
Abstract
Aphelids are little-known phagotrophic parasites of algae whose life cycle and morphology resemble those of the parasitic rozellids (Cryptomycota, Rozellomycota). In previous phylogenetic analyses of RNA polymerase and rRNA genes, aphelids, rozellids and Microsporidia (parasites of animals) formed a clade, named Opisthosporidia, which appeared as the sister group to Fungi. However, the statistical support for the Opisthosporidia was always moderate. Here, we generated full life-cycle transcriptome data for the aphelid species Paraphelidium tribonemae. In-depth multi-gene phylogenomic analyses using several protein datasets place this aphelid as the closest relative of fungi to the exclusion of rozellids and Microsporidia. In contrast with the comparatively reduced Rozella allomycis genome, we infer a rich, free-living-like aphelid proteome, with a metabolism similar to fungi, including cellulases likely involved in algal cell-wall penetration and enzymes involved in chitin biosynthesis. Our results suggest that fungi evolved from complex aphelid-like ancestors that lost phagotrophy and became osmotrophic.
Similar content being viewed by others
Introduction
Fungi are an extremely ecologically successful group of organisms involved in the saprophytic degradation of organics, with some parasitic species. They feed by osmotrophy or direct uptake of dissolved organics through their chitin cell-wall1. Traditionally, fungi encompassed several lineages of filamentous species, most of which belong to the Dikarya (Ascomycota and Basidiomycota), and a group of flagellated species known as chytrids (Chytridiomycota)2. The first molecular phylogenetic analyses based on one or a few genes confirmed the monophyly of Dikarya but also revealed that some deep-branching fungal lineages were not monophyletic, notably the chytrids and the zygomycetes2,3. In parallel, environmental studies have uncovered a wide diversity of sequences belonging to new lineages of unicellular eukaryotes branching at the base of the classical fungi. One of those lineages (known by several names: Rozellida, Cryptomycota, Rozellomycota) includes the flagellated phagotrophic unicellular parasite Rozella allomycis, which branches as a sister lineage to classical fungi in phylogenetic trees4,5. Another group corresponds to the Microsporidia, well-known fast-evolving parasites, whose deep phylogenetic branches are progressively being populated by new, slowly-evolving species6. The third clade of organisms that appear to relate to these lineages is the aphelids (Aphelida)7. While some authors include all of these protist lineages as fungi (fungi sensu lato)8,9, others do not7. Delimiting what defines a fungus is not easy, especially when the phylogenetic relationships among fungi-related protists remain undetermined1. Here, we refer to fungi as encompassing the classical core fungi (filamentous and chytrid fungi), which feed by osmotrophy through chitinous cell walls.
Aphelids constitute a group of diverse, yet poorly known, parasites of algae7,10,11. Their life cycle and morphology resemble those of zoosporic fungi (chytrids) and rozellids (Cryptomycota/Rozellosporidia), another species-rich group of parasites of fungi and oomycetes4,5. Unlike fungi, which are osmotrophs, aphelids and rozellids are phagotrophs, feeding on the host’s cytoplasm. Combined RNA polymerase and rRNA gene trees12 suggested that aphelids and rozellids relate to Microsporidia, extremely reduced parasites with remnant mitochondria unable to perform phagotrophy13. Accordingly, aphelids, rozellids and Microsporidia were proposed to be monophyletic and form a sister group to fungi called Opisthosporidia7. However, the limited phylogenetic signal of those genes combined with fast-evolving sequences from microsporidians have resulted in incongruent tree topologies, showing either rozellids12,14 or aphelids15 as the earliest-branching lineages of Opisthosporidia. Furthermore, the support for the monophyly of Opisthosporidia was always moderate, leaving the relative order of emergence of the highly diverse fungal relatives unresolved. To improve the phylogenetic signal for an accurate placement of aphelids in the opisthokont branch containing fungi, nucleariids, rozellids, and Microsporidia (usually referred to as Holomycota), we have generated the first full life-cycle transcriptome data for one aphelid species, Paraphelidium tribonemae, to our knowledge11. Phylogenomic analyses of this almost-complete transcriptome including, in particular, data from the Rozella allomycis genome16, show that Opisthosporidia are paraphyletic and that P. tribonemae branches as the sister lineage to Fungi. Comparison of gene sets involved in metabolism and major cellular functions strongly suggests that fungi evolved from complex aphelid-like ancestors that were phagotrophic.
Results
Aphelids occupy a deep pivotal position and are the sister group to fungi
To generate the aphelid transcriptome and because P. tribonemae has a complex life cycle (Fig. 1), we maximized transcript recovery by constructing two cDNA libraries corresponding to young and old enrichment cultures of its host, the yellow-green alga Tribonema gayanum, infected with the aphelid. Accordingly, transcripts corresponding to zoospores, infective cysts, trophonts, plasmodia, sporangia and sporocysts were represented11. After paired-end Illumina HiSeq2500 sequencing, we assembled a metatranscriptome of 68,130 contigs corresponding to the aphelid, its host and bacterial contaminants. After a sequential process of supervised cleaning, including comparison with a newly generated transcriptome of the algal host, we obtained a final dataset of 10,439 protein sequences that were considered free of contaminants. The final predicted proteome (Paraphelidium tribonemae version 1.5; Supplementary Data 1) was 91.4% complete according to BUSCO17. We found no stop codons interrupting coding sequences. Therefore, in contrast to Amoeboaphelidium protococcarum, for which the TAG and TAA stop codons appear to encode glutamine12, P. tribonemae does not possess these modifications from the canonical genetic code.

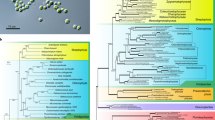
Phylogenomic analyses and cell cycle of the aphelid Paraphelidium tribonemae. a Bayesian phylogenetic tree based on single-copy protein domains for 49 species (SCPD49) inferred using a CAT-Poisson model. Statistical supports indicated at some crucial nodes correspond, from left to right, to PhyloBayes Bayesian posterior probabilities and IQ-TREE ML ultrafast-bootstrap support using the C60 model. Branches with maximum support values (pp = 1 and ubs = 100%) are indicated by black circles. The support for the monophyly of Paraphelidium and fungi (blue star) using the three datasets (SCPD, BMC and GBE) with the 49 species and after removing the fastest-evolving microsporidian sequences (36 species) is shown in Table 1. b Schematic cell cycle of P. tribonemae. Briefly, infecting cysts (red wall), deliver an ameboid trophont to an algal filament cell via an infection tube; the trophont engulfs the algal cytoplasm by phagocytosis, leaving a growing residual body (dark red particle); after nuclear and cell division, a mature sporangium releases amoeboflagellated zoospores (occasionally ameboid only) that get out the algal cell wall and close the life cycle11. c Evolution of IQ-TREE ML ubs support for the monophyly of aphelids and fungi (A + F) and the monophyly of Opisthosporidia (O) as a function of the proportion of fast-evolving sites removed from the dataset. All the phylogenomic trees can be seen in Supplementary Fig. 1
Because resolving the phylogenetic relationships in this part of the eukaryotic tree is challenging due to the occurrence of lineages with very different evolutionary rates, notably the fast-evolving Microsporidia, we used three different datasets and carried out a battery of phylogenomic analyses including and excluding the fastest-evolving species and amino acid positions from our datasets. The three datasets have been previously used to study the phylogeny of animals and related protists (Holozoa, the sister group to Holomycota) within Opisthokonts (SCPD)18, Microsporidia (including highly conserved genes for this fast-evolving group, BMC)19, and Microsporidia and related deeper-branching lineages (maximizing the putative orthologs in the whole clade, GBE)15. First, we incorporated P. tribonemae orthologs to a dataset of 93 single-copy protein domains (SCPD dataset) previously used for phylogenomic analyses of basal-branching opisthokonts18, updating it with several microsporidian species including the early-branching Mitosporidium daphniae20, the metchnikovellid Amphiamblys sp.15, and the recently released genome of Paramicrosporidium saccamoebae21. We then generated Bayesian inference trees reconstructed under the CAT-Poisson mixture model22 and maximum likelihood (ML) trees reconstructed under C60, the closest mixture model to CAT23, for a taxon sampling including 49 species (22,976 positions). The two phylogenomic analyses yielded the same topology. We recovered the monophyly of all opisthokont lineages previously reported18,24. In contrast with previous analyses based on 18S rRNA and RNA polymerase genes12,14, Opisthosporidia appeared as paraphyletic and the aphelid was placed as the sister lineage to fungi (Fig. 1). However, the ML bootstrap support was moderate (ultrafast bootstrapping; 96% ubs) and the Bayesian probability was low (0.66 pp; even if the two Bayesian chains converged in terms of likelihood, the consensus topology was not distinctively supported by the other chain). To limit potential long-branch attraction artefacts derived from the inclusion of fast-evolving Microsporidia, we built a reduced dataset without the thirteen fastest-evolving microsporidian species (SCPD36, 36 species; 24,413 positions). Bayesian and ML trees again reproduced the same topology (Supplementary Fig. 1) with higher pp (0.88) and ubs (95%) values, but still not fully supporting the consensus topology (Fig. 1; Table 1).
