
Abstract
Chemical functionalization of low-dimensional nanostructures has evolved as powerful tool to tailor the materials’ properties on demand. For two-dimensional transition metal dichalcogenides, functionalization strategies are mostly limited to the metallic 1T-polytype with only few examples showing a successful derivatization of the semiconducting 2H-polytype. Here, we describe that liquid-exfoliated WS2 undergoes a spontaneous redox reaction with AuCl3. We propose that thiol groups at edges and defects sites reduce the AuCl3 to Au0 and are in turn oxidized to disulfides. As a result of the reaction, Au nanoparticles nucleate predominantly at edges with tuneable nanoparticle size and density. The drastic changes in nanosheet mass obtained after high loading with Au nanoparticles can be exploited to enrich the dispersions in laterally large, monolayered nanosheets by simple centrifugation. The optical properties (for example photoluminescence) of the monolayers remain pristine, while the electrocatalytic activity towards the hydrogen evolution reaction is significantly improved.
Similar content being viewed by others

Introduction
Layered transition metal dichalcogenides (TMDs) represent a diverse, emerging source of two-dimensional (2D) nanostructures with broad application potential in areas from optoelectronics to energy.1,2,3,4,5,6,7,8,9,10 Numerous production techniques to obtain 2D-materials exist, with exfoliation in liquid particularly attractive due to its ease, scalable production of bulk quantities, and the possibility of further solution processing.11 Several distinct exfoliation techniques in liquid are available, with the simplest being liquid-phase exfoliation (LPE).12,13 LPE produces colloidally stable dispersions, for example, by sonicating bulk crystals in appropriate solvents or aqueous media with stabilizers. It is an inexpensive, scalable technique giving access to large quantities of solution processable nanosheets.12,13 In LPE, the semiconducting 2H-polytype is retained which is interesting for optoelectronics, whereas chemical exfoliation (CE) (often achieved after intercalation with n-BuLi)14 yields the metastable metallic 1T-polytype. While many proof of concept applications have been recently demonstrated (from printed photodetectors and transistors to catalyst electrodes or batteries),3,4,6,11,15,16 significant bottlenecks remain that impede the realization of real life applications.
Firstly, the bulk production of high-quality, monolayer (ML)-rich samples is extremely challenging. This is because broad size and thickness distributions are obtained in LPE. Size selection can be achieved by centrifugation, but MLs are small and the procedures are extremely time consuming.17,18,19
Secondly, chemical functionalisation to further tailor the materials’ properties on demand is currently in its infancy.20,21,22 With few exceptions,23,24 the covalent chemistry is currently limited to CE–1T–TMDs, which are of less interest in optoelectronic applications due to its lack of visible optical absorption or emission. In addition to the functionalisation routes involving the addition of organic molecules,20,21 it has been shown that CE–1T–TMDs react with AuCl3 even in the absence of additional reducing agents.25,26,27,28 As a result, Au0 is produced and nucleates to form Au nanoparticles (AuNPs). Such TMD–Au hybrids are interesting for a number of applications ranging from rechargeable batteries,27 to biosensing,29,30 optoelectronics,31,32,33 and catalysis.34,35,36
Finally, as in any material, defects largely control the properties of the 2D nanosheets.37 Depending on the type of defect, they are not necessarily detrimental, but may be beneficial for a given application. For example, disulfides located at nanosheet edges (i.e., defects) constitute the catalytically active sites for the hydrogen evolution reaction (HER).4 This illustrates that it will be crucial to control and tailor defects in 2D materials to maximize performance in a given application. Despite recent progress in this area, we are far away from a comprehensive understanding of defect engineering in TMDs.37
Here we present a route towards defect engineering via redox chemistry that addresses each of these three challenges associated with the advancement of LPE-TMDs. We show that LPE–2H–WS2 undergoes a spontaneous reaction with aqueous AuCl3 to form AuNPs that predominantly nucleate at edge sites. The observation of the covalent Au–S bond for the functionalized nanosheets contrasts with previous reports featuring a CE–1T–TMD where excess electrons present on the TMD were acting as the reducing agent. We suggest that thiol groups residing at edge and defects sites of the 2H–WS2 are the reducing agent in our case. Interestingly, large AuNPs merge at edges and terraces of incompletely exfoliated nanosheets. Such large-mass nanosheets can be removed by centrifugation yielding highly ML-enriched samples with tuneable lateral nanosheet sizes. After purification, AuNP edge-decorated WS2 nanosheets are obtained and optical properties of the 2H–WS2 are retained, while the catalytic activity in the electrocatalytic HER is drastically enhanced. Taken together, the developed methods provide an important step forward for ML enrichment, chemical functionalisation, and defect engineering in LPE TMDs.
Results and discussion
Figure 1 illustrates the reaction and the WS2–Au nanostructures obtained depending on the reaction conditions. 2H–WS2 nanosheet dispersions were produced by LPE in aqueous sodium cholate (SC) and separated into fractions containing large (l-WS2), medium (m-WS2) and small (s-WS2) nanosheets by liquid cascade centrifugation (LCC).18 The mean lateral size (<L>) and layer number (<N>) were determined spectroscopically (SI section 2). The main manuscript focuses on l-WS2 (<L>=115 nm, <N>=8); for additional data see SI. Initially, aqueous AuCl3 was added at different molar AuCl3:WS2 stoichiometry ratios (i.e., per WS2 formula unit) ranging from 1:1 to 8:1. A spontaneous reaction is evident from a color change of the dispersion from yellow–green to brown, red, or purple depending on the AuCl3:WS2 stoichiometry ratio. The work-up involved centrifugation to remove unreacted AuCl3 and large aggregates (see methods) and yielded nanosheet dispersions denoted as WS2–Au–x stock (Fig. 1) with x indicating the molar excess of AuCl3 over WS2 prior to the reaction.

