of lipolytic genes on days 2 and 8 of differentiation was upregulated (Fig. 7a, b). The biphasic regulation of lipogenic or lipolytic pathways may be due to ATF3 regulating cell signalling in the context dependent manners. ATF3 homodimers have been found to act as transcriptional repressors in adipogenic/lipogenic regulation14,44; however, whether the ATF3 heterodimeric complex with c-Jun (or JunD) acts as a transcriptional activator of lipolytic pathways is still unclear. We found c-Jun expression reduced after i.p. administration of ST32da to mice (Fig. 9f) but no difference in c-Jun expression after oral ST32da treatment (Fig. 10g). In addition, ST32da had a similar effect in promoting lipolysis and repressing lipogenesis in vitro (Fig. 8e). ST32da also suppressed adipogenic/lipogenic gene expression and upregulated lipolytic gene expression (Fig. 9h) in adipose tissue as well as reduced WAT depots in both subcutaneous WAT (iWAT) and visceral WAT (eWAT, mWAT, and rWAT) in HFD-fed obese mice (Fig. 9b). Increased lipolysis in adipocytes may promote free-fatty acid release and worsen lipid accumulation in insulin-sensitive organs such as the liver and muscle, thus impairing insulin sensitivity45. However, this was not found with our ATF3 inducer in vivo. In our case, increased lipolysis was accompanied by WAT browning. BAT is an efficient modulator of triglyceridemia and considered a major plasma lipid-clearing organ in rodents46. Therefore, WAT browning can induce lipid clearance and promote UCP1-related mitochondrial and β-oxidation gene activation, thus lowering serum TG levels (Fig. 10d and Supplementary Fig. 7a) while increasing insulin sensitivity. Therefore, ATF3 inducers can induce insulin sensitivity and enhance body-weight loss by promoting a positive balance between both lipolysis/lipogenesis and BAT/WAT in adipocytes.
ChREBP deficiency reduced lipogenesis and glycolysis in C57BL/6J mice47. Recent studies showed that patients who take anti-diabetic or anti-lipid drugs, including metformin, atorvastatin, DHA/EPA, and 3-hydroxybutyrate, show reduced serum ChREBP levels48. Such drugs may exert their glucose- and lipid-lowering effect by modulating ChREBP trans-activity. Although ChREBP has been reported to link lipogenesis to insulin sensitivity in adipocytes49, whether ChREBP suppression would be beneficial in treating obesity is unclear. The ATF3−/− mice we studied showed increased gene expression of ChREBP in iWAT after a HFD for 6 weeks (Fig. 7e). These adipogenic/lipogenic gene levels were markedly suppressed in ATF3-overexpressing 3T3-L1 cells (Fig. 7a, b). Our in vitro promoter assay and ChIP assay further confirmed that ATF3 regulates ChREBP by directly binding to the promotor region and suppressing ChREBP expression.
Overexpression of ChREBP induces SCD1, the enzyme responsible for the conversion of saturated fatty acids to monounsaturated fatty acids29. Although Scd1 depletion can upregulate basal thermogenesis, thereby resulting in epididymal WAT browning30,50, Scd1 deletion in liver and/or adipose tissue alone is insufficient to protect mice against HFD-induced obesity51. Our results showed that ATF3 could suppress SCD1 expression (Figs. 7b and 9h). However, overexpression of SCD1 can inhibit ATF3-induced BAT/beige-related gene expression (Fig. 7k), so SCD1 may act downstream of the ATF3-ChREBP repression signalling, thus confirming that ATF3 promotes WAT browning and enhances energy expenditure by repressing the ChREBP–SCD1 axis.
Over the past few years, several molecules have been tested and marketed as anti-obesity drugs. They are of three classes: centrally acting medications impairing dietary intake (serotonergic drugs) and increasing proopiomelanocortin neuron activity; medications that act peripherally to impair dietary absorption (orlistat, which inhibits lipase); and medications that increase energy expenditure, whose effect is mediated by the central nervous system (mirabegron: beta 3 adrenergic receptor agonist). The ideal way may help increase energy expenditure but only if this effect can be achieved via a direct effect on peripheral tissues without involving the central nervous system. Therefore, developing a drug that increases even minor amounts of functional BAT in the human adipose tissue could be a new, valuable approach to treating and preventing obesity and its metabolic complications.