To resolve the phylogenetic position of the aphelid with higher support, we analyzed two additional datasets previously used for phylogenomic analyses of Microsporidia with the same two sets of 49 and 36 opisthokont species. The resulting datasets were called BMC49 and BMC36 for a set of 53 protein markers19 which, after concatenation, yielded 15,744 and 16,840 amino acid positions, respectively; and GBE49 and GBE36 for datasets with 259 protein markers15, which resulted in 84,810 and 96,640 amino acids, respectively. For all datasets, Bayesian and ML trees recovered the monophyly of Paraphelidium and fungi (Supplementary Fig. 1) and, for the two datasets without fast-evolving Microsporidia, posterior probabilities and ML bootstrap support were maximal (Fig. 1, Table 1, Supplementary Data 2). In addition, we carried out alternative topology tests for the three datasets containing 49 species. All of the alternative topology tests supported P. tribonemae as the sister group to fungi (AU test p-value significantly excluded if <0.1), and both BMC49 and GBE49 significantly excluded the Opisthosporidia monophyly (AU p-value 0.0039 and 0.0000, respectively; Supplementary Data 2). Finally, to minimize systematic error in ML analyses due to the inclusion of fast-evolving sites in our protein datasets, we progressively removed the 5% fastest-evolving sites and reconstructed the phylogenomic trees. This resulted in the increase of support for the monophyly of fungi and the aphelid, which became very high (>98% ubs; see Supplementary Data 2) until 50% (SCPD dataset) or up to 80% (GBE dataset) of sites were removed, when phylogenetic signal was expected to be insufficient to resolve such deep phylogeny. Remarkably, the Opisthosporidia monophyly was never supported (Fig. 1c).
Collectively, our phylogenomic analyses support the monophyly of aphelids and fungi, although the branch joining the two groups is very short. Opisthosporidia, united by their ancestral phagotrophy, appear as paraphyletic.
Enzymes involved in cell-wall synthesis and degradation
Despite secondary losses in some fungi, the presence of chitin in cell walls was long considered a typical fungal trait1. However, chitin is also present in the cell wall of many other protists across the eukaryotic tree, implying that the machinery for chitin synthesis/remodeling originated prior to the radiation of fungi and other eukaryotic lineages1. Microsporidia and Rozella are also able to synthesize chitin16,25 but, unlike fungi, which possess chitin cell walls during the vegetative stage, they produce chitin only in cysts or resting spores26. Staining with fluorescently-labeled Wheat Germ Agglutinin (WGA) showed that Paraphelidium also possesses chitin in the wall of infecting cysts, but not in zoospores or, unlike fungi, in the vegetative stage (i.e., the trophonts) (Fig. 2a, b). In agreement with this observation, we identified homologs of chitin synthases, chitin deacetylases, chitinases and 1,3-beta-glucan synthases in the Paraphelidum transcriptome (Supplementary Fig. 2a–d). Specifically, we detected seven homologous sequences (including all alternative transcripts such alleles or splice variants) of division II chitin synthases18,25,27,28 in Paraphelidium corresponding to at least six distinct peptides (Rozella contains only four16). Three of them clustered with class IV chitin synthases, including Microsporidia and Rozella homologs (Supplementary Fig. 2a). The remaining four sequences branched within class V/VII enzymes28, two of them (probably corresponding to a single polypeptide) forming a deep-branching group with fungal, mostly chytrid, sequences (Supplementary Fig. 2a). Class V enzymes include a myosin motor thought to intervene in polarized fungal hyphal growth that has been hypothesized to take part in the formation of the germ tube in aphelids and rozellids20. Class V chitin synthases were lost in Microsporidia (with the exception of Mitosporidium, still retaining, like Rozella, one homolog), endowed instead with highly specialized polar tube extrusion mechanisms20. Neither spore wall nor polar tube proteins specific to Microsporidia29 occurred in the Paraphelidium transcriptome. Therefore, our data (Supplementary Fig. 2a; Supplementary Data 1 and 5) lend credit to the hypothesis that class V chitin synthases are involved in germ tube polar growth.

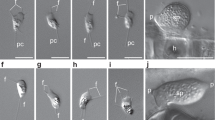
Zoospores and chitin-bearing infective cysts of Paraphelidium tribonemae. a Filament of Tribonema gayanum infected by P. tribonemae under optical microscopy. Many cysts can be seen attached to the filament surface, many (small) zoospores can be seen surrounding the filament before encystment; trophonts are present within Tribonema cell-wall delimited compartments. b The same filament as in a stained with fluorescent wheat germ agglutinin (WGA, red color) showing the presence of chitin in infective cysts (arrows show some examples) but not the zoospores (asterisks show some examples) under epifluorescence microscopy. c False-colored scanning-electron microscopy image of a filament infected by several P. tribonemae cysts (pedunculated rounded structures). The algal host filament is colored in green and parasite cysts in pink. Note that one cyst germ tube is penetrating the host cell by the space between the two algal cell-wall halves (white arrowhead) and two cysts are broken (black arrowheads), showing the penetration channel. d Ameboid zoospore (infrequent). e Amoeboflagellated zoospore. Scale bar: A = 5 µm, B–F = 1 µm. Phylogenetic trees related to chitin and other cell-wall synthesis and degradation-related enzymes are shown in Supplementary Fig. 2. Zoospore motility can be seen in Supplementary Video 1. For original fluorescent microscopy images, see Supplementary Data 7
Among the rest of chitin-related enzymes, we identified twelve sequences (at least five different homologs) of chitin deacetylase30,31 (Supplementary Fig. 2b). We detected at least three class II chitinase homologs (eight total sequences), which are ancestral in opisthokonts, containing the Glyco_hydro_18 (PF00704) domain, and a class I chitinase (CTSI) with a Glyco_hydro_19 (PF00182) domain32. The latter included the catalytic site, an N-terminal predicted signal peptide and a transmembrane region, suggesting an extracellular chitinase activity. CTSI has a peculiar phylogenetic distribution in eukaryotes, occurring only in Viridiplantae, Fungi, Opisthosporidia and Ecdysozoa (Supplementary Fig. 2c). Rozella contains two homologs and Microsporidia at least one; they have N-terminal signal peptides and are predicted to localize extracellularly but lack transmembrane domains. In our phylogenetic tree, opisthosporidian sequences appeared scattered within metazoan and fungal sequences. This might be the result of hidden paralogy and/or horizontal gene transfer (HGT) (Supplementary Fig. 2c). Regardless of its evolutionary origin, aphelid CTSI might be involved in the self-degradation of resting spore and cyst wall chitin. This might happen both, during their release from chitin-containing resting spores or at the tip of the germ tube during infection, as previously suggested for Rozella16 (Fig. 1).
Although not found in chytrids, 1,3-beta-glucan is also considered an idiosyncratic fungal cell-wall component1. Surprisingly, we identified two 1,3-beta-glucan synthase (FKS1) homologs (four sequences), with split Glucan_synthase (PF02364) and FKS1_dom1 (PF14288) domains (fused for the phylogenetic analysis) (Supplementary Fig. 2d). The presence of FKS1, absent in Rozella and Microsporidia, in aphelids traces its origin back to the ancestor of fungi and aphelids.
To feed on the algal cytoplasm, aphelids need to traverse the algal cell wall but, so far, the specific penetration mechanism, whether mechanical (germ-tube penetration through the gap between the two algal cell-wall halves) or enzymatic (digestion of algal cell-wall components) was uncertain7,11. Scanning electron microscopy (SEM) observations showed both, clear cases of mechanical penetration between the two algal cell-wall halves, and also infecting cysts scattered on the algal cell-wall surface (Fig. 2c). SEM images additionally confirmed WGA-epifluorescence observations of multiple parasitoid cysts co-infecting the same host cell (Fig. 2a–c). Multiple infections, implying the coexistence of distinct trophonts feeding within the same algal cell-wall delimited compartment, open the intriguing possibility for aphelids to engage in sexual reproduction within the algal host. Our Paraphelidium transcriptome has some genes involved in meiosis (e.g., MutS protein homolog 4), but not all (Supplementary Data 1). However, the transcriptome is not complete and, at the same time, many of the detected genes are also involved in more general recombination (e.g., meiotic recombination protein DMC1) and DNA repair (e.g., multiple helicases homologous to HFM1) processes. Therefore, whether the aphelid has the potential to carry out meiosis and whether it might indeed occur at the trophont stage will require future study. Tribonema cell walls contain cellulose II based on 1,6-linked glucan (alkali soluble cellulose), 1,3 and 1,4-linked xylose, 1,3-linked rhamnose and, mostly, 1,3; 1,4; and 1,6-linked glucose33. We performed sequence similarity searches of known fungal cellulases34 using the database mycoCLAP35, which contains functionally characterized proteins, to search for these enzymes in aphelids, followed by phylogenetic analyses. In support of an enzymatic algal cell-wall penetration, we identified various cellulases in the Paraphelidium transcriptome belonging to glycoside-hydrolase families GH3 and GH5. We detected three homologs of the GH3 cellulase beta-glucosidase/xylosidase36, which is not found in other opisthosporidians but is present in fungi, amoebozoans, several opisthokonts and other protists, as well as bacteria. Our phylogenetic analysis shows that the three aphelid sequences are most closely related to deep-branching opisthokont protists (respectively, Capsaspora, choanoflagellates, nucleariids) (Supplementary Fig. 2e). Additionally, we identified at least three GH5 cellulase37 homologs (seven sequences in total) in P. tribonemae, which were related to GH5 widespread in fungi (GH5_11, GH5_12 and GH5_24) (Supplementary Fig. 2f). Collectively, these observations strongly suggest that these cellulases are involved in the alga cell-wall penetration, but direct proof will only be obtained by purifying or heterologously expressing those cellulases and testing their activity in vitro.