Schematic representation of the reaction and purification. Liquid-exfoliated WS2 dispersions were first reacted with aqueous AuCl3 solution. An initial work-up by centrifugation was performed to remove unreacted AuCl3 and large aggregates to obtain polydisperse mixtures of Au-decorated nanosheets, denoted as ‘stock’. The decoration density with Au nanoparticles was dependent on the initial molar AuCl3:WS2 stoichiometry as indicated by reaction pathway a and b. The stock dispersions were subjected to liquid cascade centrifugation to isolate Au-decorated few-layered nanosheets (reaction pathway A) or predominately monolayers (reaction pathway b)
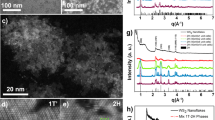
Transmission electron microscopy (TEM) on the l-WS2–Au1 and l-WS2–Au4.5 stock dispersions shows that the nanosheets are decorated with AuNPs predominantly found at edges and terraces of incompletely exfoliated nanosheets (Fig. 2a, b). Statistical TEM analysis (SI section 3) of the l-WS2–Au4.5 stock shows that ~280 small NPs are found per μm2 on the basal plane and ~1300 per μm2 nucleate around terraces. When comparing perimeter and terraces, ~150 NPs per μm are located around the edges and 100 per μm on the terraces. Similar results are obtained for the l-WS2–Au1 stock, albeit with a lower decoration density around edges/terraces and slightly higher density on the basal plane (Table S2). The AuNPs are polydisperse in size and shape. For l-WS2–Au1 (Fig. 2a), spherical particles with diameters of 1.5–2.5 nm are found along with larger though more disordered morphologies (10–30 nm). In l-WS2–Au4.5, both small and larger AuNPs have increased in size to 1.5–4 nm and 20–100 nm, respectively (Figures S7-S8). These large AuNPs (Fig. 2b) probably result from the merging of small AuNPs or Ostwald ripening.

Basic characterization of the stock dispersions. a–b Representative TEM images of the l-WS2-Au stock dispersions after addition of AuCl3. Scale bar 40 nm. a Molar AuCl3:WS2 stoichiometry of 1:1, b molar AuCl3:WS2 stoichiometry of 4.5:1. c Normalized extinction spectra of large WS2 (l-WS2) after addition of varying amounts of AuCl3. A clear contribution from Au nanoparticles (550–1000 nm) is discernible. Numbers in the figure legend indicate the molar AuCl3:WS2 concentration fraction. d Second derivative of the WS2 A-exciton for a subset of the samples in c. With increasing AuCl3 concentration the monolayer (ML) peak at ~2.03 eV becomes more pronounced compared with the few-layer (FL) peak. e Plot of extinction intensity ratio of 560 nm/410 nm as function of molar AuCl3:WS2 stoichiometry (expressed as fraction of excess AuCl3). Data for large and small (s-WS2) is shown. f Plot of normalized WS2 concentration as a function of molar AuCl3:WS2 stoichiometry. Inset: TEM image of a large aggregate, scale bar 40 μm. g Plot of mean nanosheet thickness as a function of molar AuCl3:WS2 stoichiometry. h Plot of the WS2 monolayer volume fraction as a function of molar AuCl3:WS2 stoichiometry. i Fitted XPS S2p and j Au4f core level spectra of the initial l-WS2 reference dispersion and the l-WS2Au4.5 stock dispersion showing the formation of Au0 covalently bound to WS2
Extinction spectroscopy has evolved as a powerful tool for the characterization of LPE TMDs as the spectral profile strongly depends on the nanosheet size and thickness due to edge and confinement effects.18,38 In particular, the A-exciton position (610–620 nm) can be quantitatively linked to the nanosheet layer number and the fine structure of the A-exciton to determine ML content via analysis of the second derivative (SI section 2). This is due to an exponential decrease of the A-exciton energy with increasing layer number18 which gives rise to a well-separated A-exciton position in ML (~611 nm, 2.03 eV) WS2 from that associated with the few-layer (FL) nanosheets. Such metrics enable the simultaneous determination of <N>, <L> and ML content.
Extinction spectra of the WS2–Au stock dispersions show the characteristic excitonic transition of 2H–WS2 (Fig. 2c) in the case of low AuCl3:WS2. At higher AuCl3:WS2 (>2:1), these are overlaid with a contribution from native Au products resembling previous Au(III) reductions (SI section 4). The WS2 A-exciton can be more clearly observed in the second derivative spectra (Fig. 2d), with systematic changes occurring with increasing AuCl3:WS2. Specifically, as the AuCl3:WS2 is increased, the overall peak magnitude is reduced suggesting a lower WS2 concentration, but critically, the contribution of the ML increases.
Since WS2 absorbance is highest below 500 nm and AuNP absorbance is predominantly in the 500–600 nm range,39 peak intensity ratios can be used to qualitatively express the relative AuNP to WS2 content. In Fig. 2e, the extinction intensity ratio, Ext560nm/Ext410nm, is plotted as a function of AuCl3:WS2 for both s-WS2 and l-WS2. Ext560nm/Ext410nm increases before plateauing at AuCl3:WS2 of ~4:1, after which the AuNP absorbance is significantly red-shifted; this suggests formation of larger AuNPs. In turn, the WS2 concentration estimated from the A-exciton (SI section 2) drops (Fig. 2f) with increasing AuCl3:WS2. This can be explained by the formation of large aggregates that are removed during the work-up. Critically, much of the removed WS2 appears to be multi-layer flakes, as the mean WS2 layer number dropped (<N> determined from spectroscopic metrics, Fig. 2g) and consequently, the ML volume fraction increased (ML V f, Fig. 2h). This suggests that the reaction with AuCl3 may be used to enrich WS2 monolayers, with ML V f reaching close to 100% for s-WS2 and up to 50% for l-WS2.
WS2–Au stock dispersions were subjected to X-ray photoemission spectroscopy (XPS) to identify the composition of the samples and the binding state of the Au to the WS2. Figure 2i, j shows the high resolution S2p and Au4f spectra collected on the reference l-WS2 nanosheets and the l-WS2–Au4.5 stock. In the S2p core level spectra of the reference sample (Fig. 2i, top), a single doublet with well resolved S2p3/2 and S2p1/2 peaks is observed and is attributed to 2H–WS2. In the WS2–Au4.5 stock (Fig. 2i, bottom), an additional doublet can be discerned at a 1 eV lower binding energy and is attributed to S–Au bonds.40,41 In contrast to the WS2 reference sample (Fig. 2j, top), a clear signature of Au0 (at a binding energy of 84 eV) is detected in the Au4f core level spectra of the l-WS2–Au4.5 stock (Fig. 2j, bottom). A minor contribution shifted by ~1.5 eV to higher binding energies is attributed to the formation of Au–S bonds in agreement with previous reports.42,43 The W4f core level spectra remain unchanged suggesting that WS2 was not damaged (more XPS, see SI section 5).
