
Abstract
The ability to monitor, in real time, the mechanical forces on tendons after surgical repair could allow personalized rehabilitation programmes to be developed for recovering patients. However, the development of devices capable of such measurements has been hindered by the strict requirements of biocompatible materials and the need for sensors with satisfactory performance. Here we report an implantable pressure and strain sensor made entirely of biodegradable materials. The sensor is designed to degrade after its useful lifetime, eliminating the need for a second surgery to remove the device. It can measure strain and pressure independently using two vertically isolated sensors capable of discriminating strain as small as 0.4% and the pressure exerted by a grain of salt (12 Pa), without them interfering with one another. The device has minimal hysteresis, a response time in the millisecond range, and an excellent cycling stability for strain and pressure sensing, respectively. We have incorporated a biodegradable elastomer optimized to improve the strain cycling performances by 54%. An in vivo study shows that the sensor exhibits excellent biocompatibility and function in a rat model, illustrating the potential applicability of the device to the real-time monitoring of tendon healing.
Similar content being viewed by others


Main
In the USA alone, around 14 million people per year suffer from tendon, ligament and joint injuries1. After injury, tissues in the body undergo changes in their native biomechanical properties so as to repair themselves. This is true for both hard tissue (bones) and soft tissue (tendons, skin, muscles). The objective of surgery and rehabilitation is to restore the tissues to their pre-injury function, with biomechanical properties as close as possible to native properties2. A diagnostic tool that measures the biomechanical properties of the repair site in real time would represent a significant step towards improved assessment of healing and the development of personalized rehabilitation strategies3.
Current clinical practice for monitoring tissue rehabilitation includes magnetic resonance imaging (MRI) or ultrasound, which provide a snapshot of tissue density and inflammation4. Implantable sensors could give continuous information about tissue strain during rehabilitation protocols, as well as during a patient's daily activities, allowing activities to be tailored based on what the tissue can tolerate. Previously described implantable sensors have limited biocompatibility or have been designed for laboratory biomechanics studies rather than clinical practice4,5.
Implantable sensors for rehabilitation require the measurement of both strain and pressure with high sensitivity and fast response time. The level of strain and strain rate are the most important parameters to characterize the biomechanical properties of soft tissues and their healing stage5. The sensor must be able to measure typical tendon strains (<10%) without impeding the natural movement of the tendon4,5,6. Moreover, it should be able to measure the pressure exerted on repaired sites, which directly impacts the healing profile4,5. Finally, a sensor entirely made of biodegradable materials would avoid the need for surgical extraction7,8.
Previously described degradable sensors have either had insufficient sensing performance or use materials with unproven biocompatibility9,10,11,12,13. In addition to biocompatibility, an important feature of our design is the ability of the sensor to discriminate between strain and pressure measurements. Moreover, we have designed a new type of flexible strain sensor stacked with a pressure sensor to reduce the overall sensor footprint. Our sensor, we believe, satisfies all of the requirements for tissue rehabilitation monitoring, including biodegradability, biocompatibility and an ability to discriminate strain and pressure stimuli with excellent sensitivity.
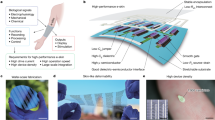
After tendon repair, several rehabilitation protocols are used with the objective of finding a balance between active exercise (to prevent tendon adhesions) and immobilization (to protect the integrity of the repair and avoid tendon rupture)14. Early active motion protocols show promising results. However, in current clinical practice, to avoid rehabilitation-related injuries, protocols with predefined long timescales and large security margins are applied, resulting in slow and costly rehabilitation protocols, which negatively impact the quality of life of the patient. The sensor proposed here could be used to assess real-time tissue healing, allowing personalization of a rehabilitation protocol (Fig. 1a)14,15.