Primary metabolism reminiscent of free-living lifestyles
Analysis of the Rozella allomycis genome showed that, like microsporidian parasites, it has considerably reduced metabolic capabilities16. To comparatively assess the metabolic potential of aphelids, we investigated the presence of orthologous groups related to eight primary metabolism categories (Gene Ontology) in the transcriptome of Paraphelidium, using eggNOG annotation38. We thus identified 1172 orthologous groups in Paraphelidium and a set of 41 eukaryotic species including representatives of major fungal lineages, opisthokont protists and other eukaryotic parasites (Supplementary Data 3). Based on their orthologous group distribution, we built a dissimilarity matrix that was analyzed by Principal Coordinate Analysis. The first axis clearly separated Paraphelidium from Microsporidia, Mitosporidium, Paramicrosporidium and Rozella, the latter two positioned near one another and having an intermediate position similar to other protist parasites (e.g., Trypanosoma, Leishmania, Toxoplasma) (Fig. 3a and Supplementary Fig. 3a). Paraphelidium was positioned at the same level as fungi, Capsaspora, Corallochytrium and Parvularia, along axis 1. However, axis 2 separated Paraphelidium and fungi from the rest of eukaryotes. These relationships were further visualized in a cluster analysis of pairwise species comparisons (Supplementary Fig. 3b). The PCoA suggested that Paraphelidium has a rich metabolic gene complement, which was made evident by the orthologous group presence/absence heatmap showing that aphelids have a metabolic potential fully comparable to that of (especially chytrid) fungi (Fig. 3b).

Complexity of Paraphelidum tribonemae metabolism and cytoskeleton-trafficking-phagotrophy-related proteome. a Principal coordinate analysis (PCoA) and b binary heat-map and species clustering based on the presence/absence of 1172 orthologous genes belonging to 8 primary metabolism Gene Onthology categories across 41 eukaryotic genomes/transcriptomes. c PCoA and d binary heat-map and species clustering based on the presence/absence of 695 KEGG orthologs related to cytoskeleton, membrane-trafficking and phagotrophy, which were selected from 11 KEGG categories. Species are color-coded according to their taxonomic assignment
The most distinctive metabolic categories when comparing Paraphelidium and other Opisthosporidia were energy production and conversion, followed by amino acid, nucleotide and lipid transport and metabolism. In all metabolic categories, the aphelid clustered with fungi, and more specifically with chytrids, and sometimes with other free-living opisthokonts (e.g., nucleariids, Capsaspora). By contrast, Rozella always clustered with Mitosporidium and Paramicrosporidium either together with other Microsporidia or with other parasitic protists (Supplementary Fig. 3c–f). The only exception corresponded to Paramicrosporidium which, for energy production and conversion, clustered with nucleariids (Supplementary Fig. 3c), in agreement with their rich energy-related gene set21.
To check whether these differences between Paraphelidium and Rozella affected particular metabolic pathways, we compared the annotated proteins in the two organisms based on KEGG annotation39. A comparison of the two species in the KEGG general metabolic pathway map showed that, even accounting for the possibility that we missed some genes in the Paraphelidium’s transcriptome (e.g., mitochondrial-encoded proteins), the aphelid map contained 200 orthologous groups more than Rozella (548 vs. 348 orthologous groups) (Supplementary Fig. 3g). In agreement with previous observations, major differences were observed in energy production and conversion, and amino acid, nucleotide and lipid transport and metabolism. In particular, contrary to Rozella, which lacks most subunits of the mitochondrial electron transport chain complex I (NADH dehydrogenase; ETC-I)16, Paraphelidium possesses a practically complete ETC-I as inferred from the nuclear encoded transcripts (the P. tribonemae transcriptome is biased against mitochondrial transcripts, which lack polyA) (Supplementary Fig. 3h). Paraphelidium also possesses wide capabilities related to nucleotide (e.g., purine, uridine, and inosine biosynthesis) and amino acid (serine, threonine, methionine, lysine, ornithine, histidine, shikimate pathway) metabolism, which Rozella has lost (Supplementary Fig. 3i). Likewise, the aphelid has also retained pathways for phosphatidylcholine, cholesterol and fatty acid biosynthesis that were subsequently lost in Rozella16. Most carbohydrate metabolic pathways were conserved in the two species, except for the galactose synthesis and degradation that was lost in Rozella (Supplementary Data 3).
By contrast, compared to Rozella, and under the assumption that our transcriptome is rather complete, the aphelid seems to lack several enzymes involved in catecholamine biosynthesis (Supplementary Data 3). However, some of these are also absent from Capsaspora, Monosiga, Salpingoeca, or Spizellomyces. These compounds are likely involved in cell–cell communication in microbes40, e.g., parasite-host signaling, suggesting that they might have a role in rozellid parasitism. The aphelid seems to lack other parasite-specific proteins, such as crinkler, nucleoside H+-symporters or ATP/ADP-antiporters, which occur in Rozella and/or Microsporidia16.
These observations suggest that Paraphelidium has a complex metabolic profile, being functionally closer to free-living protists than to parasites and having affinities with fungi and, to a lesser extent, nucleariids and holozoan protists.
Distinct and ancestral-like phagotrophy-related machinery
Like rozellids, aphelids are phagotrophs, but their global metabolism resembles that of osmotrophic fungi. What does their phagocytosis-related proteome look like? The core phagocytic molecular machinery, already present in the last eukaryotic common ancestor41, is involved in multiple dynamic cell processes (endocytosis, exocytosis, autophagy, protein recycling, etc.). Structurally, the phagocytic machinery encompasses various endomembrane organelles (phagosomes, lysosomes, peroxisomes, endoplasmic reticulum, Golgi apparatus), and multiple membrane-trafficking components (signaling pathways, transporters, specific enzymes, cytoskeleton elements, motors, etc.)42. To examine phagotrophy-related genes in Paraphelidium and the 41 additional eukaryotes used for comparison, we built a presence/absence matrix of 695 KEGG orthologs from 11 functional categories (5 KEGG maps and 6 BRITE categories) that aimed at including all necessary proteins to perform phagotrophy; i.e., phagolysosome biogenesis, membrane trafficking and the actin cytoskeleton42 (Supplementary Data 4). A PCoA showed that Paraphelidium and Rozella are much closer to one another in terms of phagotrophy- than metabolism-related genes, grouping with fungi and far from Microsporidia (Fig. 3c and Supplementary Fig. 4a). This pattern was also evident in the presence/absence matrix (Fig. 3d and Supplementary Fig. 4b). In both, the two species clustered with early-branching fungi: chytridiomycetes (Spizellomyces, Batrachochytrium, Gonapodya) and blastocladiomycetes (Allomyces, Catenaria, Blastocladiella). The clustering of Rozella and Paraphelidium with fungi based on 11 phagotrophy-related KOs (695 orthologs) might seem puzzling, since fungi are osmotrophs. However, this clustering is not unexpected, since their phagotrophic function likely involves many genes also involved in other cellular processes (e.g., cytoskeletal movement, endocytosis) that they share with fungi. Only a subset of genes in Rozella and Paraphelidium (less than 100 genes), seem to be specific and slightly different between Rozella and the aphelid. These genes are those likely responsible, together with the genes that they share with fungi, for the phagotrophic function. Furthermore, since Paraphelidium and Rozella have similar, but not identical, protein sets involved in phagosome, lysosome and endocytic processes, different protein subsets specialized in each lineage for phagocytosis function from their ancestral phagocytic ancestor (Supplementary Fig. 4c–e).