XPS evidences that, in contrast to previous results, the AuNPs are covalently attached to the sulfur in 2H-WS2, thereby confirming that the WS2 participates in the redox reaction with AuCl3. We suggest that thiols residing at edges (and terraces of incompletely exfoliated nanosheets) reduce the AuCl3 to Au0 and are in turn oxidized to disulfides. However, it is possible that the surfactant SC also serves as reducing agent. To test this, a series of reference experiments were performed (SI section 6) showing that this is indeed the case, but only when WS2 is present in the reaction mixture. For example, as the SC concentration is increased, the post reaction WS2 concentration drops for a given AuCl3:WS2 stoichiometry ratio, but with no improved ML enrichment below the critical micelle concentration of SC. We suggest that, in the presence of WS2, the Au0 nucleates to small AuNPs at the WS2 edges. The further growth of nucleated NPs on the WS2 is fed by Au0 produced from the reaction of SC with AuCl3. On few-layered WS2 nanosheets, the likelihood of individual AuNPs merging is higher for geometric reasons (Fig. 1), which results in the growth of larger AuNPs. At high AuCl3:WS2, these products become large enough to be removed in the low speed centrifugation work-up leading to enrichment of (Au-decorated) WS2 MLs in the stock.
To facilitate further characterization of the products and gain insight into the properties of 2H-WS2-Au, purification procedures were elaborated to remove the large AuNPs more completely. This allowed us to extract either few-layered WS2–Au nanosheets (via reaction pathway A in Fig. 1) or ML WS2–Au nanosheets (via reaction pathway B in Fig. 1). Importantly, we find that the mean lateral size of the WS2 is maintained after the reaction and purification. Hence, ML-enriched samples with readily tuneable lateral size can be obtained by varying <L> in the initial dispersion. Purification was achieved by LCC with details depending on the WS2 size in the parent dispersion prior to the reaction (Methods and SI section 7.1).
TEM of the purified samples shows that WS2 is decorated with predominantly small AuNPs and that the larger, polydisperse AuNPs were indeed removed (Fig. 3a–f and SI section 7.2). Dark-field scanning TEM coincident with electron energy loss spectroscopy performed in parallel with the current work elucidated plasmon damping at the physiochemical WS2–Au bond, providing an important first step in understanding energy transfer and edge states in this hybrid material.44 XPS still shows evidence of a covalent Au–S bond in the purified samples (SI section 7.3). For the purified l-WS2–Au1 sample, some of the 10–20 nm diameter AuNPs are still present (wide-view image Fig. 3a). The energy dispersive X-ray (EDX) spectrum (Fig. 3b) shows clear signatures of W, S and Au, in addition to Na, O and C (from the surfactant) and Cu (from the TEM grid holder). A higher resolution TEM image (Fig. 3c) of the l-WS2–Au1 illustrates that the WS2 nanosheet edges are decorated with small (~2 nm) AuNPs in addition to the 10–20 nm AuNPs. The fast Fourier transform diffraction pattern (Fig. 3c, inset) on such a nanosheet shows the signature of both the WS2 and AuNP crystal lattice (SI section 7.2.1). In the purified l-WS2–Au4.5 sample, larger AuNPs were almost completely removed (Fig. 3d, e, SI section 7.2.2) with only ~2–4 nm AuNPs predominantly around WS2 edges. This is because the centrifugation-based purification is extremely efficient in this case: most of the AuNPs have merged to very large Au nanostructures on few-layered nanosheets when using high AuCl3:WS2. These have significantly higher mass than the ML-WS2 only decorated sparsely with the small AuNPs around the edges leading to an efficient separation of the species. High-resolution TEM (Fig. 3f and SI section 7.2) show the intact crystal lattice of both WS2 and AuNPs.

Microscopic characterization of the purified l-WS2–Au. a Wide view TEM image of purified l-WS2–Au1. Scale bar 100 nm. AuNPs with diameters of 2–20 nm are observed decorating nanosheet edges and terraces. b EDX spectrum acquired on the nanosheets in a showing signatures of W, S, Au. c Magnified TEM image of a representative nanosheet in the purified l-WS2–Au1 sample. Scale bar 20 nm. Inset: Fast Fourier Transform. d, e Wide view TEM image of the purified l-WS2–Au4.5 sample. Scale bar 50 nm d and 20 nm e Only very small Au nanoparticles (1.5–4 nm) are observed. f High-resolution TEM image confirming the high crystallinity of the samples. Scale bar 2 nm. g–j AFM of the purified l-WS2–Au4.5. g AFM topography image (scale bar 250 nm) of a WS2 nanosheet with the corresponding line profile (bottom). h Histogram of apparent nanosheet thickness. i Histogram of apparent nanosheet length showing a mean WS2 length of 140 nm. Inset: representative AFM image (scale bar 250 nm). j Plot of nanosheet length vs. AFM height. Each data point represents one individual nanosheet
To confirm the ML enrichment, the purified samples were subjected to statistical atomic force microscopy (AFM, Fig. 3g–j and SI section 7.4). Typical apparent nanosheet thicknesses obtained from line profiles (Fig. 3g) are in the range of 3–6 nm. The histogram of the measured apparent thickness for >200 nanosheets (Fig. 3h) shows that the majority of nanosheets have an apparent height <8 nm. Note that one layer of LPE-TMDs typically has an apparent AFM height of 1.9 nm.18,38 In this study, decoration of nanosheets with AuNPs increases their apparent thickness. The thinnest nanosheets showed an apparent height of 3 nm leading to a rapid fall off of the otherwise lognormal shaped histogram. Considering this, AFM corroborates the finding of the ML enrichment. Importantly, the length histogram with typical lognormal shape obtained from the statistical AFM analysis (Fig. 3i) shows that the ML-enriched nanosheets of l-WS2 are significantly larger (<L> = 140 nm) than in other reported procedures, where they are typically <70 nm.17,18 Furthermore, this emphasizes that the WS2 lateral size in the final, ML-enriched purified l-WS2-Au dispersion (140 nm) is similar to the initial l-WS2 dispersion (115 nm) prior to the reaction. A plot of nanosheet length vs. thickness (Fig. 3j) reveals no correlation between length and thickness, in contrast to typical LPE samples,18,38 illustrating the different mechanism of the ML enrichment procedure.
