
Abstract
A single coating formulation for multifunctional composites, such as a gas barrier against both oxygen and water vapour, is the holy grail for the packaging industry. Since the last decade, graphene has been touted as the ideal barrier material in composites due to its morphology and impermeability to all gases. However, this prospect is limited by either poor dispersion of graphene or excess surfactants to aid the dispersion, both leading to shortcuts that allow gas permeation through the composite. Here, we demonstrate a combined gas barrier with starch-graphene composite films made from a single formulation of surfactant-free starch nanoparticle-stabilized graphene dispersion (2.97 mg mL−1). Hence, the incorporated graphene reduces the permeability of both the oxygen and the water vapour by over 70% under all the relative humidity conditions tested. Moreover, these films are foldable and electrically conductive (9.5 S m−1). Our surfactant-free approach of incorporating graphene into an industrially important biopolymer is highly relevant to the packaging industry, thus offering cost-effective and water-based solution depositions of multifunctional composite films for wide-ranging applications, such as gas barriers in food packaging.
Similar content being viewed by others

Introduction
Plastic pollutants from non-biodegradable products are accumulating in the ocean at an alarming rate and pose a global challenge1,2,3. Their adverse impact on the environment and its ecosystems is driving the research developments towards replacing petrochemical-based polymers with alternatives that are bio-based and biodegradable. For decades, starch has been industrially extracted from plant-based sources on large scale and used in a wide range of industry sectors, such as food, papermaking, and pharmaceuticals4. The abundant, biodegradable, and renewable nature of starch makes it an attractive matrix for composites with competitive barrier performance. Despite all these advantages, films made from starch alone are brittle and sensitive to moisture, thus limiting their industrial use in packaging applications. To overcome these drawbacks, starch has been combined with different fillers that can add specific properties to the composite films5.
Over the last decade, graphene has emerged as a promising filler that can add multifunctional properties from a single material. Graphene is a two-dimensional material with a hexagonal network of sp2-hybridized carbon atoms that gives rise to its unique combination of electrical, mechanical, and thermal properties. Moreover, the high density of electrons in its aromatic rings has been touted to prevent the permeation of all gases6,7. Until recently, it was found that hydrogen can, under certain events, permeate through extensive ripples on monolayer graphene, while a bilayer is indeed impermeable8. Consequently, when graphene sheets are homogeneously distributed within a polymer matrix, the gas molecules are forced to take tortuous pathways around the sheets in order to permeate through the composite film. Therefore, graphene sheets with a high aspect ratio and parallel orientation in-plane with the film are two key factors that can increase the tortuosity, thus improving the gas barrier properties. In contrast, inhomogeneous distribution of graphene sheets, usually due to poor dispersion of graphene, impair the barrier by opening up shortcuts that allow gas permeation through the composite. For this reason, the graphene sheets must remain well-dispersed when incorporated into the polymer matrix.
The starch, as a bio-based and biodegradable polymer matrix, is composed of polysaccharide chains that are closely linked together by intra- and intermolecular hydrogen bonds. Their partial crystallinity provides a native oxygen barrier in dried films9, while their hygroscopic nature impairs the water vapour barrier due to swelling10. To date, efforts to improve the water vapour barrier by incorporating hydrophobic fillers have, on the other hand, impaired the native oxygen barrier or kept it unchanged at best11,12,13. In general, hydrophobic materials make poor oxygen barriers, while conversely, hydrophilic materials make poor water vapour barriers. This trend originates from the chemical nature of oxygen molecules (non-polar) and water molecules (polar). These mutually opposing material limitations of the oxygen and the water vapour barrier, respectively, make the engineering of a combined gas barrier from a single coating formulation an interesting challenge. Graphene, being both hydrophobic and impermeable to all gases under ambient conditions, is therefore a promising barrier material in this regard. Hence, given that graphene with low oxygen content is mixed well within the polymer matrix, the increased tortuosity is expected to improve the overall gas barrier. However, graphene is not dispersible in aqueous systems due to its low surface energy (46.7 mN m−1) compared to the high surface tension of water (72.3 mN m−1)14. To minimize the large mismatch, a wide range of surfactants have been explored to lower the surface tension and stabilize graphene in aqueous systems15,16,17. On the other hand, a general challenge of using surfactants is their tendency to migrate within the film towards the interfaces (film/air or film/substrate) during the film formation18,19,20. These events create defects inside the film and at the interfaces, thus impairing the barrier by forming shortcuts21.
To the best of our knowledge, Ma et al. reported improved water vapour barrier properties of starch-based films by 43.7% with graphene oxide (GO, 4.0 wt%) and by 34.6% with reduced GO (RGO, 8.0 wt%) compared with the pure starch films22. Zheng et al. grafted starch onto RGO during the reduction process of GO and achieved a comparable improvement by 26.7% with less RGO (1.774 wt%)23. In general, the relatively lower performance by RGO compared with GO was attributed to its poor dispersibility in water and within the starch matrix. Ashori et al. observed a similar trend by graphene, with which the water vapour barrier was improved at most 24.1% with graphene (3.0 wt%) and then impaired by graphene aggregation24. Ge et al. reported improved oxygen barrier properties by 66.0% with surfactant-stabilized RGO (1.0 wt%)25. Nonetheless, while the oxygen barrier was improved, the water vapour barrier of the same film was impaired due to complications from the excess surfactant. To address the poor dispersibility in water and within the starch matrix, we previously developed a surfactant-free aqueous graphene dispersion (0.48 mg mL−1) using starch nanoparticles (SNPs) as dispersing agent26. Thus far, while the gas barrier properties of starch films with different carbon materials have been investigated, a combined gas barrier against both the oxygen and the water vapour has not been reported.