a, The sensor can be attached to a tendon for real-time healing assessment, allowing the rehabilitation protocol after a tendon repair to be personalized for each patient14,15. b, Concepts used for strain and pressure sensing. Strain sensing: On application of strain, the two thin-film comb electrodes slide relative to each other, resulting in variation of the capacitance. The range of 0–15% for strain sensing is chosen based on the fact that in vivo the strain exerted on tendons is lower than 10% (refs 4,5,6). Pressure sensing: On application of pressure, variation of the distance between the top and bottom electrodes results in variation of the capacitance. The dielectric layer, made of a thin, highly compressible, regularly microstructured rubber, enables the sensor to have high pressure sensitivity and a fast response time, improving the sensitivity by several orders of magnitude compared to previously published work based on an air gap approach9,8. c, Materials and overall assembly of the fully biodegradable strain and pressure sensor. The biodegradable elastomer PGS (poly(glycerol sebacate))17 is used as a dielectric layer for the capacitor constituting the pressure sensor8. It is also used in the strain sensor architecture as a stretchable non-sticking layer, allowing the electrodes to slide relative to each other. The biodegradable elastomer POMaC (poly(octamethylene maleate (anhydride) citrate)) is used for the strain sensor and packaging18. POMaC is a soft stretchable biodegradable elastomeric biomaterial synthesized from citric acid, maleic anhydride and 1,8-octanediol, which is able to mimic the mechanical properties of a wide range of soft biological tissues18. PLLA is the substrate layer for the magnesium electrodes. d, Picture of the assembled sensor.
Biodegradable sensor concept and fabrication
Our flexible sensor design incorporates two vertically stacked sensors to allow the independent discrimination of strain and pressure (Fig. 1b). This is a unique aspect of our technology, because strain gauges and nanocomposite-based strain sensors mounted on a flexible membrane are intrinsically sensitive to both strain and pressure. The strain is determined by measuring the capacitance change between two thin-film comb electrodes sliding relative to each other. They are sandwiched between two stretchable elastomer layers. Meanwhile, pressure is measured with a thin, flexible capacitor with our previously reported microstructured elastic dielectric layer for high sensitivity8,16, while both substrates supporting the top and bottom electrodes, respectively, are bonded on only one side to the stretchable package. This design allows the pressure sensor to be free of the influence of strain.
Our work presents the implementation of fully stretchable and biodegradable organic materials as a biomechanical sensor. We primarily use organic materials because they offer the advantages of versatility in molecular tuning for desirable degradation kinetics, easy processing and mass production capabilities. We select materials that are well established for their excellent biocompatibility upon degradation, potentially reducing the timeline for clinical translation. In contrast, the cytotoxicity of carbon nanotubes (CNTs) may prevent the use of degradable CNT-PLLA (polylactic acid) composites in biomedical implants12,13. Moreover, as illustrated in Fig. 1c and Supplementary Fig. 1, assembly of the sensor is a simple benchtop process involving lamination and packaging with a UV-cured sealant. This simple process may be scaled readily.
The key elements of our material design are the two biodegradable elastomers poly(glycerol sebacate) (PGS)17 and poly(octamethylene maleate (anhydride) citrate) (POMaC)18. Both materials were initially developed for tissue engineering applications inside the body. PGS is approved by the US Food and Drug Administration (FDA) for biomedical use, while POMaC has been subject to extensive biocompatibility studies, demonstrating its cell and tissue biocompatibility comparable to that of PLLA control19,20. In addition to their established biocompatibility upon degradation, they are excellent candidates for this application in terms of their mechanical properties and degradation characteristics, which can be tuned by varying the polymerization conditions17,18. The electrodes are made of Mg evaporated on top of a biodegradable polymer substrate (PLLA). The biodegradable metal Mg was chosen because of its easy processing, biocompatibility and rapid rate of hydrolysis. It has been in use for more than a decade in clinical trials in applications such as biodegradable stents21, and is considered an essential nutrient (a daily allowance of 0.7 g)22. On degradation of the sensor, body fluids will eventually penetrate through the packaging layer, resulting in the corrosion of Mg and the formation of highly soluble magnesium oxides that will be evacuated via natural paths23.
POMaC elastomer biodegradation and resistance to cycling
The desired material properties for the POMaC elastomer are defined based on two key sensor requirements: (1) the sensor must be stretchable with a low tensile modulus in order to avoid limiting motion or hindering the healing process; (2) the sensor must be resistant to cycling without breaking (to support repeated rehabilitation exercises) when exposed to physiological conditions5.
The target initial tensile modulus for POMaC is defined to be ~0.5 MPa, soft enough for high mechanical compliance and stiff enough for easy sensor assembly and manipulation. As a reference, the tensile modulus of human hand flexor tendons is 200–300 MPa (ref. 24). As illustrated in Fig. 2a, after synthesis of pre-POMaC, the POMaC elastomer can be further polymerized following different paths, resulting in PPOMaC (UV irradiation polymerization), EPPOMaC (UV irradiation followed by oven post-polymerization) or EPOMaC (oven post-polymerization)18. Different mechanical properties and biodegradation profiles are expected for PPOMaC, EPPOMaC and EPOMaC when exposed to physiological media, since the crosslinking mechanisms and chemical bonds involved are different18.