With our ATF3-specific promoter screening approaches, we isolated 19 compounds that upregulate ATF3 expression (Supplementary Table 1). In addition to ST32da, other compounds, such as ST32db and ST32c, inhibited ChREBP promoter activity (Fig. 8c). ST32da was given to the HFD-fed obese mice i.p. or orally. Both intraperitoneal and oral delivery of ST32da could reduce adipose depot weights in iWAT, mWAT, rWAT and eWAT (Figs. 9b and 10b). Because eWAT is the main visceral depot in mice and is more susceptible to developing chronic inflammation than mWAT or iWAT52, our results indicate that both i.p. and oral ST32da have similar anti-obesity effect on visceral WAT reduction. On comparing the results of oral ST32da and orlistat administration, we found that oral ST32da administration had a better anti-obesity effect than orlistat with prolonged HFD (more than 12 weeks) (Fig. 10a). In addition, suppression of adipogenic/lipogenic gene expression, WAT browning, and BAT activation were greater with oral ST32da than orlistat treatment (Fig. 10g–j). Oral ST32da treatment also lowered GOT/GPT serum levels most efficiently, so oral ST32da administration is safe and does not affect the liver or renal function (Fig. 10d–f). A recent study showed that sulfuretin, a known phytochemical ATF3 inducer, could also counteract weight gain and improve glucose tolerance in HFD fed mice53. Our drug, ST32da, similar to sulfuretin, had the specificity of ATF3 induction (Supplementary Fig. 8). However, ST32da and sulfuretin are distinctive chemical entities, both possess completely different structures. Therefore, whether ST32da may also activate BMP, mTOR, MAPK and Wnt/β-catenin signaling pathways like sulfuretin54 requires further investigation. Finally, ATF3 is an adaptive response transcription factor widely expressed in various organs24,25,37,38,39. In our study, we used traditional whole-body–knockout mice. Whether ATF3 adipocyte-specific knockout in mice receiving ST32da will have the same effect as in ATF3-knockout mice requires further exploration.
Interactions between secretory factors from adipose tissues (including adipokines and batokines) and the nervous system (including central nervous system, innervating adipose tissues) play key roles in maintaining energy metabolism and promoting survival in response to metabolic challenges55. Also, the hypothalamus plays important roles in regulating brown fat activity by regulating the sympathetic nervous system activity56. In pancreas- and hypothalamus-specific ATF3 knockout (PHT-ATF3-KO) mice, ATF3 played an important role in the control of glucose and energy metabolism by regulating agouti-related protein (Agrp), which increases food intake and reduces energy expenditure57. Thus, hypothalamic ATF3 is involved in adjusting glucose and energy metabolism by regulating the sympathetic nervous system activity in mice. However, i.p. or oral treatment with ST32da in mice could not increase thermoregulation under acute cold stress, which suggests that ST32da may not pass through the blood–brain barrier.
Strategies to enhance the fat-burning power of BAT in human adipose tissue are important in treating obesity and currently comprise a major research field for many laboratories. Our ongoing studies revealed that ST32da treatment inhibited human primary pre-adipocyte differentiation and promoted browning (Supplementary Fig. 10). In the future, subcutaneous WAT harvested from human tissue biopsies may be first treated with ST32da and then expanded and induced to differentiate into brown adipocytes before their implantation as an autologous transplantation, thus enhancing the energy expenditure and improving glucose metabolism and insulin resistance in patients with obesity. In conclusion, ST32da, as an ATF3 inducer, could restore the positive balance of lipolysis/lipogenesis and increase the amount of functional BAT without central nervous system repression on dietary intake. ATF3 inducer could be a novel class of anti-obesity drug to treat diet-induced obesity and related metabolic disorders.