In this work, we report a substantial reduction of both the oxygen permeability (OP) and the water vapour permeability (WVP) simultaneously with starch-graphene composite films made from a single formulation. For this formulation, the SNP-graphene dispersion was first optimized in terms of concentration, morphology, and colloidal stability. The SNP-graphene sheets were then incorporated into a starch matrix made from the same material as the SNPs, thus maximizing the compatibility between the incorporated SNP-graphene sheets and the starch matrix.
Results and discussion
Preparation of the aqueous graphene dispersion
In order to incorporate graphene with low oxygen content (Supplementary Table 1) into a starch matrix, we developed a surfactant-free aqueous graphene dispersion using SNPs as dispersing agent (experimental details described elsewhere)26. This approach of tailoring the dispersing agent and the polymer matrix from the same material maximizes the compatibility between the incorporated filler and the matrix, thus facilitating homogeneous composites with multifunctional properties.
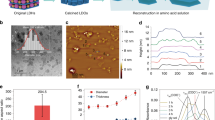
In this work, we optimized the SNP-graphene dispersion in terms of graphene concentration and colloidal stability (Fig. 1). In the absorption spectra (Fig. 1a), an optimized SNP-graphene dispersion (blue solid line) shows a sharp absorption peak at 273 nm and an absorbance of 1 au in the visible range after the graphene was stabilized by the SNPs (yellow solid line). The absorbance is related to the graphene concentration, while the peak position reflects an effect of the π → π* transitions of conjugated aromatic rings. In contrast, a non-stabilized graphene reference (grey dashed line) that was processed under the same conditions without the SNPs, shows a relatively featureless absorption spectrum due to aggregation and precipitation. The SNP-graphene dispersion was optimized in three steps. First, the effects of sonication time on the SNP-graphene dispersion were investigated with fixed concentrations of starting graphene powder (3 mg mL−1) and SNPs (60 mg mL−1). When the sonication time was extended from 0 to 60 min, the absorption peak gradually emerged from 254 to 273 nm, while the absorbance increased from 0 to 1 au at wavelength 660 nm, thus indicating a higher relative concentration of dispersed graphene (Fig. 1b). The relative graphene concentration was determined from the absorbance at 660 nm using the attenuation coefficient 3384 m−1 mg−1 mL26. Accordingly, the dispersed graphene concentration increased efficiently with the sonication time up to 30 min, after which the concentration reached a plateau between 30–60 min. Second, the particle size of the SNP-graphene sheets, on the other hand, decreased with the sonication time (Fig. 1c). Consequently, the sonication time of 60 min was chosen to maximize the dispersed graphene concentration and stability. The graphene concentration after centrifugation was determined to be 2.97 mg mL−1 (Supplementary Fig. 1). Third, the stability of the optimized SNP-graphene dispersion was evaluated over time under the storage conditions of 4 °C and 23 °C, respectively (Fig. 1d). When the dispersion was compared under these conditions, we found that the stability of the SNP-graphene sheets is temperature-dependent. The dispersion stored at the lower temperature (4 °C) dropped 89.2% of its original graphene concentration after 1 month. This drop can be attributed to starch retrogradation, a process whereby the polysaccharide chains re-associate. The rate of retrogradation is faster at 4 °C than at 23 °C27. Consequently, the dispersion at the room temperature (23 °C) remained stable during the same period. The colloidal stability at room temperature can offer long-term storage at a low cost for industrial applications.

a UV–vis absorption spectra of SNPs (yellow solid line), non-stabilized graphene reference (grey dashed line), and SNP-graphene (blue solid line). Inset: Photographs of the SNPs (1), the non-stabilized graphene reference (2), and the SNP-graphene (3). b UV–vis absorption spectra of the SNP-graphene with sonication time. c Dispersed graphene concentration (light blue line + squares) and median particle size (blue line + squares) with sonication time. d Colloidal stability at 4 °C (light blue line + squares) and 23 °C (blue line + squares). e Particle size distributions. f Zeta potential distributions.