a, POMaC polymerization paths. In EPOMaC, the crosslinking is a polycondensation without photocrosslinking, resulting in ester bond crosslinked POMaC. In PPOMaC, exposure to UV irradiation results in photocrosslinking with free-radical polymerization through the vinyl-carrying carbons. In EPPOMaC, post-polymerization results in further crosslinking of the polymer through the available free functional groups of citric acid to yield ester bond crosslinked photocrosslinked POMaC18. b, Schematic of strain cycles applied on POMaC samples to test their fatigue life (number of cycles before rupture). The applied cycling strain is initially 0–60% and increases every hours (720 cycles), resulting eventually in sample rupture. PPOMaC produced with 48 h UV exposure results in samples with a target tensile modulus of 0.5 MPa. However, because of the sample's sticky gel aspect making the manipulation challenging, PPOMaC is not further considered for sensor application. EPPOMaC produced with 20 min UV exposure followed by 48 h oven post-polymerization also results in a tensile modulus of 0.5 MPa. EPPOMaC exhibits a 33% increase in fatigue life compared to PPOMaC. EPPOMaC produced with 48 h oven post-polymerization results in samples with a tensile modulus of 0.3 MPa, below the 0.5 MPa target value. The fatigue life is found to be approximately three times higher in EPOMaC than in EPPOMaC. This is explained by the lower tensile modulus of EPOMaC, resulting in a more elastic, less brittle material. c, POMaC elastomer in vitro degradation study. When POMaC is exposed to physiological media, the tensile modulus decreases at a rate of −11% per week for EPPOMaC and 14% per week for EPOMaC. The difference in degradation rates is explained by additional non-hydrolysable UV links that maintain the mechanical cohesion in EPPOMaC as compared to EPOMaC. Cycling tests are performed on EPPOMaC and EPOMaC after in vitro degradation, showing the superiority of EPPOMaC compared to EPOMaC, with 54% higher fatigue life. This result is found for materials with identical tensile moduli (EPPOMaC incubated for 6 weeks and EPOMaC for 4 weeks, both with a tensile modulus of 0.14 MPa). This condition allows for comparison of the cycling performances. d, Stress–strain characteristics for EPOMaC and EPPOMaC for various incubation durations.
An in vitro degradation study was performed over 8 weeks, with samples incubated in phosphate buffered saline (PBS) solution at 37 °C. The fatigue life (number of cycles before rupture) was tested using the scheme in Fig. 2b, and the tensile moduli upon degradation are shown in Fig. 2c,d. This study shows the superiority of EPPOMaC compared to other polymerization conditions, with 54% longer fatigue life. The slower degradation rate of the tensile modulus in EPPOMaC as compared to EPOMaC (11% and 14% per week, respectively) is explained by additional non-hydrolysable UV-induced crosslinks retaining the mechanical cohesion longer in EPPOMaC.
Biodegradable strain and pressure sensor characterization
Sensor performance is illustrated in Fig. 3. Figure 3a shows the strain sensor response curves from five consecutive linear loading–unloading cycles. After applying a constant strain of 10% and releasing to 5% for more than 20,000 cycles, minimum and maximum capacitance (Cmin and Cmax) values increase by 11% and 8%, respectively (Fig. 3g). Moreover, there is a one order of magnitude increase in sensitivity to strain as compared to previous reports for transient strain sensors11. Indeed, for an applied strain of 15%, the relative capacitance change of our sensor is ΔC/C0 = ~50%, where C and C0 are the capacitances with and without applied strain and ΔC = |C − C0|. As a comparison, degradable Si-based strain gauges exhibited a signal change of ~5% for the same applied strain11.

a, Strain response curve for five consecutive linear loading–unloading cycles (applied strain of 0–15%). The hysteresis is observed to be negligible. b, Pressure response curve from six consecutive cycles (applied pressure of 0–100 kPa). Again, negligible hysteresis is observed. c,d, Evaluation of the smallest detectable strain (c, signal-to-noise ratio SNR = ~2.1, no averaging applied, real-time measurement) and the smallest detectable pressure (d, the signal corresponds to the SNR detection limit, again with no averaging applied, real-time measurement). e,f, Sensor response time to applied strain (e) and applied pressure (f). g,h, Cycling tests and stability of the strain response (g, applied strain 5–10%) and the pressure response (h, applied pressure 15–45 kPa).