In order to gain more insights into the diversification of the actin cytoskeleton toolkit of fungi and opisthosporidians, we analyzed the evolution of myosin motor proteins. The myosin toolkit of Paraphelidium contains a mixture of classical fungal families and others previously identified in holozoans (animals and their unicellular relatives; Supplementary Data 5). We recovered diversified class I myosins in Paraphelidium, Spizellomyces and nucleariids (Supplementary Fig. 4g), with paralogs of Ic/h and Ik families, previously described only in holozoans43. Paraphelidium, nucleariids and Gonapodya also possess homologs of class XV myosins, formerly thought holozoan-specific43. In addition, the aphelid not only possesses homologs of the V/VII myosin-motor family associated to chitin-synthase (see above; Supplementary Fig. 2a), but also myosins of If (pan-eukaryotic), II (amorphean), and XXII (opisthokont) classes, which clustered with previously described fungal homologs (Supplementary Data 5; Supplementary Fig. 4f). Thus, compared with the ancestral opisthokont myosin complement43, Paraphelidium retains all but one myosin class (VI), with homologs of all myosin families present in fungi (If, II, V, XVII - chitin synthase) plus four additional families (Ic/h, Ik, XV and XXII) that were lost in eumycotan fungi (i.e., fungi to the exclusion of chytrids). This suggests an independent step-wise simplification of the myosin complement in fungi, Rozella and Microsporidia, with Paraphelidium, nucleariids and chytrids retaining ancestral classes.
Recently, it has been proposed that WASP and SCAR/WAVE genes, encoding activators of branched actin assembly proteins44 and hence related to the actin cytoskeleton, are essential to build filopodia and for the cell motility called alpha-motility45. The aphelid zoospore motility involves filopodia (Supplementary video 1), resembling that of chytrid fungi45. Likewise, its transcriptome contains homologs of WASP and SCAR/WAVE proteins (Supplementary Data 6). Interestingly, although Rozella also contains WASP and SCAR/WAVE homologs, filopodia have not been described in its zoospores46,47,48. Therefore, either Rozella has some form of not-yet-identified filopodia or the presence of WASP and SCAR/WAVE does not necessarily imply the development of filopodial movement45. If this is the case, the Rozella genes might instead be involved in phagotrophy in this organism.
Discussion
From an evolutionary perspective, the current situation in the holomycotan branch (including Fungi) of the eukaryotic super-group Opisthokonta mirrors that of the Holozoa (including Metazoa), where the discovery of deeply-branching unicellular protists that possess genes thought unique to animals continues to challenge previous evolutionary schemes about the emergence of Metazoa24,49. Thus, the discovery that a highly diverse set of environmental sequences formed a deeply-branching clade that appeared as sister group to fungi and that included the parasite Rozella allomyces (named rozellids4, and subsequently Cryptomycota5, Rozellomycota50, or Rozellosporidia51, triggered the discussion of what fungi actually are1,52. This debate was further nourished by the discovery that aphelids, another highly diverse group of parasites of algae10, formed a large clade with rozellids and Microsporidia based on rRNA and RNA-polymerase genes7,12,51,53. This seemingly monophyletic group was named Opisthosporidia and branched as sister group to classical fungi (the clade including from chytrids and their relatives to the Dikarya52) in phylogenetic trees7. Lately, many mycologists include the three opisthosporidian lineages, Aphelida, Rozellosporidia and Microsporidia, within Fungi1,9,16,54. Some authors even incorporate as fungi the free-living phagotrophic chitin-lacking nucleariids, thus pushing the limits of fungi to incorporate all holomycotan (possibly a confusing name) lineages, despite asserting that the kingdom Fungi is characterized by osmotrophic nutrition across a chitinous cell wall8. However, unlike fungi, aphelids and rozellids (the deepest branches in the holomycotan clade to the exclusion of nucleariids) are phagotrophs, lack a chitin cell wall at the vegetative stage and are endobiotic with a unique mode of penetration into their hosts7,11,52,53. Also, because Rozella has a reduced genome, a typical trait of streamlined parasites, some authors inferred a parasitic nature for the ancestor of fungi16.
The study of the Paraphelidium tribonemae transcriptome clarifies some of this controversy. Our multi-gene phylogenetic analyses place the aphelids as a pivotal group branching very deeply in the Opisthosporidia/Fungi clade. However, contrary to previous phylogenetic analyses based on only a few genes, Opisthosporidia do not seem to be monophyletic. Aphelids strongly emerge as the sister group to fungi to the exclusion of rozellids and Microsporidia (Fig. 1). Because Rozella and aphelids now appear as paraphyletic lineages that share similar ancestral characteristics (e.g., alternate life cycle of zoospores, cyst, phagotrophic ameboid vegetative stages), our results parsimoniously suggest that fungi evolved from ancestors that were similar to aphelids. Because of its deep pivotal position, the comparative study of the Paraphelidium transcriptome allows better inference of ancestral states for fungi and the now paraphyletic Opisthosporidia clade (Fig. 4). Paraphelidium has a complex metabolism resembling that of free-living chytrids but feeds by phagotrophy like free-living nucleariids and holozoan protists (Fig. 3). This suggests that aphelids are ‘borderline’ parasites (or parasitoids, according to some definitions) that have not undergone the reduction process that characterizes Rozella and all members along the microsporidian branch. Being more gene-rich and close to the root separating fungi-aphelids from rozellids-microsporidia, aphelids may have retained more ancestral features13,15,16. These features support a free-living opisthosporidian ancestor that had a complex life cycle including chitin-containing resting cysts, amoeboflagellate zoospores and a phagotrophic amoeba stage possibly specialized in endobiotic predation (Fig. 4). By contrast, the fungal ancestor was a free-living osmotroph that had amoeboflagellate zoospores and chitin in the vegetative stage. From their aphelid-like opisthosporidian free-living ancestor, the fungal lineage lost phagotrophy, acquiring its ecologically successful osmotrophy whereas, on the rozellid line, endobiotic phagotrophic predation shifted into obligate parasitism with more complex life cycles, highly specialized morphologies and genome and metabolic reduction. The analysis of additional genomes/transcriptomes for other aphelids, rozellids and deep-branching fungi should help establishing a solid phylogenomic framework to validate and refine this evolutionary scenario.

Early evolution of fungi and related lineages. Schematic representation of evolutionary relationships between fungi, rozellids and Microsporidia, within the holomycotan branch of Opisthokonts. Inferred key ancestral features and life cycle stages are depicted at the corresponding ancestral nodes
Methods
Cultures
Paraphelidium tribonemae was maintained in an enriched culture with its host Tribonema gayanum in mineral medium or in Volvic™ at room temperature in the presence of white light11. The culture was progressively cleaned from other heterotrophic eukaryotes by micromanipulation and transfer of infected host filaments to uninfected T. gayanum cultures.
WGA staining, epifluorescence, and scanning electron microscopy
To detect chitin in different cell cycle stages of Paraphelidium tribonemae, we incubated actively growing cultures with 5 µg/mL WGA conjugated to Texas Red (Life Technologies) for 10 min at room temperature. After rinsing with Volvic™ water, cells were then observed under a LEICA DM2000LED fluorescence microscope with an HCX_PL_FLUOTAR 100×/1.30 oil PH3 objective. Pictures were taken with a LEICA DFC3000G camera using the LEICA Application Suite v4.5 and edited with ImageJ (http://imagej.nih.gov/ij/). For SEM observations, we transferred 1-week-old cultures to a Petri dish containing mineral medium and two coverslips. Cells were let to settle overnight before fixation (1% OsO4 and 1% HgCl2 for 45 min). After washing (3 × 10 min in distilled water), samples were dehydrated in ethanol series (30, 50, 70, 90, 96, and 100%) for 10 min each. After critical-point drying and sputter-coating with platinum, cells were visualized with a Zeiss Sigma FE-SEM at 1 kV acceleration voltage.
Transcriptome sequencing
To obtain a complete representation of the aphelid transcriptome across different cell-cycle stages, we extracted RNA from a young culture (3 days after inoculation) rich in aphelid zoospores and cysts, and eight old cultures (5–7 days after inoculation) with few remaining T. gayanum living cells and rich in aphelid plasmodia, resting spores and zoospores. To minimize algal overrepresentation, we maintained the cultures in the dark. Total RNA was extracted with the RNeasy mini Kit (Qiagen), quantified by Qubit (ThermoFisher Scientific) and sent to Eurofins Genomics (Germany) for de novo transcriptome sequencing. Two cDNA Illumina libraries for, respectively, the young and old enrichment cultures were constructed after polyA mRNA selection and paired-end (2 × 125 bp) sequenced with Illumina HiSeq 2500 Chemistry v4.