The lateral dimension of graphene sheets is one of the key factors in improving the gas barrier properties of composites. A graphene sheet with a wider lateral size (micrometer range) provides longer gas diffusion paths in the composites than that with a narrower size (nanometer range)28. The median particle size (Dv50) of the SNP-graphene sheets (blue area) measured 3.4 ± 0.0 µm after the sonication process with starch (Fig. 1e). The non-stabilized graphene reference without starch (grey area), in contrast, spontaneously aggregated and showed a broader particle size distribution with a median size of 53.6 ± 0.4 µm. This indicates that the SNP-graphene sheets were stabilized by the SNPs (yellow area) with the median size 0.3 ± 0.0 µm. To gain insight into the stability of the SNP-graphene sheets, we measured the zeta potential (Fig. 1f). The mean zeta potential of the SNP-graphene sheets (blue area) was −22.9 ± 4.8 mV, slightly lower than that of the pure SNPs (yellow area) at −17.6 ± 3.9 mV29. According to the Derjaguin–Landau–Verwey–Overbeek theory, a higher magnitude of zeta potential between two charged surfaces contributes to a greater electrostatic repulsion. For comparison, the zeta potential of surfactant-stabilized graphene sheets can vary from −64 mV (anionic) to 57 mV (cationic)16. Interestingly, graphene sheets that are stabilized by uncharged surfactants (non-ionic) can also have a non-zero zeta potential as low as −24 mV30. In our work, the SNP-graphene sheets demonstrated long-term stability despite the relatively low magnitude zeta potential (−22.9 mV), thus suggesting that additional stabilization mechanism by steric hindrance could be contributing to the stability of the SNP-graphene sheets. Moreover, we found that the SNPs reduced the surface tension of water from 72.3 down to a minimum of 56.9 mN m−1 (Supplementary Fig. 2), thus making the dispersion of graphene with relatively high solid content in water (~3 mg mL−1) more energetically favourable. Unala et al. observed the same effect using other relevant polysaccharides, such as chitosan and pullulan31. Overall, the SNPs play an important role in the colloidal stability of graphene in water.
Fabrication of the composite films
The next step was to fabricate composite films by incorporating the SNP-graphene sheets into the starch matrix. In the early stage, we observed that the starch matrix formulation resulted in wrinkly and brittle films after drying (Fig. 2a). To maintain the mechanical integrity of the films for packaging applications, we added sorbitol as a plasticizer. We found that the starch films improved elasticity and became foldable after the sorbitol content was increased from 0 to 38 wt% or more (Supplementary Fig. 3). Furthermore, the mechanical integrity of the films was also maintained after additional 0.75 to 3.0 wt.% graphene, and thus demonstrating a compatible interface between the incorporated SNP-graphene sheets and the starch matrix. To investigate the compatibility further, we selected the composite film with the highest graphene content, henceforth denoted starch/SNP-graphene (3.0 wt% graphene, 36.4 wt% sorbitol, and 60.6 wt% total starch).

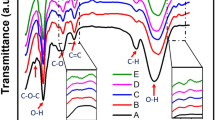
a Photographs of the starch film, the starch/sorbitol film, and the starch/SNP-graphene film before (top row) and after folding (bottom row). b FT–IR spectra of the films c TGA curves of the films and a starting graphene powder reference (grey dashed line), alongside their corresponding d DTG curves.
The physicochemical interaction of the components in the starch/SNP-graphene composite film was studied by Fourier transform infrared (FT–IR) spectroscopy (Fig. 2b). When comparing the starch/SNP-graphene film (blue solid line) with the starch control samples (yellow solid line, green solid line), we observed an additional peak emerged at 1572 cm−1 (C = C stretching) and attribute it to vibrations from conjugated aromatic rings of the graphene. The starch peaks were found at 995 cm−1, 1078 cm−1 (C—O—H bending), and 1149 cm−1 (C—O, C—C stretching, and partially C—O—H contributions)32, while the sorbitol peaks at 871 cm−1, 885 cm−1, 939 cm−1, 1000 cm−1 (O—H in-plane and out-of-plane bending), and 1047 cm−1, 1088 cm−1 (C—O—H stretching)33. The broad peak around 3303 cm−1 (O—H stretching of free, intra- and intermolecular interactions) was observed from both the sorbitol and the starch. This broad peak shifted towards the shorter wavenumbers from 3303 down to 3285 cm−1 (−18 cm−1) after sorbitol was added to the starch matrix, thus indicating stronger hydrogen bonding interactions between the sorbitol and the starch. The same peak was further downshifted to 3281 cm−1 (−22 cm−1) after the SNP-graphene was incorporated. These relative peak shifts, as a result of changes in intermolecular vibrations, are attributed to the formation of stronger hydrogen bonding between the sorbitol and the total starch within the matrix34. In addition, when the graphene content in the film was increased by 3.0 wt%, the relative peak intensity of the starch peak at 995 cm−1 (C—O—H) was substantially reduced, thus indicating weaker absorption from the starch. This was apparent by the relatively higher peak intensity of the 3303 cm−1 (O—H) over the 995 cm−1 (C—O—H). Overall, our results indicate that the sorbitol plasticized the starch film by replacing the starch-starch interactions within the starch matrix, while the SNP-graphene was incorporated without adverse effects on the film.