To examine the optical response of 2H–WS2 following AuNP-decoration, the purified samples were subjected to extinction, Raman and photoluminescence (PL) spectroscopies. The extinction spectra (Fig. 4a) confirm the lower AuNP content compared to the stock dispersion, in agreement with TEM. The second derivative of the A-exciton (Fig. 4b) shows an increased ML content (especially for purified l-WS2–Au4.5) and a blue-shift of the FL component compared to the reference, confirming that thicker nanosheets were removed. In the case of purified l-WS2–Au4.5, the ML V f increased from <0.5% to >50% with minimal loss of MLs during the purification process (SI section 7.1).

Optical properties of purified (monolayer-enriched) l-WS2–Au. a Normalized extinction spectra of purified l-WS2–Au1 and l-WS2–Au4.5. b Second derivative of the A-exciton of the extinction spectra confirming the monolayer enrichment (legend in A). c Raman spectra of the same l-WS2 dispersions (532 nm excitation wavelength). The photoluminescence of the monolayer WS2 is centered at ~2500 cm-1 Raman shift. Inset: Focus on the Raman modes. d Excitation-emission contour plots of the purified l-WS2–Au4.5 dispersion. PL from ML-WS2 is observed at 612 nm (vertical signal). The band shifting in emission wavelength with λ exc is the Raman mode of water
Raman/PL spectra measured on the surface of a dispersion (532 nm excitation) of purified l-WS2–Au samples show no changes in the Raman modes compared to the reference, thus confirming the WS2 remained intact in its 2H-polytype (Fig. 4c). At Raman shifts of 2000–3000 cm−1 (595–633 nm) the PL of WS2 is clearly discernible in the case of purified WS2-Au4.5. Since only ML TMDs show appreciable PL,45,46 this is consistent with ML enrichment. No changes in the PL peak shape or position are observed from the measurement in liquid dispersion suggesting that the AuNPs have little effect on the optical properties of the TMD in the dispersion. This can be rationalized by large areas of the WS2 basal plane not being in contact with the AuNPs due to the preferential decoration on WS2 edges (SI section 3). Significantly, the PL is widely retained in thin films of WS2–Au (SI section 7.5) suggesting that ordered restacking of the WS2 is partially prevented by the AuNPs which can be interesting for integrating these nanosheets into optoelectronic devices. Interestingly, an increased emission from trions is observed in the thin films of the purified WS2–Au. This could be a result of the random restacking which forces AuNPs to be in close proximity with basal plane areas that are not decorated in the individual nanosheets. Photoluminescence excitation (PLE, Fig. 4d) contour plots of purified l-WS2–Au4.5 further confirm that the optical properties of the 2H–WS2 are pristine across the spectral region of 325–550 nm in excitation. However, optoelectronic interaction between WS2 and Au would be expected in the spectral region where AuNP absorbance is strongest (530–600 nm). Unfortunately, this spectral region is not accessible due to the necessity of using a cut-off filter on the emission side due to light scattering from the nanomaterial dispersions. Changes in the A-exciton PL intensity can be entirely attributed to a variation in the ML content across samples (SI section 7.5). To confirm that MLs of various lateral sizes can be enriched in this way, additional data on s-WS2–Au is shown throughout the SI.
The covalent Au-S bond formation observed in XPS proves that the WS2 nanosheets indeed undergo a redox-reaction with AuCl3. Importantly, such a covalent bond is not observed when AuCl3 is reacted with CE–1T–WS2 25,26,27,28 and therefore provides evidence that the chemistry behind the AuNP formation is different compared to previous reports. In light of this evidence we propose that, to some extent (see SI section 6), thiols residing at the edges of LPE–2H–WS2 reduce the AuCl3 to Au0 and are themselves oxidized to disulfides.
To corroborate this hypothesis, we have compared the electrocatalytic activity of the WS2–Au nanosheet material (both stock and purified) with that of WS2 nanosheets that were not subject to reaction with AuCl3 (called ‘reference’ sample). Thin films were prepared by airbrush spraying the dispersion onto carbon substrates and characterized as catalyst electrodes towards H2 evolution using linear sweep voltammetry in the presence of 0.5 M H2SO4 electrolyte.
The performance of porous catalyst electrode depends on the quality of the active site themselves and the number of these sites made available by the electrode structure. A recent study by Mcateer et al.47 provides a simple model that takes into account the major factors that influence the catalytic activity of edge-site active nanosheet-based thin films, summarized by Eq. 1.
This relation indicates that the exchange current density (i.e., the reaction rate at zero overpotential) firstly depends on the geometric and microstructural aspects; the film thickness and porosity (t and P, respectively). It also depends on the characteristics of the constituent flakes that make up the film; the mean flake edge length, flake length to width aspect ratio and ML thickness (L, k and d 0, respectively). Together, these determine the area of edges made available by a given flake constituent of the film. Finally, the catalytic activity of each flake depends on the density of catalytic sites present along the flake edge and the intrinsic activity of each sites, termed the turnover frequency at zero overpotential (B and R 0, respectively). Thus, within the context of defect engineering in nanosheet catalysts, it will be useful to convert as many edge sites as possible into the catalytically active form (i.e., maximize B). In TMDs, it is known that disulfides are catalytically active site for the HER reaction.6,7, 47,48,49 Thus, if all other terms present in Eq. 1 are held constant, we would expect that by converting thiol sites into the active disulfide form the catalytic performance of the electrode should increase.
To investigate this question, the above-mentioned variables were controlled as much as possible such that differences in the measured exchange current density can be mainly attributed to differences in the density of the catalytic sites following the reaction with AuCl3. We chose m-WS2 (SI section 7.7) for this comparison as a trade-off between the population of catalytically-active edge sites and readily accessible nanosheet mass enabling the preparation of replicate samples. For this dispersion type the mean lateral dimension for the flakes was found to be <L>=67 nm. The flake aspect ratio tends to be fairly constant for a given TMD produced by LPE.38 Equal masses of WS2 per unit area were achieved through depositing equal volumes of concentration-adjusted dispersions to ensure the films have similar thicknesses. Profilometry measurements determined that all electrodes were ~100 nm thick. One variable that is both difficult to measure accurately (without preparing very thick films) is the film porosity. This quantity depends mainly on the deposition conditions (which were held constant), so that porosity should be comparable across the electrodes studied.