Methods
Animal studies
ATF3–/– mice were kindly provided by Dr. Tsonwin Hai as described58. The ATF3–/– allele was backcrossed into C57BL/6 mice for at least seven generations before the experiments. To assess metabolic parameters, 4-week-old male wild-type and ATF3−/− mice were fed a chow diet (normal diet, ND, 8 % kcal from fat) or high-fat diet (HFD, 45% kcal from fat). Body weight, serum TG level, glucose tolerance, and insulin sensitivity were measured. To monitor the effects of i.p. or oral ST32da treatment, 6-week-old wild-type mice were fed a HFD with or without i.p. ST32da or oral ST32da for 12 and 16 weeks. Body weights and food intake were measured every week throughout the experiments. Metabolic parameters such as insulin sensitivity, glucose tolerance and liver weight were measured at the end of the treatment. Body composition was measured by using a TD-NMR analyzer (Minispec LF-50; Bruker Optics). All procedures were performed according to the protocols approved by the Institutional Animal Care and Utilization Committee, Academia Sinica, Taipei, Taiwan.
Glucose tolerance test and insulin tolerance test
Glucose tolerance test, mice were fasted overnight for 16 h before receiving an i.p. administration of 1.5 g glucose kg−1 body weight in saline. Plasma glucose levels were measured from the tail blood at 0, 15, 30, 45, 60, and 120 min after glucose injection. Mice underwent insulin tolerance test after 4 h of fasting. Mice were i.p. injected with 0.75 Unit insulin kg−1 body weight in saline. Plasma glucose levels were measured from the tail blood at 0, 15, 30, 45, and 60 min after insulin injection. Plasma glucose levels collected from tail veins were determined by using a commercially available glucose meter (OneTouch Ultra blood glucose meter, LifeScan, Milpitas, CA).
Cold exposure
For testing resistance to cold exposure, mice were individually caged and exposed to 4 °C with free access to water. Core body temperature was monitored by using a rectal thermometer (KN-91, NATSUME) at the beginning and every 2 h after the start of cold exposure. Images were taken after cold exposure with use of an infrared thermographic camera (F30s, NEC Avio Infrared Technologies, Tokyo)
Indirect calorimetry measurements
Mice were measured after 9 weeks on a HFD by using an 8chamber LabMaster Calorimetry Module (TSE‐Systems GmbH) with one mouse per chamber. After acclimatization for 72 h, the O2 consumption (VO2, ml h−1 kg−1), CO2 production (VCO2, ml h−1 kg−1), respiratory quotient (ratio of VCO2/VO2) and energy expenditure were determined. VO2 and VCO2 were recorded every 1 for 48 h. Energy expenditure was calculated as the product of the calorific value of oxygen (3.815 + 1.232 × respiratory quotient) and the volume of O2 consumed.
Analysis of serum parameters
The levels of serum BUN, creatinine, glucose, TG, GOT, and GPT were measured by sing Spotchem EZ SP 4430 (ARKRAY, Kyoto, Japan).
Adipokine arrays
The serum levels of adipokines were analyzed by using Proteome Profiler Mouse Adipokine Array Kit (R&D Systems, Minneapolis, MN, USA). Blots were developed by using an enhanced chemiluminescence and a Fuji Film Imaging System (Application Note LAS-4000; Fuji film, Tokyo). Densitometry of blots involved use of the Gel-Pro Analyzer software.
Histology, adipocyte size and adipocyte number
For hematoxylin and eosin (H&E) staining, dissected tissues of kidney, heart and adipose tissue were fixed in 4% paraformaldehyde overnight at 4 °C and paraffin-fixed before sectioning and staining. The stained sections of WAT were analyzed by using Image J. For adipocyte size measurements, 20 consecutive fat cells of the gonadal fat pad from mice were selected for measurement of area. The adipocyte number of the gonadal fat pad was calculated as the fat pad volume divided by the average fat cell volume.
Production of recombinant AAV carrying ATF3
This procedure was described previously24. Full-length ATF3 was obtained by PCR amplification from a human complementary cDNA library and cloned into XbaI/HindIII sites of the pAAV-MCS vector. A three-plasmid cotransfection method was used to produce the AAV virus59. The plasmids used in transfection included the AAV-CMV-ATF3 plasmid with the gene driven by the CMV promoter, which carried the promoter-driven transgene flanked by AAV inverted terminal repeats; the helper plasmid, which contained helper genes from the adenovirus; and the pseudotyped AAV packaging plasmid containing the AAV8 serotype capsid gene coupled with the AAV2 rep gene. The AAV8-GFP (control) or AAV8-ATF3 (experimental group) was purified twice by caesium chloride gradient ultracentrifugation, and the titers of vector genome particles were determined as described60. The recombinant viruses with 1 × 1012 viral particles in 30 µl phosphate buffered saline (PBS) were injected into a mouse-tail vein after 5 and 7 weeks of HFD feeding.