The thermal properties of the starch/SNP-graphene composite film were further studied by thermogravimetric analysis (TGA) and corresponding derivative thermogravimetric (DTG) under nitrogen atmosphere (Fig. 2c, d). The TGA curve of a graphene powder reference (grey dashed line) was relatively flat in the entire temperature range 25–700 °C, thus indicating a high degree of carbon purity35. The pure starch film (yellow solid line), in contrast, showed three decomposition regions. In general, the region 25–170 °C (I) is the weight loss of weakly adsorbed water, 200–400 °C (II) is the depolymerization of the starch backbone and accounts for the largest weight loss, and finally 400 °C (III) and higher is the formation of carbonaceous residues36. Since graphene was relatively inert during the heating process, its mass contributed to a larger residual weight in the starch/SNP-graphene film (blue solid line). Therefore, the graphene content in the film was determined to be 2.6 wt% (slightly lower than the estimated 3.0 wt%) based on the increased residual weight of the starch/sorbitol film (green solid line) with additional graphene at 700 °C. Among the films, the water content was the lowest in the starch/SNP-graphene (5.2%), then the starch/sorbitol (6.4%), and the highest in the pure starch (9.5%). This trend indicates that the graphene and the starch-sorbitol interaction reduced the water uptake of the starch. The thermal stability of the starch/SNP-graphene film was investigated by the maximum peak (Tmax) in the DTG curves at which the rate of weight loss is maximum (Fig. 2d). When the sorbitol and the SNP-graphene were incorporated into the starch matrix, the Tmax of the film was increased from 314 to 326 °C (12 °C).
Graphene distribution in the composite films
To gain insight into the distribution and the structural configuration of the incorporated SNP-graphene sheets within the starch matrix, we performed scanning electron microscope (SEM) analysis on the cross-section of the composite films (Fig. 3). Along with the cross-section of each film (Fig. 3a, d, g), we observed continuous distribution of graphene sheets located deep inside the film as well as partly protruded out from the plane. At higher magnification (Fig. 3b, e, h), these graphene sheets inside the film appeared outstretched with a high aspect ratio reflecting the size dimensions of the sheets in the dispersion (lateral size ~3.4 µm/thickness ~5 nm26). Furthermore, the preferred orientation of the sheets was in-plane with the film and thereby faced perpendicular to the gas diffusion direction. The high aspect ratio and the parallel orientation of the graphene sheets can contribute to a dense network of graphene with increased tortuosity. Indeed, the tortuosity inside the film increased consistently with the graphene content (0.75 wt%, 1.5 wt%, and 3.0 wt%). In addition, the film with the highest graphene content (3.0 wt%) also showed a graphene-rich region (~8 µm thickness) at the bottom, most likely due to the gravitational effect during the film formation (Supplementary Fig. 4c). To support the SEM analysis, we performed Raman mapping over the cross-section to confirm the chemical distribution of graphene (blue) and starch (yellow) in the films (Fig. 3c, f, i). From the lowest graphene content to the highest (0.75–3.0 wt%), the relative Raman intensity of the starch was reduced, while the intensity of the graphene, in contrast, was enhanced in proportion to the graphene content and eventually dominated the entire area. Although the total starch fraction in the film was larger (60.6 wt%) than the graphene fraction (3.0 wt%), the backscattered light from the starch was relatively weaker, and thereby almost diminished in the Raman spectra. In addition, at the highest graphene content (3.0 wt%), the intensity was higher in the graphene-rich region of the film, consistent with the SEM analysis. We also found evidence of graphene-matrix interaction based on the G peak and the D peak in two distinctly different Raman spectra of the graphene inside the film (Supplementary Fig. 4). Despite being the same incorporated SNP-graphene, the buried graphene sheets located deep inside the film reflected matrix compression-induced effects on the Raman spectra37, while the protruded graphene sheets in the graphene-rich region, in contrast, behaved similarly to a graphene reference. The G peak position was estimated based on a statistical histogram of the entire Raman map and was 1583 ± 1.7 cm−1 for the graphene reference, 1581 ± 1.7 cm−1 for the protruded graphene, and 1587 ± 3.5 cm−1 for the buried graphene. Moreover, the full-width half-maximum differences between this studied graphene also reflected a strong compression-induced effect. To provide visual evidence of the statistical distribution of the stress-induced peak shifts and the degree of disorder, we reconstructed the Raman map for the G peak, the D peak, and their intensity ratio I(D)/I(G). This Raman spectroscopy study is further described elsewhere (Supplementary Fig. 5). Overall, our findings from the SEM and Raman data analysis indicate a strong interaction between the incorporated SNP-graphene sheets and the starch matrix. This was evidenced by the stress transferred from the starch matrix on the graphene, thus increasing the vibration frequency, as reflected in the Raman spectra. As a result of the compatible interaction, the graphene sheets could form a densely interconnected network that was expected to improve the gas barrier properties.

From top row to bottom row: a–c 0.75 wt%. d–f 1.5 wt%. g–i 3.0 wt%. Left column: SEM images of the film cross-section. Scale bar: 20 µm. Middle column: magnified views of the SEM images at the boxes marked with white dashed lines. Scale bar: 5 µm. Right column: raman mapping of the graphene (blue) and the starch (yellow). Scale bar: 10 µm.