Transcriptome assembly, decontamination and annotation
A total of 71,015,565 and 58,826,379 reads, respectively, were obtained for the libraries of young and old cultures. From these, we obtained 34,844,871 (young) and 25,569,845 (old) paired-end reads. After quality/Illumina Hiseq adapter trimming using Trimmomatic v0.3355, we retained 8,601,196 forward and 6,535,164 reverse reads (young) and 10,063,604 forward and 4,563,537 reverse unpaired reads (old). Resulting reads were assembled using Trinity56 with min_kmer_cov 2 and normalize_reads options. This initial assembly, Partr_v0, contained 68,130 contigs. 47 of them were identified as ribosomal RNAs using RNAMMER57 and included sequences of Paraphelidium tribonemae, its host Tribonema gayanum, chloroplasts and bacteria. No other eukaryotic ribosomal genes were detected. To decontaminate P. tribonemae from host and bacterial sequences, we used Blobtools58 and, as input files, our assembly, a read map file obtained with the fast gapped-read alignment program Bowtie259 and a formatted hit list obtained after applying diamond-blastx60 to our sequences against the NCBI RefSeq database (e-value threshold 1e−10). To build the blobDB file, we used the first 10 hits for each putative transcript with the bestsum as tax-rule criterion to get a taxonomic affiliation at species rank. Sequences with no hit (41,836 sequences, 61.4% of the assembly) and sequences affiliated to bacteria, archaea, or viruses were removed. The remaining 21,688 eukaryotic sequences were blasted against a partial REFSEQ protein database (e-value threshold 1e−10) containing sequences of 5 fungi (Spizellomyces punctatus DAOM BR117, Batrachochytrium dendrobatidis JAM81, Agaricus bisporus var. bisporus H97, Cryptococcus neoformans var. grubii H99 and Cryptococcus neoformans var. neoformans JEC21) and 6 stramenopiles (Nannochloropsis gaditana CCMP526, Phytophthora sojae, Phytophthora parasitica INRA-310, Phytophthora infestans T30–4, Aureococcus anophagefferens and Aphanomyces invadans). If the first hit was a stramenopile (potential host origin), the sequence was discarded. 13,786 transcripts appeared free of contamination. Transdecoder v2 (http://transdecoder.github.io) with—search_pfam option yielded 16,841 peptide sequences that were filtered using Cd-hit v4.661 with a 100% identity resulting in 13,035 peptides. These peptides were functionally annotated with eggNOG-mapper38 using both DIAMOND and HMMER mapping mode, eukaryotes as taxonomic scope, all orthologs and non-electronic terms for Gene Ontology evidence. This resulted in 10,669 annotated peptides for the Paraphelidium tribonemae predicted proteome (version Partr_v1). Finally, we generated transcriptomic data for the Tribonema gayanum host using the same methods and parameters for sequencing, assembly and annotation, as described above. After blasting Partr_v1 against a database of Tribonema gayanum proteins, we further excluded 219 + 11 additional proteins that were 100% or >95% identical to Tribonema proteins. After these additional cleaning steps, version 1.5 of the Paraphelidium tribonemae predicted proteome contained 10,439 proteins (Supplementary Data 1). The completeness of this new transcriptome was estimated to 91.4% with BUSCO17 using the eukaryotic ortholog database 9 (protein option).
Phylogenomic analyses
We used three different protein datasets previously used for phylogenomic analyses of Opisthokonta and, more specifically, Microsporidia that included, respectively, 93 single-copy protein domains (SCPD)18, 53 proteins (BMC)19 and 259 proteins (GBE)15. We updated these datasets with sequences from the Paraphelidium tribonemae transcriptome, thirteen derived Microsporidia as well as Mitosporidium daphniae20, Paramicrosporidium saccamoebae21 and Amphiamblys sp15. Some stramenopile taxa such Ectocarpus siliculosus62, Thalassiosira pseudonana63 or Phytophthora infestans (GenBank NW_003303758.1) were also included to further prevent any previously undetected Tribonema gayanum contamination. Markers were aligned with MAFFT v764, using the method L-INS-i with 1000 iterations. Unambiguously aligned regions were trimmed with TrimAl65 with the automated1 algorithm, then approximate ML trees were inferred using FastTree66, all visually inspected in Geneious67 to check for possible contamination. Contamination-free alignments were relieved from distant outgroup taxa. For in-depth analyses, we used opisthokont, mainly holomycotan, sequences, including apusomonad and breviate sequences as outgroup. We then made phylogenomic analyses including or excluding thirteen fast-evolving microsporidian parasites (49 or 36 species). Sequences of the selected taxa were concatenated using Alvert.py from the package Barrel-o-Monkeys (http://rogerlab.biochemistryandmolecularbiology.dal.ca/Software/Software.htm#Monkeybarrel). This resulted in alignments containing the following number of amino acids as a function of dataset and taxon sampling: 22,976 (SCPD49); 15,744 (BMC49); 84,810 (GBE49); 24,413 (SCPD36); 16,840 (BMC36); 96,640 (GBE36). Bayesian phylogenetic trees were inferred using PhyloBayes-MPI v1.568 under the CAT-Poisson evolutionary model, applying the Dirichlet process and removing constant sites. Two independent MCMC chains for each dataset were run for >15,000 generations, saving one every 10 trees. Phylogenetic analyses were stopped once convergence thresholds were reached after a burn-in of 25% (i.e., maximum discrepancy < 0.1 and minimum effective size > 100 using bpcomp). We also applied ML phylogenetic reconstruction using IQ-TREE69 with the profile mixture model C6023. Statistical support was obtained with 1000 ultrafast bootstraps70 and 1000 replicates of the SH-like approximate likelihood ratio test71. To alleviate the local computational burden, many of these analyses were carried out using CIPRES Science Gateway72. Trees were visualized with FigTree (http://tree.bio.ed.ac.uk/software/figtree/). To test if the best topology obtained was significantly better than other alternatives (Supplementary Data 2), we tested whether constrained alternative topologies could be rejected for different datasets. We used Mesquite73 to constrain topologies representing the opisthosporidian monophyly (aphelids + rozellids + microsporidians) and the monophyly of aphelids with either blastocladiomycetes, chytridiomycetes or chytridiomycetes + blastocladiomycetes. The constrained topologies without any branch-length were reanalyzed with the -g option of IQ-TREE and the best-fitting model for each dataset. The resulting trees were then concatenated and AU tests were performed for each dataset with the -z and -au options as described in the advanced documentation of IQ-TREE. To minimize systematic error due to the inclusion of fast-evolving sites in our protein datasets, we progressively removed the fastest evolving sites at steps of 5% sites removed at a time. Among-site evolutionary rates were inferred using IQ-TREE -wsr option and its best-fitting model for each dataset (Supplementary Data 2). A total of 19 subsets were created for each dataset. We then reconstructed phylogenetic trees using IQ-TREE with the same best fitting model as for the whole dataset. To know how supported were alternative topologies in bootstrapped trees, we used CONSENSE from the PHYLIP package74 and interrogated the.UFBOOT file using a Python script (M Kolisko, pers. comm).
Homology searches and phylogenetic analyses of specific proteins
Protein sets used in this study were obtained in May 2017 from the NCBI protein database (https://www.ncbi.nlm.nih.gov/protein/), except the following: Chromosphaera perkinsii, Corallochytrium limacisporum and Ichthyophonus hoferi genome data, obtained from the ref. 24; Spizellomyces punctatus, Gonapodya prolifera, Batrachochytrium dendrobatidis, Allomyces macrogynus, Catenaria anguillulae and Blastocladiella britannica, retrieved from MycoCosm portal, Joint Genome Institute* (http://www.jgi.doe.gov/); Parvularia atlantis (previously Nuclearia sp. ATCC50694, from https://doi.org/10.6084/m9.figshare.3898485.v4); and Paramicrosporidium saccamoebae, from NCBI in January 2018. [* These sequence data were produced by the US Department of Energy Joint Genome Institute http://www.jgi.doe.gov/ in collaboration with the user community]. Query sequences to retrieve chitin and related cell-wall proteins (chitin synthases (CHS), chitin deacetylases (CDA), and chitinases (CTS)) were obtained from the study of Rozella allomycis16 except for 1,3-beta glucan synthase (FKS1), which was obtained from Saccharomyces cerevisiae S288C. We used sequences from Aspergillus fumigatus as initial queries for cellulases34 and also opisthokont homologs retrieved by BLASTp against the Paraphelidium transcriptome. Homology was confirmed by searching for the identified sequences in the eggnog38 annotation file (Supplementary Data 1), performing reverse BLAST against the non-redundant NCBI database and HMMER analyses75. Cell-wall proteins, notably chitinases and cellulases, were also annotated using CAZyme identification tools (http://csbl.bmb.uga.edu/dbCAN/annotate.php). We produced comprehensive alignments including sequences retrieved from previous studies and sequences identified by BLAST in GenBank or in the mycoCLAP database35 with the MAFFT web server64 applying the progressive L-INS-i algorithm (except for the multidomain CHS sequences for which E-INS-i was used). Alignments were trimmed from gaps and ambiguously aligned sites using Trimal65 with the automated1 algorithm. ML trees were inferred using IQ-TREE web server69 with the LG + G4 evolutionary model and visualized with FigTree. In the case of myosins, protein sequences were retrieved by querying the HMM profiles of the myosin head domain (Pfam accession: PF00063) against our database of Paraphelidium tribonemae proteins, using the hmmersearch utility of HMMER (http://hmmer.org) and the profile-specific gathering threshold cut-off. For each retrieved homolog, the protein segment spanning the Pfam-defined myosin head domain was aligned against a pan-eukaryotic alignment of myosins43, using MAFFT64 with the G-INS-i algorithm optimized for global sequence homology. The alignment was run for up to 10³ cycles of iterative refinement and manually examined, curated and trimmed. The ML phylogenetic reconstruction of the trimmed alignment (997 sequences, 400 sites) was done with IQ-tree69 under the LG model with a 10-categories free-rate distribution, selected as the best-fitting model according to the IQ-TREE TESTNEW algorithm as per the Bayesian information criterion. The best-scoring tree was searched for up to 100 iterations, starting from 100 initial parsimonious trees; statistical supports for the bipartitions were drawn from 1000 ultra-fast bootstrap70 replicates with a 0.99 minimum correlation as convergence criterion, and 1000 replicates of the SH-like approximate likelihood ratio test. We classified the myosin homologs of Paraphelidium tribonemae according to the phylogenetic framework of Sebé-Pedrós et al43. To characterize the classified homologs (Supplementary Data 5), we recorded the protein domain architectures of the full-length proteins using Pfamscan and the 29th release of the Pfam database76.