Plasmid constructs
The ATF3 sequence was subcloned into pcDNA3.1. The −3.6 kb ATF3-luc was generated by PCR amplification of the target region from genomic DNA and inserting into KpnI/NheI sites of the pGL4.17 vector (Promega). The 2.9-, 2.7-, 2.5-, 2-, and 1-kb ChREBP-luc promoters were generated by PCR amplification of the target region from genomic DNA and inserting into KpnI/EcoRV sites of the pGL4.17 vector (Promega). The 2.9-, 2- and 1-kb FABP4-luc promoters were generated by PCR amplification of the target region from genomic DNA and inserting into KpnI/NheI sites of the pGL4.17 vector (Promega). The SCD1 expression vector was from GeneCopoeia.
Cell culture
3T3-L1 cells and human pre-adipocyte cells were used in this study. 3T3-L1 cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% calf serum, 100 U ml−1 penicillin, and 0.1 mg ml−1 streptomycin. Human pre-adipocyte cells were maintained in pre-adipocyte growth medium. For transient transfection assays, cells were seeded at 1.15 × 104 cells cm−2. After 24 h, cells were transiently transfected with pcDNA-ATF3 and shRNA-ATF3 by using Maestrofectin transfection reagent (Omics Bio). After 48-h incubation, transfection efficiency was determined by real-time PCR and western blot analysis. For 3T3-L1 differentiation experiments, 2 days after 3T3-L1 cells reached confluence (referred to as day 0), 3T3-L1 cells were induced to differentiate in culture medium supplemented with 5 μg ml−1 insulin, 0.5 mM 3-isobutyl-1-methylxanthine, 1 μM dexamethasone for 2 days and then maintained in culture medium supplemented with 5 μg ml−1 insulin. For human pre-adipocyte differentiation experiments, 2 days after the human pre-adipocyte cells reached confluence (referred to as day 0), human pre-adipocytes were induced to differentiate in adipocyte differentiation medium for 14 days. For analyzing the effect of ST32da on cell differentiation, 3T3-L1 cells were treated with ST32da for 8 days of differentiation and human pre-adipocytes were treated with ST32da 14 days of differentiation.
Oil-red O staining
After 8 and 14 days of pre-adipocyte differentiation in 3T3-L1 cells and human preadipocytes, respectively, differentiated adipocytes were washed twice with PBS and fixed for 1 h in 10% formalin. Cells were then stained with Oil-red O working solution for 1 h. Cells were washed four times with distilled water before microscopy analysis. Stained Oil-red O was eluted with 100% isopropanol (v/v) and quantified by measuring the optical absorbance at 500 nm.
Western blot analysis
Cells were washed in cold PBS and lysed in RIPA buffer on ice. Adipose tissues (100 mg) were homogenized by using a tissue ruptor (Qiagen). Cell lysates and tissue homogenates were centrifuged at 14,000 rpm for 15 min at 4 °C and the infranatant was collected. Protein concentrations were determined by Bradford method. Proteins were resolved by SDS-PAGE and transferred to a PVDF membrane (Merck Millipore). The following antibodies were used: anti-ATF3 (cat# sc-188; 1:500; Santa Cruz Biotechnology), anti-UCP1 (cat# ab10983; 1:1000; Abcam), anti-ChREBP (cat# 81958; 1:1000; Abcam), anti-SCD1 (cat# 19862; 1:1000; Abcam), anti-adiponectin (cat# ab22554; 1:1000; Abcam), anti-actin (cat# sc-47778; Santa Cruz Biotechnology), anti-GAPDH (cat# ab8245; 1:10000; Abcam).
Real-time PCR
Total RNA was extracted from cultured cells or adipose tissues by using Trizol reagent (Invitrogen) and RNA was reverse transcribed to cDNA with the iScript cDNA Synthesis Kit (Biorad). Real-time quantitative PCR analysis involved using the ABI PRISM 7700 Sequence Detection System (Applied Biosystems, Grand Island, NY) with SYBR green (Biorad). Sequences of primers used for real-time PCR are in Supplementary Tables, 2 and 3.