Barrier performance of the composite films
To determine the OP and the WVP of the starch/SNP-graphene composite films, we measured the transmission rates of oxygen and water vapour through the films, respectively (Fig. 4). The measurements were performed under the conditions 23 °C and 50% relative humidity (RH), and in addition 80% RH for the oxygen transmission. When the graphene content in the films was increased from 0 to 3.0 wt%, the OP was substantially reduced from 80 down to 21 cm3 µm m−2 day−1 (74.3% reduction) under 50% RH and from 13693 down to 4086 cm3 µm m−2 day−1 (70.2% reduction) under the higher humidity condition 80% RH (Fig. 4a). In general, as the humidity increases, the moisture uptake of the starch accelerates and causes the film to swell, thus impairing the gas barrier properties9. Nonetheless, our results showed that the gas barrier, even under the higher humidity condition, improved consistently with the graphene content. The low amount of graphene present in the films could reduce the moisture uptake of the starch and consequently limit the swelling. With the same films, the increase in graphene content was also accompanied by a substantial reduction of the WVP from 1193 down to 260 g µm m−2 day−1 (78.2% reduction) under 50% RH (Fig. 4b). Hence, the reduction of both the OP and the WVP indicates that the incorporated SNP-graphene, when increased in content, successively established a denser network of graphene sheets within the starch matrix, as confirmed by the SEM and Raman analysis. Overall, the combined gas barrier properties could be correlated with the increased tortuosity in the films. To the best of our knowledge, simultaneous reduction of both the OP and the WVP with the same film made from a single coating formulation is unusual, in particular with starch-based composites. Thus far, efforts to combine these two gas barriers by incorporating carbon materials have reduced the permeability of only one of the gases, while the other gas is allowed (Table 1). Similarly, most commercial polymers, including the petrochemical-based, are also limited either as an oxygen barrier (e.g. ethylene vinyl alcohol (EVOH)) or a water vapour barrier (e.g. low-density polyethylene (LDPE) and polypropylene (PP)). Among the common petrochemical-based, only polyvinylidene chloride (PVCD) could function as both the oxygen and the water vapour barrier. However, its use in the industry is being discouraged by more stringent regulations due to carcinogenic effects. To compare the barrier performance between materials, Wang et al.38 and Wu et al.39 suggested a barrier classification (from “poor” to “very high”) based on the gas permeability values. Accordingly, when comparing our starch/SNP-graphene film with the commercial plastics, the oxygen barrier performance is rated as “very high”, while the water vapour barrier performance as “high”. Specifically, our OP is comparable with that of EVOH and PVDC (~20 cm3: µm m−2 day−1), while our WVP with that of LDPE and PP (~300 g µm m−2 day−1). These combined gas barrier values make our film superior to most biodegradable films, and when optimized, could be competitive with the conventional packaging films. Currently, to achieve combined gas barrier properties that meet the requirement for food packaging on an industrial scale, multi-layered films made from different coating formulations of barriers are needed39. Therefore, the single formulation of bio-based and biodegradable starch incorporated with graphene is an attractive approach for combined gas barriers with competitive performance.

a OP at 23 °C 50% RH (light red bars) and 80% RH (red bars). b WVP at 23 °C 50% RH (blue bars). Error bars represent the standard deviation.
Motivated by the interconnected graphene network within the starch/SNP-graphene composite films leading to the improved gas barrier performance, we also anticipated improved electrical conductivity as a corollary result. Indeed, when the graphene content in the film was 3.0 wt%, the electrical conductivity of the film was substantially improved from being non-conductive to highly conductive at 9.5 S m−1 (Supplementary Table 2). To the best of our knowledge, the obtained electrical conductivity value is among the highest compared with similar starch-based composite films reported in the literature. For instance, Bin-Dahman et al.40 achieved 2.2 S m−1 with graphene (25.0 wt%) and Ma et al.22 achieved 1 S m−1 with RGO (1.0 wt%).
Based on these results, we introduce the following approach to maximize the compatibility between the graphene and the starch matrix (Fig. 5). First, graphene powder was dispersed in water using a dispersing agent made from the same material as the chosen starch matrix. Second, the SNPs improved the dispersion of graphene in water without the aid of surfactants, and thus eliminating common surfactant-related complications that impair the composite films. In addition, the SNPs also facilitated the incorporation of graphene into the starch matrix. Third, from a single drop of this coating formulation, the accompanying SNP-stabilized graphene sheets spread homogeneously within the starch matrix during the drying process and self-assembled into outstretched sheets with high aspect ratio and parallel orientation. This structural configuration explained the simultaneous improvement of both the oxygen barrier and the water vapour barrier, as well as the high electrical conductivity. We expect that the same approach can be applied with other relevant biopolymers, such as cellulose and chitosan. Here, our focus was mainly to demonstrate the feasibility of a single formulation for composite films that resolves a seemingly contradictory material challenge of simultaneously reducing both the OP and the WVP by over 70%. Further optimization in the future work will be focused on achieving gas barrier values that meet the current and future demands of the packaging industry.

(1) The starch/SNP-graphene composite film is based on a surfactant-free aqueous dispersion of graphene using SNPs. (2) The SNPs on the graphene surface facilitate the incorporation of graphene into the starch matrix. (3) A single drop of this coating formulation can be drop-cast into multifunctional composite films.