Figure 3b shows the pressure sensor response curves from six consecutive cycles for sensors with POMaC packaging for encapsulation. After applying a constant pressure of 45 kPa and releasing to 15 kPa for more than 30,000 cycles, Cmin increased only by 5% and Cmax decreased by only 2%. Moreover, the pressure sensitivity is 0.7 ± 0.4 kPa−1 in the low-pressure regime (p < 1 kPa) and 0.13 ± 0.03 kPa−1 at higher pressures (5 < p < 10 kPa). These values are in good agreement with our previous results8. These pressure sensitivities are one to three orders of magnitude higher than previously published degradable sensors9. Consecutive pressure measurements with increasing maximum pressures (Supplementary Fig. 2) indicate that the device output is highly reproducible even with large applied pressures up to 430 kPa. The high sensitivity for small applied strains and pressures is illustrated in Fig. 3c,d, respectively, where strains as low as 0.4% and pressures as low as 12 Pa (corresponding to a grain of salt) are successfully measured. In addition, the sensor can measure strain and pressure independently from each other, as illustrated in Supplementary Fig. 3. The fast response time of our device in the millisecond range is illustrated in Fig. 3e,f for strain and pressure sensing, respectively. Moreover, the strain and pressure response curves of the sensor can be reproducibly cycled thousands of times. The fast response time and cycling durability satisfy the requirements for real-time biomonitoring for orthopaedic rehabilitation.
Sensor in vivo function and biocompatibility
In vitro and in vivo studies were performed to verify that the biodegradable strain and pressure sensor will be functional (Fig. 4) and well tolerated inside the body (Fig. 5). The sensor, designed to stay operational in vivo for more than 2 weeks, slowly degrades after its period of use when immersed in physiological media (Supplementary Fig. 4). This period corresponds to the duration of interest for monitoring tendon healing, with rehabilitation protocols starting as early as 24 h post-operatively14,15. The performance of the sensor immersed in PBS solution at 25 °C was assessed over 5 weeks and compared to a reference sensor made of non-biodegradable materials (Supplementary Fig. 5). Stable operation over 2 to 3 weeks is observed, with sensitivity comparable to the reference sensor, until the degradation of the Mg electrodes occurs (Supplementary Fig. 5b-e). As shown in Fig. 4a, the sensor can be successfully used to measure the physiological strain signal on a real tendon.

a, Top, Sensor fixed on a tendon mounted in a stress–strain Instron microtester, allowing for measurement of the tensile modulus of the tendon, 201 MPa, which is in good agreement with literature values (0–14% strain, strain rate 0.25 mm s−1)24. Bottom, The signal measured with the strain sensor is compared to the reference microtester signal, showing a linear relation, with deviation from linear fit better than R2 = 89% (R2 = coefficient of determination). In vivo study: b, The biodegradable strain and pressure sensor was subcutaneously implanted on the back of a Sprague–Dawley rat. c, Strain and pressure signals applied on the implanted sensor in vivo. d, Pressure signal recorded after 2 weeks (top left) and 3.5 weeks (top right) after sensor implantation. The corresponding baseline shows respiration of the animal recorded with the pressure sensor after 2 weeks (bottom left) and 3.5 weeks (bottom right) in vivo. e, Strain signal recorded after 2 weeks (top left) and 3.5 weeks (top right) after sensor implantation. The corresponding baseline shows the respiration of the animal recorded with the strain sensor, after 2 weeks (bottom left) and 3.5 weeks (bottom right) of sensor implantation.

a, Results of immunohistochemistry. After one week, the number of CD68-positive cells—indicating an inflammatory reaction—is larger in tissues surrounding the POMaC sample than in the tissues surrounding silicone sample. However, after 3 weeks and 8 weeks of implantation, there is no statistically significant difference between POMaC and silicone (control) samples, indicating good biocompatibility of POMaC in vivo. b, Results of paraffin sections for H&E staining. A fibrous capsule of comparable width is formed around both the POMaC and silicone (control) samples at 1, 3 and 8 weeks.