In vitro promoter assay
For in vitro promoter assay, 3T3-L1 cells were transfected with or without pGL4.17-ChREBP (or pGL4.17-FABP4) and pcDNA-ATF3 for 24 h before harvesting for luciferase assay. Firefly luciferase activity was determined and normalized to Renilla luciferase activity. Data shown represent mean and SEM from three independent experiments.
Stable transfection
pcDNA-ATF3 and pGL4.17-ATF3 vectors were stably transfected into 3T3-L1 pre-adipocytes by using Maestrofectin transfection reagent (Omics Bio). Transfected cells were selected in 1000 µg ml−1 G418 for 4 weeks, then several stable clones were established. pcDNA-ATF3 stable clones were confirmed by real-time quantitative PCR and western blot analysis. The pGL4.17-ATF3 stable clones were confirmed by luciferase activity and compared to pGL4.17 control stable clones. Furthermore, pGL4.17-ATF3 stable clones were used for further drug screening.
Chromatin immunoprecipitation assay
3T3-L1 cells were fixed in 1% formaldehyde and chromatin immunoprecipitation (ChIP) was performed according to the Upstate protocol (Millipore). Chromatin was immunoprecipitated with anti-ATF3 antibody (Santa Cruz Biotechnology). The purified DNA was detected by standard PCR. Primers are in Supplementary Table 4.
Measure serum levels of adiponectin, ICAM1, and resistin
Serum levels of adiponectin, ICAM1 and resistin were measured using an ELISA (Abcam, cat# ab226900 for mouse adiponectin ELISA kit; cat# ab252355 for mouse ICAM1 ELISA kit; cat# ab205574 for mouse resistin ELISA kit) according to the manufacturer’s instructions.
Microarray data sources
We use microarray experiment data obtained from GEO (Gene Expression Omnibus, https://www.ncbi.nlm.nih.gov/geo/) at NCBI, including nonalcoholic fatty liver disease (accession no. GDS4881, liver from obese individuals) and morbidly obese subjects (accession no. GDS3679, adipose tissue from obese individuals; accession no. GDS368, muscle tissue from obese individuals; accession no. GSE69039, monocytes from obese individuals).
Statistical analyses
Values are expressed as means ± SEM from at least three experiments. The statistical significance was analyzed usingANOVA followed by the Tukey test for the in vivo experiments. Samples from the patients were analyzed using the rank sum test. A value of p < 0.05 was considered statistically significant.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All data generated or analyzed during this study are included in this published article. Full blots are shown in Supplementary Information. The source data underlying plots presented in main figures are shown in Supplementary Data 1.
References
Tang, Q. Q. & Lane, M. D. Adipogenesis: from stem cell to adipocyte. Annu. Rev. Biochem. 81, 715–736 (2012).
Kajimura, S. & Saito, M. A new era in brown adipose tissue biology: molecular control of brown fat development and energy homeostasis. Annu Rev. Physiol. 76, 225–249 (2014).
Rothwell, N. J. & Stock, M. J. A role for brown adipose tissue in diet-induced thermogenesis. Nature 281, 31–35 (1979).
Bachman, E. S. et al. betaAR signaling required for diet-induced thermogenesis and obesity resistance. Sci. (New Y., N. Y.) 297, 843–845 (2002).
Feldmann, H. M., Golozoubova, V., Cannon, B. & Nedergaard, J. UCP1 ablation induces obesity and abolishes diet-induced thermogenesis in mice exempt from thermal stress by living at thermoneutrality. Cell Metab. 9, 203–209 (2009).
Frontini, A. & Cinti, S. Distribution and development of brown adipocytes in the murine and human adipose organ. Cell Metab. 11, 253–256 (2010).
Rosen, E. D. & Spiegelman, B. M. What we talk about when we talk about fat. Cell 156, 20–44 (2014).
Frontini, A. et al. White-to-brown transdifferentiation of omental adipocytes in patients affected by pheochromocytoma. Biochim Biophys. Acta 950-959, 2013 (1831).