In conclusion, simultaneous improvements of both the oxygen and the water vapour barrier properties of starch-based composite films were achieved by incorporating SNP-stabilized graphene sheets from a single coating formulation. At relative low graphene content (3.0 wt%), the graphene reduced both the OP and the WVP by over 70% under all the conditions tested (50 and 80% RH). In addition, the films measured a high electrical conductivity of 9.5 S m−1, while maintaining mechanical flexibility. For this coating formulation, we optimized a surfactant-free aqueous dispersion of SNP-graphene sheets with graphene concentration up to 2.97 mg mL−1, median particle size 3.4 µm, and stability for at least 1 month. The SNPs improved the dispersion of graphene in water and facilitated the incorporation into the starch matrix. SEM and Raman data analysis confirmed a densely interconnected network of outstretched graphene sheets with a high aspect ratio and parallel orientation. This structural configuration explained the combined gas barrier performance and high electrical conductivity. These findings will have a scientific impact on facilitating compatible graphene into composites with commercial potential for wide-ranging applications, such as gas barriers in food packaging.
Methods
Materials and reagents
Graphene powder (AV-70-1-1.5) was purchased from Avanzare Innovacion Tecnologica, S.L. (Logroño, Spain). This graphene grade is a highly reduced graphene oxide with low oxygen content (<1.5%), as provided by the supplier. Unmodified corn starch powder (S4126) containing 27% amylose and 73% amylopectin and D-Sorbitol (S1876) were purchased from Sigma-Aldrich. Milli-Q water was used for all the aqueous dispersions.
Preparation of the aqueous graphene dispersion
SNP-graphene dispersions were prepared by probe sonication using a fixed starch:graphene mass ratio of 20:1. In general, starch powder (3 g) was dissolved in water (50 mL) and heated at 95 °C for 30 min under stirring at 1000 rpm. The gelatinized starch was further ruptured by sonication for 3 min to reduce the viscosity and obtain a homogeneous solution of SNPs (Sonics Vibra-Cell VCX 750, tip diameter: 13 mm, frequency: 20 kHz, and amplitude: 50%). A fixed amount of starting graphene powder (0.15 g) was added to the SNPs and dispersed by additional sonication for 60 min under cooling. Finally, any residual graphene aggregates and impurities in the resulting SNP-graphene dispersion were removed by centrifugation at 1000 rpm for 10 min. The final SNP-graphene dispersion was used for all characterizations and composite film formulations.
Fabrication of the composite films
The starch/SNP-graphene composite films were prepared by solvent-casting after mixing SNP-graphene dispersions (filler) into starch/sorbitol blends (starch matrix). For the starch matrix, the starch:sorbitol mass ratio was fixed to 5:3. In general, starch powder (1.95 g) and sorbitol powder (1.17 g) were dissolved in water (50 mL) and heated at 95 °C for 30 min under stirring at 1000 rpm. This gelatinized blend was drop-cast into petri dishes (8 mL) and then dried at 25 °C for 3 days to form starch films for reference. For the starch/SNP-graphene films, the sorbitol content was adjusted based on the total starch in the final film to maintain plasticization. For instance, the composite film formulation (64 mL) of 8 film replicates with the highest graphene content (3.0 wt%) was prepared by adding the SNP-graphene dispersion (40 mL) of graphene (0.12 g) and SNPs (2.40 g) to the starch matrix (24 mL) of starch (0.03 g) and sorbitol (1.46 g). Similarly, a series of starch/SNP-graphene films (0.75 wt%, 1.5 wt%, and 3.0 wt%) were prepared by the same approach. After the drying process, all the final films had a consistent weight (0.5 g) and diameter of 8.5 cm.
Aqueous graphene dispersion characteristics
The ultraviolet–visible (UV–vis) absorption spectra were recorded in quartz cuvettes using a UV–vis spectrophotometer (Lambda 650, PerkinElmer). Each sample was diluted by a factor of 100 and then equilibrated for 1 min prior to measurement. The relative graphene concentration was determined from the optical absorbance at wavelength 660 nm, according to Beer–Lambert’s law A = αlc. In this expression, the absorbance (A) is proportional to the attenuation coefficient (α), light path length (l), and concentration (c). In this work, the attenuation coefficient of 3384 m−1 mg−1 mL was used. The absorbance in the stability study was recorded in polystyrene cuvettes. Six dilutions were measured and then averaged. Particle size analysis was performed using a laser diffraction-based particle size analyzer with a Hydro SV measurement cuvette (Mastersizer 3000, Malvern). Each sample was added to the cuvette under stirring at 500 rpm until the obscuration level reached 5%. Five measurements were recorded and then averaged. The volume-weighted particle size distributions are calculated by the software using Mie theory. Different refractive index (RI) and absorption (A) were used for the graphene (RI: 2.73, A: 1.36) and the starch (RI: 1.53, A: 0.01), respectively. The zeta potential of the SNP-graphene sheets was determined using a Zetasizer (Nano ZS, Malvern). The SNP-graphene dispersion was diluted by a factor of 300 (∼0.01 mg mL−1) and then equilibrated in a folded capillary cell for 30 s prior to measurements. Six measurements were recorded and then averaged. Each sample measured a pH of 6.8 using a standard pH meter.
Physicochemical properties of the composite films
Spectroscopic analysis of the chemical interactions between graphene and starch was performed using an FT–IR spectrometer in attenuated total reflectance mode (Spectrum One, PerkinElmer). Sixteen scans were collected at a resolution of 4 cm−1 and then averaged. The resulting spectra were baseline corrected and normalized in the OriginPro 2020 software. TGA analysis was performed in 70 µL alumina crucibles using a thermogravimetric analyzer (TGA 2 STARe System, Mettler Toledo). The dried weight was consistent at 2 mg for all samples. The measurements were recorded under a nitrogen atmosphere at a constant heating rate of 10 °C min−1.