Sensors were implanted subcutaneously on the backs of Sprague–Dawley rats (Fig. 4b). Strain and pressure stimuli were applied in vivo as illustrated in Fig. 4c. Corresponding signals were successfully recorded after 3.5 weeks of implantation, as shown in Fig. 4d,e for pressure and strain sensing, respectively. The high sensitivity of our device both in strain and pressure assessment is illustrated in these figures, where a zoom on the baseline signal shows the recording of the respiration of the animal. The degradation behaviour of both POMaC and PGS is based on surface erosion rather than bulk erosion18,20,25. This mechanism allows the POMaC packaging to provide efficient protection against penetration of body fluids inside the sensor, preventing premature degradation of the Mg electrodes.
Moreover, the animals tolerated the presence of the sensor with no long-term adverse inflammatory reaction, as quantitatively assessed in Fig. 5. Both immunohistochemistry and H&E staining show the good biocompatibility of POMaC after 8 weeks of implantation, with no statistically significant difference between POMaC and silicone (control) sample.
Conclusions
The high-sensitivity, fast time response and biodegradability of our sensor means that it could also be of value in biomedical applications beyond orthopaedic rehabilitation monitoring. For example, it could be relevant to cardiovascular patches26 and reconstructive surgery27, where the monitoring of mechanical deformations and pressures in real time in vivo will allow for refined and personalized medicine. Future research will consist of developing a wireless system made entirely of biodegradable materials, including the circuit used for wireless transmission of measured signals though the skin.
Methods
Sensor fabrication
Synthesis of POMaC top and bottom encapsulation layers
The synthesis of PPOMaC and EPPOMaC was performed as described in ref. 18. Briefly, maleic anhydride (Fluka, CAS 108-31-6), citric acid (Sigma-Aldrich, CAS 77-92-9) and 1,8-octanediol (Sigma-Aldrich, CAS 629-41-4) were mixed in a three-necked round-bottom flask with a molar feed ratio of 3:2:5, respectively. The flask content was heated at an initial temperature of 160 °C and stirred under a nitrogen atmosphere. After the mixture had melted, the temperature was set to 140 °C and it was continuously stirred under nitrogen for 3 h. To remove any of the unreacted monomers and oligomers, the prepolymer was dissolved in tetrahydrofuran (THF, ~5 g in 20 ml), and purified by dropwise precipitation into 2 l of deionized water. Photocrosslinked POMaC networks (PPOMaC) were formed by crosslinking through free-radical polymerization. The photoinitiator 2-hydroxy-4′-(2-hydroxyethoxy)-2-methylpropiophenone (CAS 106797-53-9, 0.06 g) was dissolved in ethyl acetate (1 ml) and mixed with pre-POMaC (6 g) using a speed mixer at 3,000 r.p.m. for 3 min. The solution was then poured into a PTFE container. After solvent evaporation, the sample was first exposed to a 365 nm UV light lamp (25 W) for 20 min. It was then cured in an oven at 80 °C for 48 h to complete the polymerization process to give EPPOMaC, which was used as the encapsulation layer (thickness 0.8 mm).
Preparation of POMaC elastomers
In this study, three different POMaC networks were investigated, with polymerization protocols similar to those described in ref. 18. Photocrosslinked POMaC (PPOMaC) was produced by exposure to UV irradiation, where the free-radical polymerization was initiated to crosslink pre-POMaC through vinyl-carrying carbons (48 h UV exposure). Ester bond crosslinked POMaC (EPOMaC) was produced by polycondensation without photocrosslinking (48 h oven post-polymerization at 80 °C). Finally, ester bond crosslinked photocrosslinked POMaC (EPPOMaC) was produced by further crosslinking PPOMaC through the available free functional groups of citric acid (20 min UV exposure followed by 48 h oven post-polymerization at 80 °C). The cycling tests in Fig. 2b were performed at a speed of ~5 cycles per min.
Synthesis of PGS and fabrication of microstructured dielectric layer for the pressure sensor
PGS was synthesized based on previously published methods8,17, where an equimolar mixture of glycerol and sebacic acid was reacted at 120 °C under nitrogen for 24 h, resulting in a viscous uncrosslinked PGS prepolymer. Fabrication of the PGS microstructured dielectric layer is similar to that in our previous report8. Briefly, the PGS prepolymer was further polymerized in an oven at 130 °C in vacuum for 24 h. Afterwards, the highly viscous prepolymer was cured between the PDMS mould and a bare silicon wafer, both treated with a non-adhesive layer evaporated in vacuum ((tridecafluoro-1,1,2,2-tetrahydrooctyl)trichlorosilane, Gelest, CAS 78560-45-9). Two-dimensional arrays of square pyramids were formed into PGS from the PDMS mould. The PDMS mould itself was fabricated from a (100) Si-wafer mould that had etched arrays of pyramidal recesses. After being cured at 130 °C in vacuum for 15 h, the ~150-µm-thick PGS film was peeled off and laminated with the bottom and top electrodes. In addition to the PGS layer used as a dielectric in the pressure sensor, two additional PGS layers were used as non-sticking layers, allowing the strain sensor top and bottom electrodes to slide relative to each other. These two PGS layers were fabricated as described above, except that the PDMS mould had no square pyramids.