Sidossis, L. & Kajimura, S. Brown and beige fat in humans: thermogenic adipocytes that control energy and glucose homeostasis. J. Clin. Investig. 125, 478–486 (2015).
Townsend, K. & Tseng, Y. H. Brown adipose tissue: recent insights into development, metabolic function and therapeutic potential. Adipocyte 1, 13–24 (2012).
Lidell, M. E., Betz, M. J. & Enerback, S. Brown adipose tissue and its therapeutic potential. J. Intern Med. 276, 364–377 (2014).
Saito, M. Human brown adipose tissue: regulation and anti-obesity potential. Endocr. J. 61, 409–416 (2014).
Whittle, A. J., Lopez, M. & Vidal-Puig, A. Using brown adipose tissue to treat obesity - the central issue. Trends Mol. Med. 17, 405–411 (2011).
Jang, M. K., Kim, C. H., Seong, J. K. & Jung, M. H. ATF3 inhibits adipocyte differentiation of 3T3-L1 cells. Biochemical Biophysical Res. Commun. 421, 38–43 (2012).
Kim, H. B. et al. NFATc4 and ATF3 negatively regulate adiponectin gene expression in 3T3-L1 adipocytes. Diabetes 55, 1342–1352 (2006).
Jang, M. K., Son, Y. & Jung, M. H. ATF3 plays a role in adipocyte hypoxia-mediated mitochondria dysfunction in obesity. Biochem. Biophys. Res. Commun. 431, 421–427 (2013).
Wu, S. et al. Effect of obesity on the association between ATF3 gene haplotypes and C-reactive protein level in Taiwanese. Clin. Chim. acta; Int. J. Clin. Chem. 412, 1026–1031 (2011).
Ahrens, M. et al. DNA methylation analysis in nonalcoholic fatty liver disease suggests distinct disease-specific and remodeling signatures after bariatric surgery. Cell Metab. 18, 296–302 (2013).
MacLaren, R. E., Cui, W., Lu, H., Simard, S. & Cianflone, K. Association of adipocyte genes with ASP expression: a microarray analysis of subcutaneous and omental adipose tissue in morbidly obese subjects. BMC Med. Genom. 3, 3 (2010).
Park, J. J., Berggren, J. R., Hulver, M. W., Houmard, J. A. & Hoffman, E. P. GRB14, GPD1, and GDF8 as potential network collaborators in weight loss-induced improvements in insulin action in human skeletal muscle. Physiol. Genom. 27, 114–121 (2006).
Jung, U. J., Seo, Y. R., Ryu, R. & Choi, M. S. Differences in metabolic biomarkers in the blood and gene expression profiles of peripheral blood mononuclear cells among normal weight, mildly obese and moderately obese subjects. Br. J. Nutr. 116, 1022–1032 (2016).
Trayhurn, P. & Wood, I. S. Adipokines: inflammation and the pleiotropic role of white adipose tissue. Br. J. Nutr. 92, 347–355 (2004).
Borgeson, E. et al. Lipoxin A4 attenuates adipose inflammation. FASEB J.: Off. Publ. Federation Am. Societies Exp. Biol. 26, 4287–4294 (2012).
Lin, H. et al. Activating transcription factor 3 protects against pressure-overload heart failure via the autophagy molecule Beclin-1 pathway. Mol. Pharmacol. 85, 682–691 (2014).
Chen, H. H. et al. Urinary miR-16 transactivated by C/EBPbeta reduces kidney function after ischemia/reperfusion-induced injury. Sci. Rep. 6, 27945 (2016).
Ferrand, N. et al. WISP1/CCN4 inhibits adipocyte differentiation through repression of PPARgamma activity. Sci. Rep. 7, 1749 (2017).
Meng, J. et al. Identification of HNF-4alpha as a key transcription factor to promote ChREBP expression in response to glucose. Sci. Rep. 6, 23944 (2016).
Yu, Y., Maguire, T. G. & Alwine, J. C. ChREBP, a glucose-responsive transcriptional factor, enhances glucose metabolism to support biosynthesis in human cytomegalovirus-infected cells. Proc. Natl Acad. Sci. USA 111, 1951–1956 (2014).
Benhamed, F. et al. The lipogenic transcription factor ChREBP dissociates hepatic steatosis from insulin resistance in mice and humans. J. Clin. Investig. 122, 2176–2194 (2012).