Cross-section morphology and chemical imaging
The SEM images of the cross-section were captured using an SEM (Quanta FEG 250, FEI). The cross-section was prepared by cleaving the films after immersion in liquid nitrogen. Raman mapping of the cross-section was performed using a confocal Raman microscope (WITec alpha300 system, WITec GmbH). For each film, the Raman spectra were scanned using an excitation wavelength of 532 nm and a low laser power between 2.5–5 mW to prevent damage to the graphene. A typical integration time was in the range of 0.05–0.25 s. The Raman spectra were processed in the WITec Project 5.1 software (WITec GmbH, Ulm, Germany). For instance, cosmic ray removal, background subtraction, and then custom filters to extract the starch and the graphene.
Oxygen and water vapour transmission measurements
The oxygen transmission rate (OTR) was measured using a Mocon Oxtran instrument following the ASTM F1927-14 standard, and the water vapour transmission rate (WVTR) was measured using a Mocon Permatran instrument following the ISO 15106-1 standard. The same films (six replicates) were used for both the OTR and the WVTR measurements and then normalized to the film thickness. The films were masked to reduce the test area down to 5 cm2 and to prevent edge leakage. The test conditions were set to 23 °C with 50% RH and 80% RH, respectively.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Jambeck, J. R. et al. Plastic waste inputs from land into the ocean. Science 347, 768–771 (2015).
Lebreton, L. et al. Evidence that the Great Pacific Garbage Patch is rapidly accumulating plastic. Sci. Rep. 8, 4666 (2018).
Solberg, E. Norway’s Prime Minister: Ocean science can boost jobs and wellbeing. Nature 588, 9–9 (2020).
Le Corre, D. & Angellier-Coussy, H. Preparation and application of starch nanoparticles for nanocomposites: a review. React. Funct. Polym. 85, 97–120 (2014).
Zhao, R., Torley, P. & Halley, P. J. Emerging biodegradable materials: starch- and protein-based bio-nanocomposites. J. Mater. Sci. 43, 3058–3071 (2008).
Bunch, J. S. et al. Impermeable Atomic Membranes from Graphene Sheets. Nano Lett. 8, 2458–2462 (2008).
Leenaerts, O., Partoens, B. & Peeters, F. M. Graphene: a perfect nanoballoon. Appl. Phys. Lett. 93, 193107 (2008).
Sun, P. Z. et al. Limits on gas impermeability of graphene. Nature 579, 229–232 (2020).
Forssell, P. Oxygen permeability of amylose and amylopectin films. Carbohydr. Polym. 47, 125–129 (2002).
Bertuzzi, M. A., Castro Vidaurre, E. F., Armada, M. & Gottifredi, J. C. Water vapor permeability of edible starch based films. J. Food Eng. 80, 972–978 (2007).
Han, J. H., Seo, G. H., Park, I. M., Kim, G. N. & Lee, D. S. Physical and mechanical properties of pea starch edible films containing beeswax emulsions. J. Food Sci. 71, E290–E296 (2006).
Ghasemlou, M. et al. Physical, mechanical and barrier properties of corn starch films incorporated with plant essential oils. Carbohydr. Polym. 98, 1117–1126 (2013).
Christophliemk, H., Johansson, C., Ullsten, H. & Järnström, L. Oxygen and water vapor transmission rates of starch-poly(vinyl alcohol) barrier coatings for flexible packaging paper. Prog. Org. Coat. 113, 218–224 (2017).
Wang, S., Zhang, Y., Abidi, N. & Cabrales, L. Wettability and surface free energy of graphene films. Langmuir 25, 11078–11081 (2009).
Lotya, M. et al. Liquid phase production of graphene by exfoliation of graphite in surfactant/water solutions. J. Am. Chem. Soc. 131, 3611–3620 (2009).
Smith, R. J., Lotya, M. & Coleman, J. N. The importance of repulsive potential barriers for the dispersion of graphene using surfactants. N. J. Phys. 12, 125008 (2010).
Backes, C. et al. Production and processing of graphene and related materials. 2D Mater. 7, 022001 (2020).
Roulstone, B. J., Wilkinson, M. C. & Hearn, J. Studies on polymer latex films: II. effect of surfactants on the water vapour permeability of polymer latex films. Polym. Int. 27, 43–50 (1992).
Kientz, E. & Holl, Y. Distribution of surfactants in latex films. Colloids Surf. A Physicochem. Eng. Asp. 78, 255–270 (1993).
Tzitzinou, A. et al. Surfactant concentration and morphology at the surfaces of acrylic latex films. Prog. Org. Coat. 35, 89–99 (1999).
Aramendia, E., Barandiaran, M. J., Grade, J., Blease, T. & Asua, J. M. Improving water sensitivity in acrylic films using surfmers. Langmuir 21, 1428–1435 (2005).
Ma, T., Chang, P. R., Zheng, P. & Ma, X. The composites based on plasticized starch and graphene oxide/reduced graphene oxide. Carbohydr. Polym. 94, 63–70 (2013).