Comparative analyses of proteins involved in primary metabolism, cytoskeleton, membrane-trafficking, and phagotrophy
To get a broad view of the metabolic capabilities of Paraphelidium tribonemae in comparison with parasitic and free-living organisms, we performed a statistical multivariate analysis similar to that previously used for a similar purpose in Rozella allomycis16. We initially searched in P. tribonemae for the presence of 6469 unique orthologous groups as per eggnog38, corresponding to 8 primary metabolism categories (Gene Ontology, GO). The correspondence between GO terms and primary metabolism COGs was obtained from http://geneontology.org/external2go/cog2go, and was as follows: [C] Energy production and conversion (912 orthologs); [G] Carbohydrate transport and metabolism (1901 orthologs); [E] Amino acid transport and metabolism (714 orthologs); [F] Nucleotide transport and metabolism (351 orthologs); [H] Coenzyme transport and metabolism (375 orthologs); [I] Lipid transport and metabolism (965 orthologs); [P] Inorganic ion transport and metabolism (842 orthologs) and [Q] Secondary metabolites biosynthesis, transport and catabolism (570 orthologs). From these categories, we identified 1172 non-ambiguous orthologs in the P. tribonemae transcriptome that were shared with a set of other 41 protist species (Supplementary Data 3). These 41 protist species represent a broader taxonomic sampling as compared to the R. allomyces study16. We included 11 opisthosporidians: Paramicrosporidium saccamoebae (Par_sa), Amphiamblys sp. (Amp_sp), Antonospora locustae (Ant_lo), Encephalitozoon cuniculi (Enc_cu), Encephalitozoon intestinalis (Enc_in), Enterocytozoon bieneusi (Ent_bi), Mitosporidium daphniae (Mit_da), Nematocida parisii (Nem_pa), Nosema ceranae (Nos_ce), Rozella allomycis (Roz_al), Paraphelidium tribonemae (Par_tr); 10 fungi covering most fungal phyla: Spizellomyces punctatus (Spi_pu), Gonapodya prolifera (Gon_pr), Batrachochytrium dendrobatidis (Bat_de), Allomyces macrogynus (All_ma), Catenaria anguillulae (Cat_an), Blastocladiella britannica (Bla_br), Mortierella verticillata (Mor_ve), Coprinopsis cinerea (Cop_ci), Ustilago maydis (Ust_ma), Schizosaccharomyces pombe (Sch_po); 2 nucleariids Fonticula alba (Fon_al), Parvularia atlantis (Par_at); 7 holozoans: Salpingoeca rosetta (Sal_ro), Monosiga brevicollis (Mon_br), Capsaspora owczarzaki (Cap_ow), Chromosphaera perkinsii (Chr_pe), Corallochytrium limacisporum (Cor_li), Ichthyophonus hoferi (Ich_ho), Sphaeroforma arctica (Sph_ar); 3 amoebozoans: Acanthamoeba castellanii (Aca_ca), Dictyostelium purpureum (Dic_pu), Polysphondylium pallidum (Pol_pa), Entamoeba histolytica (Ent_hi); the free-living apusomonad Thecamonas trahens (The_tr), Naegleria gruberi (Nae_gr) and 6 other eucaryotic parasites: Toxoplasma gondii (Tox_go), Plasmodium falciparum (Pla_fa), Trypanosoma brucei (Try_br), Leishmania major (Lei_ma), Phytophora infestans (Phy_in). We annotated all 41 protein sets using eggNOG-mapper38 using DIAMOND mapping mode, eukaryotes as taxonomic scope, all orthologs and non-electronic terms for Gene Ontology evidence. Species-wise ortholog counts were transformed into a presence/absence matrix (encoded as 0/1), or binary ortholog profile. This binary ortholog profile excluded all orthologs with no identifiable orthologs in any of the 41 species used in this analysis (e.g., proteins related to exclusive prokaryotic metabolism or photosynthesis) leaving a total number of 1172 primary metabolism ortholog (Supplementary Data 3). The similarity between each species’ binary COG profile was assessed using the Pearson’s correlation coefficient (r statistic) as implemented in the R stats library77. We built a complementary species distance matrix by defining dissimilarity as 1-r., and we analyzed this dissimilarity matrix using a Principal Coordinate Analysis (PCoA) as implemented in the R ape library78, with default parameters. For each PCoA, we represented the two vectors with the highest fraction of explained variance, which were in all cases higher than the fractions expected under the broken stick model. In addition, we plotted binary OG profiles of each species in a presence/absence heatmap, produced using the heatmap.2 function in the R gplots library79. Species’ order was defined by a Ward hierarchical clustering of the aforementioned interspecific Pearson’s correlation coefficients. Ortholog order was defined by Ward hierarchical clustering on euclidean distances. All clustering and distance analyses were performed using R stats library77. We represented the raw species clustering (Pearson correlation + Ward) in a separate heatmap, scaling the color code to display positive Pearson correlation values (0–1). Finally, each sub-set of primary metabolism COGs was also analyzed separately (categories C, E, F, G, H, I, P, and Q) using the same hierarchical clustering as above to cluster species according to their metabolism gene content (based on Pearson + Ward). We carried out similar comparisons and statistical analyses for proteins involved in phagotrophy, membrane trafficking and cytoskeleton. Briefly, we looked for proteins broadly related to those systems (actin cytoskeleton, exocytosis, fungal vacuole, exosome, endosome, mitochondrial biogenesis, ER-Golgi, lysosome, autophagy, peroxisome) in the KEGG BRITE reference database (http://www.genome.jp/kegg/brite.html). We identified 849 proteins related to membrane trafficking (ko04131), 279 cytoskeletal proteins (ko04812) and 361 proteins related to phagotrophy (KEGG map04144_endocytosis, map04145_phagosome, map04146_peroxisome, map04142_lysosome, map04810_actin_regulation, map04141_autophagy and map04071_sphingolipid_signal), which were used to carry out PCoA and clustering analyses as above (Supplementary Fig. 4). Since the eggNOG annotation file did not provide specific KEGG orthologs but only KEGG maps, we used the following approach to get the KEGG ortholog profile for the 41 taxa. All proteins in 35 eukaryote genomes80 were clustered to a non-redundant set using cd-hit at a 90% identity threshold. Those 35 nr90 genomes were compared in an all-vs-all BLAST analysis with an e-value cutoff of 1 × 10−3. The all-vs-all BLAST output was clustered using the MCL algorithm with an inflation parameter of 2.0. To avoid lineage specific proteins, the resultant clusters were retained if the proteins in the cluster were derived from 3 or more organisms. Proteins from each cluster were aligned using MAFFT, the alignments trimmed with trimAl, and HMM profiles built for each alignment using HMMer3. This analysis resulted in a set of 14,095 HMMs. All proteins from the UniProt-SwissProt database were searched against the entire set of 14,095 HMMs. Each HMM was assigned a best-hit protein annotation from the UniProt-SwissProt database. KEGG ortholog identifiers (IDs) associated with each HMM were obtained from UniProtKB IDs and KEGG ortholog IDs using the mappings available from UniProtKB. Additional KEGG ortholog IDs were mapped to the HMMs by searching the HMMs against all proteins annotated to a given KEGG ortholog ID. The best hit bit score among those proteins to the HMM was compared to the best-hit bit score of the UniProtKB annotation for that HMM. If the best hit KEGG ortholog ID bit score was at least 80% of the best hit UniProtKB protein bit score, and there was no KEGG ortholog ID already associated with the UniProtKB annotation, the KEGG ortholog ID was transferred to that HMM. To build a presence-absence matrix associated with KEGG ortholog IDs, the HMMs were searched against all proteins in a genome. Best hit HMMs below threshold (total protein e-value ≤ 1 × 10−5 and a best single domain e-value ≤ 1e−4) were assigned for each protein in a genome. The best hit HMMs and their associated KEGG ortholog IDs were used for presence/absence calls. If at least one protein in a genome had a best hit to a given HMM, the associated KEGG ortholog was considered present for that genome. From a total of 1568 orthologs associated with the KEGG maps of interest, 633 KEGG ortholog IDs were identified in the UniProtKB annotations of the HMMs. An additional 62 KEGG ortholog IDs were mapped to HMMs by considering similarity of bit scores between the best UniProt/SwissProt protein hit to the HMM and the best KEGG ortholog ID protein hit to the HMM (Supplementary Data 3). Those 695 KEGG ortholog IDs were used to perform the same multivariate statistical analyses as per the metabolism section for the 41 taxa. The phagolysosome KEGG maps in Paraphelidium tribonemae and Rozella allomycis were compared using KEGG tools. To do so, we first annotated the two protein sets using BlastKOALA81 with eukaryotes as taxonomy group and genus_eukaryotes KEGG GENES database. We then uploaded our annotations in the KEGG Mapper Reconstruct Pathway and BRITE servers.