Figure 5a presents the measured linear sweep voltammograms for the m-WS2 reference, m-WS2–Au stock and purified samples, indicating that the Au modified samples indeed respond to the applied potential with a significantly greater reaction rate than films of the reference WS2. The data were converted into Tafel plots (shown in the Fig. 5a inset) and the Tafel slope and exchange current density were determined. All of the electrodes were found to possess Tafel slopes close to 100 mV/decade, typical of TMD nanosheet HER electrocatalysts.48 The applied potential was corrected for the IR drop associated with the working electrode (this includes the film resistance due to differences between the sample thicknesses or conductivity) using the impedance (at 100 kHz) measured prior to the linear sweep measurements. The obtained resistance values did not differ significantly among the samples tested suggesting only small differences in film conductivity despite the presence of the AuNPs (see SI, Table S7). This suggests that at this AuNP loading, no percolation throughout the film is achieved which would substantially alter the conductivity.

Electrocatalytic activity. a Linear sweep voltammograms comparing the activity of m-WS2–Au stock and purified samples to a m-WS2 reference dispersion not reacted with AuCl3 towards the HER. The inset presents the linear region of the data used to determine the Tafel slope and exchange current density. b Thickness normalized exchange current density of these electrode materials plotted as a function of the mean nanosheet length making up the network electrode. This plot enables comparison to other TMD-based nanomaterial networks catalysts presented in the literature
While it is common to present exchange current densities when comparing electrode materials, we emphasize that for porous electrode systems, the exchange current density will depend on the film thickness. Hence, here we present the exchange current volume (J 0/t), which should depend mostly on the term BR 0, as a function of the mean nanosheet length, as plotted in Fig. 5b. Error bars present the standard deviation across four films each. This approach also enables comparison with electrodes prepared using other TMDs from the literature (though with different mean nanosheet length, i.e., number of edge sites per area). Our reference electrode possess a J 0/t value similar to unmodified WS2 electrodes of similar lateral size prepared by Gholamvhand et al.49 Importantly, the WS2-Au electrodes possess J 0/t values five times higher than the reference dispersion and over an order of magnitude higher than many other LPE TMD-based network electrodes. This shows that the combined electrode materials and deposition conditions used result in an electrode structure with excellent performance among LPE TMDs. It is currently unclear why the stock WS2-Au presents improved performance compared with the purified sample. Future work including a systematic variation of AuNP loading level and nanosheet lateral size will be required for clarification.
Overall, these results provide strong evidence that the redox reaction with AuCl3 enables a substantial conversion of edge thiol sites (or potentially also minor defect sites on the basal plane) to the catalytically-active disulfide form and demonstrate that defect engineering of TMDs has great potential for realizing high-performance nanomaterial network-based catalytic electrodes.
In summary, we have reported evidence that edges (and defect sites) of LPE 2H–WS2 can be chemically converted and exploited in redox reactions. A spontaneous reaction with AuCl3 results in Au-decorated nanosheets with covalently bound AuNPs preferentially at edges and terraces of incompletely exfoliated WS2. By controlling the size and relative content of AuNPs, it was possible to tune the reaction products to obtain highly ML-enriched 2H–WS2 nanosheets decorated with 2–4 nm AuNPs at the edges. Alternatively, FL nanosheets with small AuNPs and slightly larger ones (10–20 nm) are also accessible. Importantly, optical properties of the 2H–WS2 are retained so that this reaction and purification sequence gives access to bulk quantities of ML 2H–TMDs with easily tuneable lateral size. It is suggested that thiols on TMDs act as reducing agents for the AuCl3 and are oxidized to disulfides as qualitatively confirmed by an enhanced electrocatalytic activity after reaction with AuCl3. Such nanosheets show great promise as catalyst electrodes in HER and potentially in other application areas such as printed (opto)electronics where large, ML-enriched nanosheet dispersions are required. In addition, further exploration of the edge and defect redox chemistry will open new routes for defect engineering in TMDs.
Methods
For schematic illustration, see SI.
Preparation of the initial WS2 dispersions
WS2 dispersions were prepared by probe sonicating the powder (Sigma Aldrich, order number 243639–50G, initial concentration 20 g/L) in an aqueous surfactant solution (SC, Sigma Aldrich, order number C1254-100G). WS2 was immersed in 80 mL of aqueous surfactant solution (C SC = 6 g/L) in a 100 mL metal beaker. The beaker was mounted in a water bath connected to a chiller system to maintain the external temperature at 5 °C. A solid flathead sonication tip was lowered to the bottom of the beaker, then raised 1 cm above the beaker bottom. The mixture was sonicated by probe sonication (Sonics VXC-500) for 1 h at 60% amplitude with a pulse of 8 s on and 2 s off. The dispersion was centrifuged in 20 mL aliquots using 50 mL vials in a Hettich Mikro 220R centrifuge equipped with a fixed-angle rotor 1016 at 5 krpm (2660g) for 1.5 h. The supernatant was discarded. The sediment was collected in 80 mL of fresh surfactant (C SC = 2 g/L) and subjected to a second sonication using the solid flathead tip for 5 h at 60% amplitude with a pulse of 6 s on and 2 s off.
To initially select nanosheets by size, LCC was used with a Beckman Coulter Avanti XP centrifuge equipped with two different rotors. For centrifugation at <15,000g, the JA25.50 fixed angle rotor and 50 mL centrifuge tubes (VWR, order number 525-0402 filled with 20 mL) were used. For centrifugation at >15,000g, the JA25.15 rotor was used with 14 mL vials (Beckman Coulter, order number 331,374 filled with 10 mL). Each centrifugation step was performed at 10 °C for 2 h. Unexfoliated WS2 was first removed by centrifugation at 400g. The sediment was discarded and the supernatant was subjected to centrifugation at 1000g. The sediment containing the fraction of larger, thicker nanosheets (denoted as l-WS2) was collected in fresh surfactant at reduced volume (10 mL, C SC = 2 g/L unless otherwise noted), while the supernatant was centrifuged at 5000g. Again, the sediment was collected (denoted as medium-sized, m-WS2) and the supernatant subjected to centrifugation at 22,000g. The supernatant was discarded and the sediment after this step, containing small, thin WS2 nanosheets (s-WS2), was collected in fresh SC. All WS2–SC dispersions were refrigerated to avoid degradation.