Zheng, P., Ma, T. & Ma, X. Fabrication and properties of starch-grafted graphene nanosheet/plasticized-starch composites. Ind. Eng. Chem. Res. 52, 14201–14207 (2013).
Ashori, A. Effects of graphene on the behavior of chitosan and starch nanocomposite films. Polym. Eng. Sci. 54, 2258–2263 (2014).
Ge, X. et al. Improved mechanical and barrier properties of starch film with reduced graphene oxide modified by SDBS. J. Appl. Polym. Sci. 134, 44910 (2017).
Zhao, W. et al. Surfactant-free stabilization of aqueous graphene dispersions using starch as a dispersing agent. ACS Omega 6, 12050–12062 (2021).
Wang, S., Li, C., Copeland, L., Niu, Q. & Wang, S. Starch retrogradation: a comprehensive review. Compr. Rev. Food Sci. Food Saf. 14, 568–585 (2015).
Yoo, B. M., Shin, H. J., Yoon, H. W. & Park, H. B. Graphene and graphene oxide and their uses in barrier polymers. J. Appl. Polym. Sci. 131, 39628 (2014).
Chang, R. et al. Green preparation and characterization of starch nanoparticles using a vacuum cold plasma process combined with ultrasonication treatment. Ultrason. Sonochem. 58, 104660 (2019).
White, B., Banerjee, S., O’Brien, S., Turro, N. J. & Herman, I. P. Zeta-potential measurements of surfactant-wrapped individual single-walled carbon nanotubes. J. Phys. Chem. C. 111, 13684–13690 (2007).
Uysal Unalan, I., Wan, C., Trabattoni, S., Piergiovanni, L. & Farris, S. Polysaccharide-assisted rapid exfoliation of graphite platelets into high quality water-dispersible graphene sheets. RSC Adv. 5, 26482–26490 (2015).
Kizil, R., Irudayaraj, J. & Seetharaman, K. Characterization of irradiated starches by using FT-Raman and FTIR Spectroscopy. J. Agric. Food Chem. 50, 3912–3918 (2002).
Quinquenet, S., Ollivon, M., Grabielle-Madelmont, C. & Serpelloni, M. Polimorphism of hydrated sorbitol. Thermochim. Acta 125, 125–140 (1988).
Yang, J., Yu, J. & Ma, X. Study on the properties of ethylenebisformamide and sorbitol plasticized corn starch (ESPTPS). Carbohydr. Polym. 66, 110–116 (2006).
Liu, W. & Speranza, G. Tuning the oxygen content of reduced graphene oxide and effects on its properties. ACS Omega 6, 6195–6205 (2021).
Zhang, X., Golding, J. & Burgar, I. Thermal decomposition chemistry of starch studied by 13C high-resolution solid-state NMR spectroscopy. Polymer 43, 5791–5796 (2002).
Frank, O. et al. Compression behavior of single-layer graphenes. ACS Nano 4, 3131–3138 (2010).
Wang, J. et al. Moisture and oxygen barrier properties of cellulose nanomaterial-based films. ACS Sustain. Chem. Eng. 6, 49–70 (2018).
Wu, F., Misra, M. & Mohanty, A. K. Challenges and new opportunities on barrier performance of biodegradable polymers for sustainable packaging. Prog. Polym. Sci. 117, 101395 (2021).
Bin-Dahman, O. A., Rahaman, M., Khastgir, D. & Al-Harthi, M. A. Electrical and dielectric properties of poly(vinyl alcohol)/starch/graphene nanocomposites. Can. J. Chem. Eng. 96, 903–911 (2018).
Nzenguet, A. M. et al. Novel bionanocomposite films based on graphene oxide filled starch/polyacrylamide polymer blend: structural, mechanical and water barrier properties. J. Polym. Res. 25, 86 (2018).
Ashori, A. & Bahrami, R. Modification of physico-mechanical properties of chitosan-tapioca starch blend films using nano graphene. Polym. Plast. Technol. Eng. 53, 312–318 (2014).
Acknowledgements
This work was financially supported by the Swedish Foundation for Strategic Research (SSF, grant no. FID-15-0105) and BillerudKorsnäs AB. The authors thank Karin Hallstensson for support with the SEM imaging, Mikael Sundin for performing the XPS analysis and Marie Ernstsson for interpreting the data, Andreas Fall and Niklas Nordgren for insightful discussions and review of the manuscript.
Author information
Authors and Affiliations
Contributions
W.Z. conceived the original idea of the aqueous SNP-graphene dispersion method and together with A.S. and A.A. designed the overall work. W.Z. prepared the SNP-graphene dispersion and formulated the composite films, carried out dispersion characterizations, interpreted the data, and wrote the manuscript through contributions of all authors. Z.B.Z. and S.J.Z. performed the electrical characterizations. T.G. and J.A.L. performed the barrier measurements. J.S. performed the Raman measurements and together with W.Z. and I.D. analyzed the data. All authors contributed to discussions at different stages and have approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhao, W., Sugunan, A., Gillgren, T. et al. Surfactant-free starch-graphene composite films as simultaneous oxygen and water vapour barriers. npj 2D Mater Appl 6, 20 (2022). https://doi.org/10.1038/s41699-022-00292-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41699-022-00292-x