Code availability
Custom scripts have been deposited in Github (https://github.com/xgrau/paraphelidium2018).
Data availability
Raw read sequences have been deposited in NCBI under accession number PRJNA402032. Paraphelidium tribonemae metatranscriptomic nucleotide contig assembly and version 1.5 of the predicted proteome are deposited in figshare82,83 under Creative Commons 4.0 licence. All trees are available as Supplementary file all_trees.txt.
References
Richards, T. A., Leonard, G. & Wideman, J. G. What defines the “Kingdom” fungi? In Heitman et al. (ed.), The fungal Kingdom. p. 57–77 (ASM Press, Washington, 2017).
James, T. Y. et al. Reconstructing the early evolution of Fungi using a six-gene phylogeny. Nature 443, 818–822 (2006).
James, T. Y. et al. A molecular phylogeny of the flagellated fungi (Chytridiomycota) and description of a new phylum (Blastocladiomycota). Mycologia 98, 860–871 (2006).
Lara, E., Moreira, D. & Lopez-Garcia, P. The environmental clade LKM11 and Rozella form the deepest branching clade of fungi. Protist 161, 116–121 (2010).
Jones, M. D. et al. Discovery of novel intermediate forms redefines the fungal tree of life. Nature 474, 200–203 (2011).
Bass, D. et al. Clarifying the relationships between microsporidia and cryptomycota. J. Eukaryot. Microbiol. https://doi.org/10.1111/jeu.12519 (2018).
Karpov, S. A. et al. Morphology, phylogeny, and ecology of the aphelids (Aphelidea, Opisthokonta) and proposal for the new superphylum Opisthosporidia. Front. Microbiol. 5, 112 (2014).
Berbee, M. L., James, T. Y. & Strullu-Derrien, C. Early diverging fungi: diversity and impact at the dawn of Terrestrial life. Annu. Rev. Microbiol. 71, 41–60 (2017).
Spatafora, J. W. et al. The fungal tree of life: from molecular systematics to genome-scale phylogenies. In Heitman et al. (ed.), The fungal Kingdom. p. 3–34 (ASM Press, Washington, DC, 2017).
Gromov, B. V. Algal parasites of the genera Aphelidium, Amoeboaphelidium, and Pseudaphelidium from the Cienkovski’s “monadinea” group as representatives of a new class. Zool. Zh. 79, 517–525 (2000).
Karpov, S. A. et al. Morphological and genetic diversity of Opisthosporidia: new aphelid Paraphelidium tribonemae gen. et sp. nov. J. Eukaryot. Microbiol. 64, 204–212 (2017).
Karpov, S. A. et al. Obligately phagotrophic aphelids turned out to branch with the earliest-diverging fungi. Protist 164, 195–205 (2013).
Corradi, N. Microsporidia: eukaryotic intracellular parasites shaped by gene loss and horizontal gene transfers. Ann. Rev. Microbiol. 69, 167–183 (2015).
Letcher, P. M. et al. Characterization of Amoeboaphelidium protococcarum, an algal parasite new to the Cryptomycota isolated from an outdoor algal pond used for the production of biofuel. PLoS One 8, 15 (2013).
Mikhailov, K. V., Simdyanov, T. G. & Aleoshin, V. V. Genomic survey of a hyperparasitic microsporidian Amphiamblys sp. (Metchnikovellidae). Genome Biol. Evol. 9, 454–467 (2017).
James, T. Y. et al. Shared signatures of parasitism and phylogenomics unite Cryptomycota and Microsporidia. Curr. Biol. 23, 1548–1553 (2013).
Simao, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Torruella, G. et al. Phylogenomics reveals convergent evolution of lifestyles in close relatives of animals and fungi. Curr. Biol. 25, 2404–2410 (2015).
Capella-Gutierrez, S., Marcet-Houben, M. & Gabaldon, T. Phylogenomics supports microsporidia as the earliest diverging clade of sequenced fungi. BMC Biol. 10, 47 (2012).
Haag, K. L. et al. Evolution of a morphological novelty occurred before genome compaction in a lineage of extreme parasites. Proc. Natl Acad. Sci. USA 111, 15480–15485 (2014).
Quandt, C. A. et al. The genome of an intranuclear parasite, Paramicrosporidium saccamoebae, reveals alternative adaptations to obligate intracellular parasitism. Elife 6, e29594 (2017).
Lartillot, N. & Philippe, H. A Bayesian mixture model for across-site heterogeneities in the amino-acid replacement process. Mol. Biol. Evol. 21, 1095–1109 (2004).
Le, S. Q., Lartillot, N. & Gascuel, O. Phylogenetic mixture models for proteins. Philos. Trans. R. Soc. Lond. B 363, 3965–3976 (2008).
Grau-Bove, X. et al. Dynamics of genomic innovation in the unicellular ancestry of animals. eLife 6, e26036 (2017).
Ruiz-Herrera, J. & Ortiz-Castellanos, L. Analysis of the phylogenetic relationships and evolution of the cell walls from yeasts and fungi. Fems. Yeast. Res. 10, 225–243 (2010).
James, T. Y. & Berbee, M. L. No jacket required—new fungal lineage defies dress code: recently described zoosporic fungi lack a cell wall during trophic phase. Bioessays 34, 94–102 (2012).
Merzendorfer, H. The cellular basis of chitin synthesis in fungi and insects: common principles and differences. Eur. J. Cell Biol. 90, 759–769 (2011).
Goncalves, I. R. et al. Genome-wide analyses of chitin synthases identify horizontal gene transfers towards bacteria and allow a robust and unifying classification into fungi. BMC Evol. Biol. 16, 252 (2016).
Vavra, J. & Lukes, J. Microsporidia and ‘the art of living together’. Adv. Parasitol. 82, 253–319 (2013).
Zhao, Y., Park, R. D. & Muzzarelli, R. A. Chitin deacetylases: properties and applications. Mar. Drugs 8, 24–46 (2010).
Viens, P., Lacombe-Harvey, M. E. & Brzezinski, R. Chitosanases from family 46 of glycoside hydrolases: from proteins to phenotypes. Mar. Drugs 13, 6566–6587 (2015).
Udaya Prakash, N. A. et al. Evolution, homology conservation, and identification of unique sequence signatures in GH19 family chitinases. J. Mol. Evol. 70, 466–478 (2010).
Cleare, M. & Percival, E. Carbohydrates of the fresh water alga Tribonema aequale. I. Low molecular weight and polysaccharides. Brit. Phycol. J. 7, 185–193 (1972).
Kubicek, C. P., Starr, T. L. & Glass, N. L. Plant cell wall-degrading enzymes and their secretion in plant-pathogenic fungi. Annu. Rev. Phytopathol. 52, 427–451 (2014).
Murphy, C., Powlowski, J., Wu, M., Butler, G. & Tsang, A. Curation of characterized glycoside hydrolases of fungal origin. Database 2011, bar020 (2011).
McAndrew, R. P. et al. From soil to structure, a novel dimeric beta-glucosidase belonging to glycoside hydrolase family 3 isolated from compost using metagenomic analysis. J. Biol. Chem. 288, 14985–14992 (2013).
Aspeborg, H. et al. Evolution, substrate specificity and subfamily classification of glycoside hydrolase family 5 (GH5). BMC Evol. Biol. 12, 186 (2012).
Huerta-Cepas, J. et al. eggNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 44, D286–D293 (2016).
Okuda, S. et al. KEGG Atlas mapping for global analysis of metabolic pathways. Nucleic Acids Res. 36, W423–W426 (2008).