Preparation of the WS2–Au stock dispersions
After the initial size selection of the liquid-exfoliated WS2 (see above), the WS2 was diluted to a concentration of ~0.5 g/L (2 mM) with aqueous sodium cholate (C SC = 2 g/L unless otherwise noted). Aqueous AuCl3 solution was added 1:1 by volume to this dispersion, yielding the desired initial molar AuCl3:WS2 stoichiometry ratios. For the initial study (Fig. 2 and SI section 4), where the AuCl3 concentration was screened, total sample volumes were 1.5 mL. For example, to yield a molar stoichiometry ratio of 2:1, 1.5 ml of a prepared 4 mM aqueous AuCl3 solution was added to 1.5 mL of the 2 mM WS2 dilution. The resultant mixture was refrigerated overnight to equilibrate and then subjected to centrifugation at 15,000g (2 h) to remove unreacted AuCl3 in the supernatant. This centrifugation was performed in a Hettich Mikro 220R centrifuge equipped with a fixed-angle rotor 1195-A in 1.5 mL Eppendorf centrifuge tubes. The sediment was redispersed in fresh SC by bath sonication for 5 min and centrifuged again at 50g (2 h) to remove large aggregates prior to further characterization and/or purification.
Purification
For the preparation of the purified WS2–Au samples, WS2–Au stock dispersions were prepared in larger quantities for a subset of the WS2 dispersions for low and high initial AuCl3 contents (molar AuCl3:WS2 stoichiometry ratios of 1 and ~4.5), respectively. Typically, 50 mL of the WS2 dispersion (C WS2 = 0.5 g/L, C SC = 0.5 g/L) were mixed with 50 mL of aqueous AuCl3 (C AuCl3 = 2 mM and C AuCl3 = 9 mM, respectively). For the purification of s-WS2–Au and m-WS2–Au, the dispersion was centrifuged at 4000g (2 h), the sediment discarded and the supernatant centrifuged at 16,000g to collect the purified WS2–Au in the sediment. In the case of l-WS2–Au, the l-WS2–Au stock was centrifuged at 1000g, the sediment discarded and the supernatant centrifuged at 3000g to collect the purified sample in the sediment in fresh 2–4 mL SC.
Characterization
Optical extinction was measured on a Varian Cary 6000i in quartz cuvettes with a pathlength of 0.4 cm in 0.5 nm increments. Samples were diluted accordingly to extinction values of <2 across the entire spectral region.
For TEM, 3 µL of WS2–Au dispersion was drop-cast onto 20 nm thick SiO2 membranes (SPI Supplies, West Chester, PA USA). Each TEM grid was pre-heated to ~105 ˚C to flash evaporate the solvent for minimal aggregation. Imaging was performed on a FEI Titan3 80–300 TEM operated at 300 kV outfitted with a sperical aberration corrector from CEOS. Images were acquired with a post-column Gatan Imaging Filter.
For XPS measurements on a Thermo Scientific ESCALAB 250Xi system, thin films were prepared by vacuum filtration onto porous mixed cellulose ester membranes (0.025 µm pore size) and transferred onto ITO substrates. The XPS measurements were performed using a XR6 monochromated Al Kα source (hν = 1486.6 eV) and a pass energy of 20 eV. Spectra were collected at three different spots for each sample and analyzed using the Avantage software. After subtraction of a Shirley background, the core-level spectra were fitted with Gaussian–Lorentzian line shapes using Marquardt’s algorithm.
AFM was carried out on a Dimension ICON3 scanning probe microscope (Bruker AXS S.A.S.) in tapping mode in air under ambient conditions using aluminium coated silicon cantilevers (OTESP-R3). The concentrated dispersions were diluted with water to yield a pale colloidal dispersion. A drop of the dilute WS2–Au dispersions (20 μL) was deposited on a pre-heated (180 °C) Si/SiO2 wafer (1 × 1 cm2) with an oxide layer of 300 nm. After deposition, the wafer was rinsed with ~5 mL of water and ~5 mL of isopropanol. Typical image sizes were 10 × 10 μm2 at scan rates of 0.5 Hz with 1024 lines per image. For the statistical analysis of the purified l-WS2-Au sample, length, width and thickness of 270 individual nanosheets was recorded. After ~200 counts, the shape of the histograms did not change further confirming that sufficient objects were included in the statistics. Extreme outliers, where the superimposed lognormal distribution had already gone to 0 were excluded. The reported mean values are the arithmetic mean.
Raman (Raman/PL) spectroscopy was performed on the surface of liquid drops of concentrated dispersions using a Renishaw InVia microscope with 532 nm excitation laser in air under ambient conditions. The Raman emission was collected by a 50×, long working distance objective lens in streamline mode and dispersed by a 2400 l/mm grating with 1 % of the laser power (<0.2 mW) focused 5 μm above the surface of the dispersion. Measurements on a minimum of five different spots showed the samples to be homogeneous under these measurement conditions. The spectra were baseline corrected and averaged after acquisition. Great care must be taken during the measurement on the surface of liquid drops, as changes in the focal plane during the acquisition will introduce an error in the PL/Raman ratio. This is often reflected in a tilted baseline or asymmetric PL due to inner-filter and reabsorption effects. It can also be visually recognized when the size of the laser spot in the optical image has changed during the measurement. In such cases, the data was discarded.
PL to obtain the contour plots was measured using a Horiba Scientific Fluorlog-3 spectrometer equipped with a Xe lamp (450 W) and a PPD-900 photomultiplier tube detector. Measurements were taken under ambient conditions with double monochromators for excitation and emission. Prior to the measurement, the samples were diluted to an optical density of 0.4 (with respect to 1 cm pathlength) at 430 nm. The quartz cuvettes with 0.4 × 1 cm dimensions were placed inside the spectrometer so that the excitation light passed through the 0.4 cm side of the cuvette. Emission was collected in a 90° angle, i.e., after having passed through the 1 cm side. Bandwidths were 5 nm with acquisition times of 0.4 s. To avoid artefacts from scattering of the nanomaterial dispersion, a 550 nm cut-off filter was placed on the emission side. The excitation was corrected for the light intensity.