Sampath, H. et al. Skin-specific deletion of stearoyl-CoA desaturase-1 alters skin lipid composition and protects mice from high fat diet-induced obesity. J. Biol. Chem. 284, 19961–19973 (2009).
Zmuda, E. J. et al. The roles of ATF3, an adaptive-response gene, in high-fat-diet-induced diabetes and pancreatic beta-cell dysfunction. Mol. Endocrinol. (Baltim., Md.) 24, 1423–1433 (2010).
Lee, Y. S. et al. ATF3 expression is induced by low glucose in pancreatic alpha and beta cells and regulates glucagon but not insulin gene transcription. Endocr. J. 61, 85–90 (2014).
Tsai, W. W. et al. ATF3 mediates inhibitory effects of ethanol on hepatic gluconeogenesis. Proc. Natl Acad. Sci. USA 112, 2699–2704 (2015).
Despres, J. P. & Lemieux, I. Abdominal obesity and metabolic syndrome. Nature 444, 881–887 (2006).
White, U. A. & Tchoukalova, Y. D. Sex dimorphism and depot differences in adipose tissue function. Biochim Biophys. Acta 1842, 377–392 (2014).
Smith, U. Abdominal obesity: a marker of ectopic fat accumulation. J. Clin. Investig. 125, 1790–1792 (2015).
Lai, P. F. et al. ATF3 Protects against LPS-Induced Inflammation in Mice via Inhibiting HMGB1 Expression. Evid. Based Complement Alternat. Med. 2013, 716481 (2013).
Teasdale, J. E. et al. Cigarette smoke extract profoundly suppresses TNFalpha-mediated proinflammatory gene expression through upregulation of ATF3 in human coronary artery endothelial cells. Sci. Rep. 7, 39945 (2017).
De Nardo, D. et al. High-density lipoprotein mediates anti-inflammatory reprogramming of macrophages via the transcriptional regulator ATF3. Nat. Immunol. 15, 152–160 (2014).
Suganami, T. et al. Activating transcription factor 3 constitutes a negative feedback mechanism that attenuates saturated Fatty acid/toll-like receptor 4 signaling and macrophage activation in obese adipose tissue. Circulation Res. 105, 25–32 (2009).
Jang, M. K. & Jung, M. H. ATF3 inhibits PPARgamma-stimulated transactivation in adipocyte cells. Biochem. Biophys. Res. Commun. 456, 80–85 (2015).
Virtanen, K. A. et al. Functional brown adipose tissue in healthy adults. N. Engl. J. Med. 360, 1518–1525 (2009).
Vijgen, G. H. et al. Brown adipose tissue in morbidly obese subjects. PloS ONE 6, e17247 (2011).
Fernandez-Verdejo, R. et al. Activating transcription factor 3 attenuates chemokine and cytokine expression in mouse skeletal muscle after exercise and facilitates molecular adaptation to endurance training. FASEB J.: Off. Publ. Federation Am. Societies Exp. Biol. 31, 840–851 (2017).
Morigny, P., Houssier, M., Mouisel, E. & Langin, D. Adipocyte lipolysis and insulin resistance. Biochimie 125, 259–266 (2016).
Calderon-Dominguez, M. et al. Fatty acid metabolism and the basis of brown adipose tissue function. Adipocyte 5, 98–118 (2016).
Iizuka, K., Bruick, R. K., Liang, G., Horton, J. D. & Uyeda, K. Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc. Natl Acad. Sci. USA 101, 7281–7286 (2004).
Iizuka, K. The transcription factor carbohydrate-response element-binding protein (ChREBP): a possible link between metabolic disease and cancer. Biochim. Biophys. Acta Mol. Basis Dis. 1863, 474–485 (2017).
Baraille, F., Planchais, J., Dentin, R., Guilmeau, S. & Postic, C. Integration of ChREBP-mediated glucose sensing into whole body metabolism. Physiol. (Bethesda, Md.) 30, 428–437 (2015).
Lee, S. H. et al. Lack of stearoyl-CoA desaturase 1 upregulates basal thermogenesis but causes hypothermia in a cold environment. J. lipid Res. 45, 1674–1682 (2004).