Roshchina, V. V. New Trends and perspectives in the evolution of neurotransmitters in microbial, plant, and animal cells. Adv. Exp. Med. Biol. 874, 25–77 (2016).
Yutin, N., Wolf, M. Y., Wolf, Y. I. & Koonin, E. V. The origins of phagocytosis and eukaryogenesis. Biol. Direct. 4, 9 (2009).
Boulais, J. et al. Molecular characterization of the evolution of phagosomes. Mol. Syst. Biol. 6, 423 (2010).
Sebé-Pedrós, A., Grau-Bové, X., Richards, T. A. & Ruiz-Trillo, I. Evolution and classification of myosins, a paneukaryotic whole-genome approach. eLife 6, 290–305 (2014).
Sebé-Pedrós, A. et al. Insights into the origin of metazoan filopodia and microvilli. Mol. Biol. Evol. 30, 2013–2023 (2013).
Fritz-Laylin, L. K., Lord, S. J. & Mullins, R. D. WASP and SCAR are evolutionarily conserved in actin-filled pseudopod-based motility. J. Cell. Biol. 216, 1673–1688 (2017).
Held, A. A. Zoospore of Rozella allomycis—ultrastructure. Can. J. Bot. 53, 2212–2223 (1975).
Held, A. A. Development of rozella in allomyces—a single zoospore produces numerous zoosporangia and resistant sporangia. Can. J. Bot. 58, 959–979 (1980).
Letcher, P. M. et al. Morphological, molecular, and ultrastructural characterization of Rozella rhizoclosmatii, a new species in Cryptomycota. Fungal Biol. 121, 1–10 (2017).
Hehenberger, E. et al. Novel predators reshape Holozoan phylogeny and reveal the presence of a two-component signaling system in the ancestor of animals. Curr. Biol. 27, 2043–2050.e2046 (2017).
Corsaro, D. et al. Microsporidia-like parasites of amoebae belong to the early fungal lineage Rozellomycota. Parasitol. Res. 113, 1909–1918 (2014).
Karpov, S. A., Torruella, G., Moreira, D., Mamkaeva, M. A. & López-García, P. Molecular phylogeny of Paraphelidium letcheri sp. nov. (Aphelida, Opisthosporidia). J. Eukaryot. Microbiol. 64, 573–578 (2017).
Cavalier-Smith, T. Early evolution of eukaryote feeding modes, cell structural diversity, and classification of the protozoan phyla Loukozoa, Sulcozoa, and Choanozoa. Eur. J. Protistol. 49, 115–178 (2013).
Karpov, S. A., Mamkaeva, M. A., Benzerara, K., Moreira, D. & Lopez-Garcia, P. Molecular phylogeny and ultrastructure of Aphelidium aff. melosirae (Aphelida, Opisthosporidia). Protist 165, 512–526 (2014).
Tedersoo, L. & Lindahl, B. Fungal identification biases in microbiome projects. Environ. Microbiol. Rep. 8, 774–779 (2016).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011).
Lagesen, K. et al. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108 (2007).
Laetsch, D. R. & Blaxter, M. L. BlobTools: Interrogation of genome assemblies [version 1; referees: 2 approved with reservations]. F1000Res. 6, 1287 (2017).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60 (2015).
Fu, L., Niu, B., Zhu, Z., Wu, S. & Li, W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152 (2012).
Cock, J. M. et al. The Ectocarpus genome and the independent evolution of multicellularity in brown algae. Nature 465, 617–621 (2010).
Armbrust, E. V. et al. The genome of the diatom Thalassiosira pseudonana: ecology, evolution, and metabolism. Science 306, 79–86 (2004).
Katoh, K., Kuma, K., Toh, H. & Miyata, T. MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 33, 511–518 (2005).
Capella-Gutiérrez, S., Silla-Martinez, J. M. & Gabaldon, T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973 (2009).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS One 5, e9490 (2010).
Kearse, M. et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649 (2012).
Lartillot, N., Lepage, T. & Blanquart, S. PhyloBayes 3: a Bayesian software package for phylogenetic reconstruction and molecular dating. Bioinformatics 25, 2286–2288 (2009).
Nguyen, L. T., Schmidt, H. A., von Haeseler, A. & Minh, B. Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2015).
Minh, B. Q., Nguyen, M. A. & von Haeseler, A. Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 30, 1188–1195 (2013).
Anisimova, M., Gil, M., Dufayard, J.-F., Dessimoz, C. & Gascuel, O. Survey of branch support methods demonstrates accuracy, power, and robustness of fast likelihood-based approximation schemes. Syst. Biol. 60, 685–699 (2011).
Miller, M. A., Pfeiffer, W. & Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. Proceedings of the Gateway Computing Environments Workshop (GCE), p. 1–8 (New Orleans, LA, 2010).
Mesquite: a modular system for evolutionary analysis. Version3.40 (2018).
PHYLIP - Phylogeny Inference Package v. 3.6 (University of Washington, Seattle, WA, 1999).
Finn, R. D., Clements, J. & Eddy, S. R. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39, W29–W37 (2011).
Finn, R. D. et al. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 44, D279–D285 (2016).
R: A language and environment for statistical computing. v. http://www.r-project.org (R Foundation for Statistical Computing, Vienna, Austria, 2017).
Paradis, E., Claude, J. & Strimmer, K. APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20, 289–290 (2004).
Warnes, G. R. et al. gplots: various R programming tools for plotting data. R. Package Version 2, 1 (2016).
Burns, J. A., Pittis, A. A. & Kim, E. Gene-based predictive models of trophic modes suggest Asgard archaea are not phagocytotic. Nat. Ecol. Evol. 2, 697–704 (2018).
Kanehisa, M., Sato, Y. & Morishima, K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 428, 726–731 (2016).
Torruella, G. et al. https://doi.org/10.6084/m9.figshare.7339469.v1 (2018).
Torruella, G. & Ruiz-Trillo, I. Transcriptome of Parvularia atlantis (Nuclearia sp. ATCC50694) https://doi.org/10.6084/m9.figshare.3898485.v4 (2017).
Acknowledgements
We are grateful to Wayne Pfeiffer and Mark Miller from the CIPRES Science Gateway (http://www.phylo.org/) for computational resources and personal assistance. We thank Victoria S. Tcvetkova for micromanipulating infected Tribonema sp. filaments in view of establishing clean aphelid cultures, Alex de Mendoza for providing CHS alignments, Philippe Deschamps, Rafael Ponce, Guillaume Reboul, Ana Gutiérrez-Preciado, Ares Rocañín-Arjó and Ricardo Rodríguez de la Vega for discussion on bioinformatics and statistics aspects, and Peter Kubicek and Bernard Hernissat for helpful discussion about cellulases. Research leading to these results received funding from the European Research Council under the European Union’s Seventh Framework Program (ERC Grant Agreement 322669 ProtistWorld). G.T. was also financed by the European Marie Sklodowska-Curie Action (704566 AlgDates). S.K. was supported by RSF grant No 16–14–10302, ZIN RAS program АААА-А17–117030310322–3 and’Jean d’Alembert’ program (Université Paris-Saclay).
Author information
Authors and Affiliations
Contributions
G.T., D.M., and P.L-G. conceived and coordinated the study. G.T. performed culture cleaning, RNA extraction, de novo transcriptome assembly, phylogenomics and comparative genomic analyses. G.T. and X.G-B. cleaned the protein set from contamination. X.G-B. and A.S-B. performed myosin comparative genomic analyses. X.G-B. carried out multivariate statistical analyses of metabolic genes. G.T. and J.A.B. carried out phagolysosome protein analyses. G.T. and S.K. maintained cultures and performed WGA staining and imaging. S.K. studied morphology and life-cycle aspects, E.V. performed SEM fixation and imaging. P.L-G. and G.T. wrote the manuscript. All authors commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Torruella, G., Grau-Bové, X., Moreira, D. et al. Global transcriptome analysis of the aphelid Paraphelidium tribonemae supports the phagotrophic origin of fungi. Commun Biol 1, 231 (2018). https://doi.org/10.1038/s42003-018-0235-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-018-0235-z
This article is cited by
-
Genomes of fungi and relatives reveal delayed loss of ancestral gene families and evolution of key fungal traits
Nature Ecology & Evolution (2023)
-
Intercomparison of Two Fluorescent Dyes to Visualize Parasitic Fungi (Chytridiomycota) on Phytoplankton
Microbial Ecology (2023)
-
Associated bacterial microbiome responds opportunistic once algal host Scenedesmus vacuolatus is attacked by endoparasite Amoeboaphelidium protococcarum
Scientific Reports (2022)
-
Divergent genomic trajectories predate the origin of animals and fungi
Nature (2022)
-
Phylogenomics of a new fungal phylum reveals multiple waves of reductive evolution across Holomycota
Nature Communications (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