Thin films were prepared by airbrush spraying to investigate the catalytic properties of m-WS2–Au samples towards the HER. Here the nanosheet concentration of m-WS2, m-WS2–Au stock and purified m-WS2–Au (both with AuCl3:WS2 = 4.5) were adjusted to 0.1 g/L by the addition of water. Equal volumes of the dispersions were deposited (using conditions described elsewhere50) onto pyrolytic carbon substrates. Linear sweep voltammograms were obtained (at 5 mV/s in 0.5 M H2SO4) from 0 to −0.6 V vs. a Gaskatel-supplied Hydroflex H2-reference electrode. The measured voltage was compensated for resistances determined by an impedance measurement (0 V vs. SHE at 0.1 MHz, 10 mV RMS). Film thickness were measured by contact profilometry (DektakXT).
Data availability
Data that supports the findings of this study is available from the corresponding authors upon reasonable request.
References
Jariwala, D., Sangwan, V. K., Lauhon, L. J., Marks, T. J. & Hersam, M. C. Emerging device applications for semiconducting two-dimensional transition metal dichalcogenides. ACS Nano 8, 1102–1120 (2014).
Mak, K. F. & Shan, J. Photonics and optoelectronics of 2D semiconductor transition metal dichalcogenides. Nat. Photon-. 10, 216–226 (2016).
Buscema, M. et al. Photocurrent generation with two-dimensional van der Waals semiconductors. Chem. Soc. Rev. 44, 3691–3718 (2015).
Voiry, D., Yang, J. & Chhowalla, M. Recent strategies for improving the catalytic activity of 2D TMD nanosheets toward the hydrogen evolution reaction. Adv. Mater. 28, 6197–6206 (2016).
Li, B. L. et al. Low-dimensional transition metal dichalcogenide nanostructures based sensors. Adv. Funct. Mater. 26, 7034–7056 (2016).
Tao, H. et al. Two-dimensional nanosheets for electrocatalysis in energy generation and conversion. J. Mater. Chem. C 16, 7257–7284 (2017).
Pumera, M., Sofer, Z. & Ambrosi, A. Layered transition metal dichalcogenides for electrochemical energy generation and storage. J. Mater. Chem. C. 2, 8981–8987 (2014).
Mendoza-Sánchez, B. & Gogotsi, Y. Synthesis of two-dimensional materials for capacitive energy storage. Adv. Mater. 28, 6104–6135 (2016).
Tan, C. et al. Recent advances in ultrathin two-dimensional nanomaterials. Chem. Rev. 117, 6225–6331 (2017).
Lu, Q., Yu, Y., Ma, Q., Chen, B. & Zhang, H. 2D transition-metal-dichalcogenide-nanosheet-based composites for photocatalytic and electrocatalytic hydrogen evolution reactions. Adv. Mater. 28, 1917–1933 (2016).
Bonaccorso, F., Bartolotta, A., Coleman, J. N. & Backes, C. 2D-crystal-based functional inks. Adv. Mater. 28, 6136–6166 (2016).
Nicolosi, V., Chhowalla, M., Kanatzidis, M. G., Strano, M. S. & Coleman, J. N. Liquid exfoliation of layered materials. Science 340, 1420 (2013).
Tao, H. et al. Scalable exfoliation and dispersion of two-dimensional materials - an update. Phys. Chem. Chem. Phys. 19, 921–960 (2017).
Chhowalla, M. et al. The chemistry of two-dimensional layered transition metal dichalcogenide nanosheets. Nat. Chem. 5, 263–275 (2013).
Kelly, A. G. et al. All-printed thin-film transistors from networks of liquid-exfoliated nanosheets. Science 356, 69–73 (2017).
McManus, D. et al. Water-based and biocompatible 2D crystal inks for all-inkjet-printed heterostructures. Nat. Nano. 12, 343–350 (2017).
Kang, J. et al. Thickness sorting of two-dimensional transition metal dichalcogenides via copolymer-assisted density gradient ultracentrifugation. Nat. Commun. 5, 5478 (2014).
Backes, C. et al. Production of highly monolayer enriched dispersions of liquid-exfoliated nanosheets by liquid cascade centrifugation. ACS Nano 10, 1589–1601 (2016).
Kang, J., Sangwan, V. K., Wood, J. D. & Hersam, M. C. Solution-based processing of monodisperse two-dimensional nanomaterials. Acc. Chem. Res. 50, 943–951 (2017).
Chen, X. & McDonald, A. R. Functionalization of two-dimensional transition-metal dichalcogenides. Adv. Mater. 28, 5738–5746 (2016).
Presolski, S. & Pumera, M. Covalent functionalization of MoS2. Mater. Today 19, 140–145 (2016).
Hirsch, A. & Hauke, F. Post graphene 2D chemistry: the emerging field of molybdenum disulfide and black phosphorus functionalization. Angew. Chem. Int. Ed. https://doi.org/10.1002/anie.201708211 (2017).
Backes, C. et al. Functionalization of liquid-exfoliated two-dimensional 2H-MoS2. Angew. Chem. Int. Ed. 54, 2638–2642 (2015).
Chen, X., Berner, N. C., Backes, C., Duesberg, G. S. & McDonald, A. R. Functionalization of two-dimensional MoS2: on the reaction between MoS2 and organic thiols. Angew. Chem. Int. Ed. 55, 5803–5808 (2016).
Kim, J., Byun, S., Smith, A. J., Yu, J. & Huang, J. Enhanced electrocatalytic properties of transition-metal dichalcogenides sheets by spontaneous gold nanoparticle decoration. J. Phys. Chem. Lett. 4, 1227–1232 (2013).
Shi, Y. et al. Selective decoration of Au nanoparticles on monolayer MoS2 single crystals. Sci. Rep. 3, 1839 (2013).