Fabrication of biodegradable metal electrodes
The electrodes were fabricated by evaporating Mg (100-µm-thick electrode) on top of a 50-µm-thick PLLA layer (Goodfellow) after having exposed the substrate surface to oxygen plasma. The electrodes were operated below the standard potential of −1.23 V (corresponding to the electrolysis of water) to avoid any unwanted redox reaction at the interface of the Mg electrodes with body fluids, and to stay within the safe water window28.
Sensor assembly
The sensor was assembled as described in Supplementary Fig. 1. The layers were laminated on top of each other, and the packaging was closed using a layer of UV-cured pre-POMaC as the sealing agent.
Characterization of the sensor
Strain and pressure response measurement set-up
The pressure measurement set-up consisted of a motorized vertical stage used in combination with a force gauge (digital force gauge series 5, Mark-10), while the capacitance of the sensor was measured with an E4980A Agilent Precision LCR meter. The strain measurement set-up consisted of a motorized horizontal stage, and the capacitance of the sensor was measured with the LCR meter. Measurements were performed in a controlled temperature and humidity atmosphere (23 ± 1 °C and 50 ± 10% relative humidity).
In vivo sensor function assessment
Implantation of sensor
Three Sprague–Dawley rats (12 weeks, 300–350 g, male, ENVIGO) were cared for in compliance with the regulations of the animal care and use committee of the Veteran Affair Palo Alto Health Care System Research Administration. Sensors were implanted in a subcutaneous paravertebral pocket under isoflurane inhalation anaesthesia. Each animal was administered a dose of enrofloxacin (Bayer) for antibiotic prophylaxis preoperatively and buprenorphine (Reckitt Benckiser Pharmaceuticals) for pain control post-operatively. The rats were monitored throughout the study. Wire and surgical sites were covered with an occlusive dressing between tests.
Sensor function
The function of the sensor was tested on days 0, 1, 3, 5, 7, 10, 14, 17, 21 and 24 after implantation. Tests were performed under isoflurane inhalation anaesthesia.
In vivo biocompatibility assessment
Implantation and harvest of materials
The biocompatibility of POMaC and silicone (control) were evaluated histologically. Nine Sprague–Dawley rats (12–14 weeks, 300–350 g, male, ENVIGO) were used for 1, 3 and 8 weeks of evaluation. Three rats for each time point evaluation underwent sham operations or material implantation surgeries. Under isoflurane inhalation anaesthesia, subcutaneous paravertebral pockets were created on the right and left sides of the upper backs of the rats. Six pockets of the three rats were randomly divided into three groups: POMaC, silicone and sham. In the POMaC and silicone groups, the tested materials were put into the pockets after sterilization. In the sham group, the wound was closed without implantation of material. The rats were monitored throughout the study. At 1, 3 and 8 weeks after implantation, rats were euthanized by CO2 inhalation, and the materials and their surrounding tissues were harvested. The samples were then cut in half longitudinally; half for paraffin sections for H&E staining and the other half for frozen sections for immunohistochemistry.
H&E analysis
After fixing the samples in 10% formalin, four 5-µm-thick paraffin sections were prepared per material at each time point, followed by H&E staining. The width of the fibrous capsules was measured at three points per section, which were selected at random on the superficial side of the capsule. The mean value of the three measurements was calculated and used for evaluations.
Immunohistochemistry for CD68
After fixing the sample in 4% paraformaldehyde (PFA), four 10-µm-thick longitudinal frozen sections were prepared per material at each time point for immunohistochemistry for CD68. After antigen retrieval using proteinase K (PK; Sigma-Aldrich), donkey serum (Sigma-Aldrich) was added to the slides as a blocking procedure. The slides were incubated with primary antibodies (rabbit polyclonal anti-CD68 antibodies (1:100; Abcam)), followed by incubation with secondary antibodies (donkey anti-rabbit IgG (H + L) whole antibodies (1:200; CFTM488 fluorescent reagents; Biotium)). The slides were viewed under a fluorescence microscope (Keyence BZ-X700; Keyence). More than six fields at ×10 magnification per section were selected at random in the area within 1 mm of the material on the superficial side. The number of CD68-positive cells in the fields was measured using ImageJ analysis software and the mean value was calculated per section.