Flowers, M. T., Ade, L., Strable, M. S. & Ntambi, J. M. Combined deletion of SCD1 from adipose tissue and liver does not protect mice from obesity. J. lipid Res. 53, 1646–1653 (2012).
Mulder, P. et al. Reduction of obesity-associated white adipose tissue inflammation by rosiglitazone is associated with reduced non-alcoholic fatty liver disease in LDLr-deficient mice. Sci. Rep. 6, 31542 (2016).
Kim, S. et al. Atf3 induction is a therapeutic target for obesity and metabolic diseases. Biochem. Biophys. Res. Commun. 504, 903–908 (2018).
Auh, Q. S. et al. Sulfuretin promotes osteoblastic differentiation in primary cultured osteoblasts and in vivo bone healing. Oncotarget 7, 78320–78330 (2016).
Zhu, Q., et al. Neuroendocrine regulation of energy metabolism involving different types of adipose tissues. Int. J. Mol. Sci. 20, 2707 (2019).
Srivastava, S. & Veech, R. L. Brown and brite: the fat soldiers in the anti-obesity fight. Front Physiol. 10, 38 (2019).
Lee, Y. S. et al. Hypothalamic ATF3 is involved in regulating glucose and energy metabolism in mice. Diabetologia 56, 1383–1393 (2013).
Hartman, M. G. et al. Role for activating transcription factor 3 in stress-induced beta-cell apoptosis. Mol. Cell. Biol. 24, 5721–5732 (2004).
Xiao, X., Li, J. & Samulski, R. J. Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. J. Virol. 72, 2224–2232 (1998).
Rabinowitz, J. E. et al. Cross-packaging of a single adeno-associated virus (AAV) type 2 vector genome into multiple AAV serotypes enables transduction with broad specificity. J. Virol. 76, 791–801 (2002).
Acknowledgements
We thank Ms. Pei-Chih King for performing mouse surgery, and the Academia Sinica animal core for mouse husbandry and behavior experiments. This work was supported by Ministry of Science and Technology [MoST 104-2314-B-303-014-MY3 to C-F.C., MoST 103-2314-B-038-016-MY3, MoST 104-2314-B-038-047-MY2, MoST 104-2314-B-303-014-MY3 to H. L., and MOST 106-2314-B-038-064-MY3 to H-H.C.], Tzu Chi General Hospital [TCRD105-39, TCRD106-03E and TCMMP108-04-02 to C-F.C.], Taipei Medical University and Taipei Medical University Hospital (104TMU-TMUH-10 to H-H.C. and H.L.), and Tzu Chi and Academia Sinica cooperation [TCAS-108-01translational research grants to C-F.C.].
Author information
Authors and Affiliations
Contributions
This study was designed by C-F.C., H.L., and H-H.C. The paper was written by C-F.C., H.L. and H-C.K. H-C.K., H.L., and H-F.L. performed most of the cell and adipose tissue experiments. H-C.K., C-F.C., and P-F.L performed the animal experiments. J-J.C. and M-J.D. performed the drug screen and compound isolation. S-W.C performed the compound purification and production. C.-C. C. and H.-H. C. performed energy expenditure measurements. C-F.C., H.L., and H-H.C. analyzed the data. All authors contributed to editing the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cheng, CF., Ku, HC., Cheng, JJ. et al. Adipocyte browning and resistance to obesity in mice is induced by expression of ATF3. Commun Biol 2, 389 (2019). https://doi.org/10.1038/s42003-019-0624-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-019-0624-y
This article is cited by
-
Constructing eRNA-mediated gene regulatory networks to explore the genetic basis of muscle and fat-relevant traits in pigs
Genetics Selection Evolution (2024)
-
Identification of novel SCD1 inhibitor alleviates nonalcoholic fatty liver disease: critical role of liver-adipose axis
Cell Communication and Signaling (2023)
-
A non-invasive mouse model that recapitulates disuse-induced muscle atrophy in immobilized patients
Scientific Reports (2023)
-
Mediation effects of thyroid function in the associations between phthalate exposure and lipid metabolism in adults
Environmental Health (2022)
-
ATF3 in atherosclerosis: a controversial transcription factor
Journal of Molecular Medicine (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