Zhang, P. et al. MoS2 nanosheets decorated with gold nanoparticles for rechargeable Li-O2batteries. J. Mater. Chem. C. 3, 14562–14566 (2015).
Topolovsek, P. et al. Thiol click chemistry on gold-decorated MoS2: elastomer composites and structural phase transitions. Nanoscale 8, 10016–10020 (2016).
Sun, H. et al. Gold nanoparticle-decorated MoS2 nanosheets for simultaneous detection of ascorbic acid, dopamine and uric acid. RSC Adv. 4, 27625–27629 (2014).
Su, S. et al. Direct electrochemistry of glucose oxidase and a biosensor for glucose based on a glass carbon electrode modified with MoS2 nanosheets decorated with gold nanoparticles. Microchim. Acta 181, 1497–1503 (2014).
Forcherio, G. T. & Roper, D. K. Spectral characteristics of noble metal nanoparticle–molybdenum disulfide heterostructures. Adv. Opt. Mater. 4, 1288–1294 (2016).
Forcherio, G. T., Benamara, M. & Roper, D. K. Electron energy loss spectroscopy of hot electron transport between gold nanoantennas and molybdenum disulfide by plasmon excitation. Adv. Opt. Mater. 5, 1600572 (2017).
Lin, J., Li, H., Zhang, H. & Chen, W. Plasmonic enhancement of photocurrent in MoS2 field-effect-transistor. Appl. Phys. Lett. 102, 203109 (2013).
Yin, Z. et al. Au nanoparticle-modified MoS2 nanosheet-based photoelectrochemical cells for water splitting. Small 10, 3537–3543 (2014).
Kang, Y. et al. Plasmonic hot electron enhanced MoS2 photocatalysis in hydrogen evolution. Nanoscale 7, 4482–4488 (2015).
Huang, X. et al. Solution-phase epitaxial growth of noble metal nanostructures on dispersible single-layer molybdenum disulfide nanosheets. Nat. Commun. 4, 1444 (2013).
Zhong, L. et al. Defect engineering of two-dimensional transition metal dichalcogenides. 2D Mater. 3, 022002 (2016).
Backes, C. et al. Edge and confinement effects allow in situ measurement of size and thickness of liquid-exfoliated nanosheets. Nat. Commun. 5, 4576 (2014).
Mie, G. Beiträge zur Optik trüber Medien, speziell kolloidaler Metallösungen. Ann. Phys. 330, 377–445 (1908).
Bandyopadhyay, S., Chattopadhyay, S. & Dey, A. The protonation state of thiols in self-assembled monolayers on roughened Ag/Au surfaces and nanoparticles. Phys. Chem. Chem. Phys. 17, 24866–24873 (2015).
Battocchio, C. et al. Gold nanoparticles stabilized with aromatic thiols: interaction at the molecule–metal interface and ligand arrangement in the molecular shell investigated by SR-XPS and NEXAFS. J. Phys. Chem. C. 118, 8159–8168 (2014).
Vitale, F. et al. Mono- and bi-functional arenethiols as surfactants for gold nanoparticles: synthesis and characterization. Nanoscale Res. Lett. 6, 103 (2011).
Gade, M. et al. Immobilization of multivalent glycoprobes on gold surfaces for sensing proteins and macrophages. Analyst 141, 2250–2258 (2016).
Forcherio, G. T. et al. Gold nanoparticles physicochemically bonded onto tungsten disulfide nanosheet edges exhibit augmented plasmon damping. AIP Adv. 7, 075103 (2017).
Mak, K. F., Lee, C., Hone, J., Shan, J. & Heinz, T. F. Atomically thin MoS2. A new direct-gap semiconductor. Phys. Rev. Lett. 105, 136805 (2010).
Mitioglu, A. A. et al. Optical manipulation of the exciton charge state in single-layer tungsten disulfide. Phys. Rev. B 88, 245403 (2013).
McAteer, D. et al. Thickness dependence and percolation scaling of hydrogen production rate in MoS2 nanosheet and nanosheet-carbon nanotube composite catalytic electrodes. ACS Nano 10, 672–683 (2016).
Gholamvand, Z. et al. Comparison of liquid exfoliated transition metal dichalcogenides reveals MoSe2 to be the most effective hydrogen evolution catalyst. Nanoscale 8, 5737–5749 (2016).
Gholamvand, Z., McAteer, D., Harvey, A., Backes, C. & Coleman, J. N. Electrochemical applications of two-dimensional nanosheets: the effect of nanosheet length and thickness. Chem. Mater. 28, 2641–2651 (2016).
Higgins, T. M. & Coleman, J. N. Avoiding resistance limitations in high-performance transparent supercapacitor electrodes based on large-area, high-conductivity PEDOT:PSS films. ACS Appl. Mater. Interfaces 7, 16495–16506 (2015).
Acknowledgements
C.B. acknowledges the German research foundation DFG under Emmy-Noether grant BA4856/2-1. The research leading to these results has received funding from the European Union’s Horizon 2020 under grant agreement n°604391 Graphene Flagship. J.R.D. and G.T.F acknowledge support from the National Science Foundation (NSF) Graduate Research Fellowship Program. This project began from support to J.R.D. from NSF’s Graduate Research Opportunities Worldwide (GROW) program in coordination with Science Foundation Ireland., T.M.H. acknowledges the European Union’s Horizon 2020 research and innovation program under the Marie Sklodowska–Curie grant agreement No. 707644. We thank Jana Zaumseil for the access to the full infrastructure of the Chair of Applied Physical Chemistry in Heidelberg.
Author information
Authors and Affiliations
Contributions
J.R.D. initiated the project, prepared samples, and performed initial optical characterization. P.L. optimized the sample preparation and purification, T.M.H. prepared films and tested HER, G.T.F and M.B. performed TEM, N.M. prepared HER substrates, D.K.R. and J.N.C. discussed the results with J.R.D. and C.B., Y.V. performed and interpreted XPS, C.B. prepared samples, performed optical characterization and AFM, designed the experiments, analyzed the data and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dunklin, J.R., Lafargue, P., Higgins, T.M. et al. Production of monolayer-rich gold-decorated 2H–WS2 nanosheets by defect engineering. npj 2D Mater Appl 1, 43 (2017). https://doi.org/10.1038/s41699-017-0045-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41699-017-0045-z