Statistical analysis
All data are expressed as mean ± standard deviation. Data were compared using an unpaired t-test. P values of <0.05 were considered statistically significant.
Reporting Summary
Further information on experimental design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The data that support the plots within this paper and other findings of this study are available from the corresponding author upon reasonable request.
References
Yang, G., Rothrauff, B. B. & Tuan, R. S. Tendon and ligament regeneration and repair: clinical relevance and developmental paradigm. Birth Defects Res C. Embryo Today 99, 203–222 (2013).
Place, E. S., Evans, N. D. & Stevens, M. M. Complexity in biomaterials for tissue engineering. Nat. Mater. 8, 457–470 (2009).
McGilvray, K. C. et al. Implantable microelectromechanical sensors for diagnostic monitoring and post-surgical prediction of bone fracture healing. J. Orthop. Res. 33, 1439–1446 (2015).
Bogaerts, S., Desmet, H., Slagmolen, P. & Peers, K. Strain mapping in the Achilles tendon—a systematic review. J. Biomech. 49, 1411–1419 (2016).
Roriz, P. et al. From conventional sensors to fibre optic sensors for strain and force measurements in biomechanics applications: a review. J. Biomech. 47, 1251–1261 (2014).
Kubo, K., Kanehisa, H., Kawakami, Y. & Fukunaga, T. Influence of static stretching on viscoelastic properties of human tendon structure in vivo. J. Appl. Physiol. 90, 520–527 (2001).
Waugh, C. M., Blazevich, A. J., Fath, F. & Korff, T. Age-related changes in mechanical properties of the Achilles tendon. J. Anat. 220, 144–155 (2012).
Boutry, C. M. et al. A sensitive and biodegradable pressure sensor array for cardiovascular monitoring. Adv. Mater. 27, 6954–6961 (2015).
Luo, M., Martinez, A. W., Song, C., Herrault, F. & Allen, M. G. A microfabricated wireless RF pressure sensor made completely of biodegradable materials. J. Micro. Syst. 23, 4–13 (2014).
Hwang, S. W. et al. A physically transient form of silicon electronics. Science 28, 1640–1644 (2012).
Xu, L. et al. 3D multifunctional integumentary membranes for spatiotemporal cardiac measurements and stimulation across the entire epicardium. Nat. Commun. 5, 3329 (2014).
Liu, Y., Chakrabartty, S., Stamatis Gkinosatis, D., Mohanty, A. K. & Lajnef, N. Multi-walled carbon nanotubes/poly(L-lactide) nanocomposite strain sensor for biomechanical implants. Biomedical Circuits and Systems Conference BIOCAS 2007 119–122 (2007).
Singh, S. & Singh Nalwa, H. Nanotechnology and health safety—toxicity and risk assessments of nanostructured materials on human health. J. Nanosci. Nanotechnol. 7, 3048–3070 (2007).
Hein, C., Wilton, P. & Wongworawat, M. D. Review of flexor tendon rehabilitation protocols following zone II repair. Crit. Rev. Phys. Rehabil. Med. 27, 11–18 (2015).
Saini, N., Kundnani, V., Patni, P. & Gupta, S. P. Outcome of early active mobilization after flexor tendons repair in zones II–V in hand. Indian J. Orthop. 44, 314–321 (2010).
Mannsfeld, S. C. B. et al. Highly sensitive flexible pressure sensors with microstructured rubber dielectric layers. Nat. Mater. 9, 859–864 (2010).
Wang, Y., Ameer, G. A., Sheppard, B. J. & Langer, R. Atough biodegradable elastomer. Nat. Biotechnol. 20, 602–606 (2002).
Tran, R. T. et al. Synthesis and characterization of a biodegradable elastomer featuring a dual crosslinking mechanism. Soft Matter 6, 2449–2461 (2010).
Zhang, B. et al. Biodegradable scaffold with built-in vasculature for organ-on-a-chip engineering and direct surgical anastomosis. Nat. Mater. 15, 669–678 (2016).
Davenport Huyer, L. et al. Highly elastic and moldable polyester biomaterial for cardiac tissue engineering applications. ACS Biomater. Sci. Eng. 2, 780–788 (2016).
Purnama, A., Hermawan, H. & Mantovani, D. Biodegradable metal stents: a focused review on materials and clinical studies. J. Biomater. Tissue Eng. 7, 868–874 (2014).
Zheng, Y. F., Gu, X. N. & Witte, F. Biodegradable metals. Mater. Sci. Eng. R. 77, 1–34 (2014).
Yin, L. et al. Dissolvable metals for transient electronics. Adv. Funct. Mater. 24, 645–658 (2014).
Huang, H., Zhang, J., Sun, K., Zhang, X. & Tian, S. Effects of repetitive multiple freeze–thaw cycles on the biomechanical properties of human flexor digitorum superficialis and flexor pollicis longus tendons. Clin. Biomech. 26, 419–423 (2011).
Rai, R., Tallawi, M., Grigore, A. & Boccaccini, A. R. Synthesis, properties and biomedical applications of poly(glycerol sebacate) (PGS): a review. Prog. Polym. Sci. 37, 1051–1078 (2012).
Kapnisi, M., Mawad, D., Rathnayake-Arachchige, D., Conway, P. & Stevens, M. Auxetic micropatterning of a cell adhesive-conductive composite for cardiac tissue engineering. Proc. San Francisco MRS LL02.02 (2015).
Bannister, M. & Ah-See, K. W. Enhanced recovery programmes in head and neck surgery: systematic review. J. Laryngol. Otol. 129, 416–420 (2015).
Pour Aryan, N, Kaim, H. & Rothermel, A. Stimulation and Recording Electrodes for Neural Prostheses (Springer, New York, USA, 2015).
Acknowledgements
C.M.B. acknowledges postdoctoral fellowship support from the Swiss National Science Foundation (Postdoc Mobility Fellowship no. P2EZP2_152118) and the European Commission (Marie-Curie International Outgoing Fellowship grant no. 622362). Part of this work was performed at the Stanford Nano Shared Facilities (SNSF), supported by the National Science Foundation under award ECCS-1542152. B.C.S. acknowledges the National Research Fund of Luxembourg for financial support (project no. 6932623).
Author information
Authors and Affiliations
Contributions
C.M.B. is the main contributor to this work, elaborating the sensor concept, developing new fabrication processes, performing all experiments for materials investigations, device characterization, data collection for in vitro and in vivo experiments, data analysis and interpretation, and drafting of the article. B.C.S. contributed to the POMaC investigation, including polymer synthesis, chemical properties characterization, data analysis and interpretation, and provided critical revision of the article. A.C. contributed to the drafting of the article and provided critical revision. Y.K. performed all surgeries related to the in vivo studies, including design of the in vivo studies, development of the protocols, sensors implantation and operation studies, and materials biocompatibility studies. He also worked on data analysis and interpretation, and contributed to the drafting and critical revision of the article. A.L. performed all surgeries related to the tendon in vitro studies, and worked on data analysis and interpretation. Z.W., J.C. and P.F. contributed to the design of the in vivo studies, working on the protocols and data interpretation. P.F. also contributed to the drafting and critical revision of the article. Z.B. contributed to developing the sensor concept, fabrication processes and materials investigation, device characterization, data interpretation and critical revision of the article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Figures 1–5
Rights and permissions
About this article

Cite this article
Boutry, C.M., Kaizawa, Y., Schroeder, B.C. et al. A stretchable and biodegradable strain and pressure sensor for orthopaedic application. Nat Electron 1, 314–321 (2018). https://doi.org/10.1038/s41928-018-0071-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41928-018-0071-7
This article is cited by
-
Review of sustainable, eco-friendly, and conductive polymer nanocomposites for electronic and thermal applications: current status and future prospects
Discover Nano (2024)
-
Fully bioresorbable hybrid opto-electronic neural implant system for simultaneous electrophysiological recording and optogenetic stimulation
Nature Communications (2024)
-
Ultra-sensitive, highly linear, and hysteresis-free strain sensors enabled by gradient stiffness sliding strategy
npj Flexible Electronics (2024)
-
Creep-free polyelectrolyte elastomer for drift-free iontronic sensing
Nature Materials (2024)
-
Highly Elastic, Bioresorbable Polymeric Materials for Stretchable, Transient Electronic Systems
Nano-Micro Letters (2024)
