
Abstract
Homologous recombination deficiency (HRD) testing has been approved by FDA for selecting epithelial ovarian cancer (EOC) patients who may benefit from the first-line poly (ADP-ribose) polymerase inhibitor (PARPi) maintenance therapy. However, the effects of HRD on the clinical outcomes of first-line chemotherapy and first-line PARPi maintenance therapy have not been rigorously evaluated in Chinese EOC patients. Here, we developed an HRD assay and applied it to two large retrospectively collected Chinese EOC patient cohorts. In the first-line adjuvant chemotherapy cohort (FACT, N = 380), HRD status significantly improved PFS (median, 15.6 months vs. 9.4 months; HR, 0.688; 95% CI, 0.526–0.899; P = 0.003) and OS (median, 89.5 months vs. 60.9 months; HR, 0.636; 95% CI, 0.423–0.955; P = 0.008). In the first-line PARPi maintenance therapy cohort (FPMT, N = 83), HRD status significantly improved PFS (median, NA vs. 12 months; HR, 0.438; 95% CI, 0.201–0.957; P = 0.033) and OS (median, NA vs. NA months; HR, 0.12; 95% CI, 0.029–0.505; P = 0.001). Our results demonstrate that HRD status is a significant predictor for PFS and OS in both first-line chemotherapy and first-line PARPi maintenance therapy, providing strong real-world evidence for conducting genetic testing and improving clinical recommendations for Chinese EOC patients.
Similar content being viewed by others


Introduction
Epithelial ovarian cancer (EOC) is the most lethal gynecologic malignancy1, and there were more than 313,959 new cases and 207,252 deaths worldwide in 20202. Based on conservative estimates, in 2015, 57,200 new cases and 27,200 deaths occurred in China3. Identification of mutation carriers among probands not only represents a great opportunity for risk-reduction interventions but also provides reassurance to noncarriers4. Furthermore, homologous recombination deficiency (HRD) assays have been used to stratify EOC patients for effective treatment5.
Recent progress in developing targeted therapies of EOC, such as poly (ADP-ribose) polymerase inhibitor (PARPi), has created a great need for genetic testing6,7,8. Four PARPi (i.e., niraparib, olaparib, rucaparib, and fluzoparib) have been approved for the maintenance treatment of patients with EOC who exhibit complete or partial response to platinum-based chemotherapy regardless of BRCA mutation status and HRD status9,10,11,12,13,14,15,16 Several HRD models have been developed, including “MyChoice CDx”17 and “FoundationFocus CDxBRCA LOH”9. However, beyond BRCA-mutated tumors, current HRD assays have not demonstrated a differentiation power in predicting patient response to PARPi and other antiangiogenic therapy to justify their routine use in the clinic5. In particular, despite several recent studies18,19,20,21,22, little is known about the real-world impact of HRD on the therapeutic effects of chemotherapy and PARPi in Chinese EOC patients.
To fill this knowledge gap, we developed an HRD assay and applied it to two large patient cohorts to evaluate the impact of HRD in first-line chemotherapy and subsequent first-line PARPi maintenance therapy in Chinese EOC patients.
Results
Development and validation of an HRD assay
To characterize the HRD status of tumor samples, we developed a customized sequencing panel named “Precision Human HRD Assay.” This HRD assay contains two sets of probes (Fig. 1a). One set of HRD-score probes (~50 K) evenly covers the whole genome (Fig. 1b) and aims to assess genomic instability on a global scale. We developed an HRD score algorithm to calculate a score for each of the three features: the loss of heterozygosity (LOH), telomeric allelic imbalance (TAI), and large-scale state transitions (LST), and the overall HRD score was the sum of LOH, TAI, and LST scores. The other set of DDR-gene probes aims to evaluate the genotype of 36 DNA damage repair (DDR) genes, among which are 28 homologous recombination repair (HRR) genes (Supplementary Table 1). To validate the analytical performance of our HRD assay, we compared our HRD score with (i) that of myChoice-Plus, a widely used commercial HRD assay (R2 = 0.983, Fig. 1c), (ii) that based on whole-exome sequencing data (WES) plus backbone (R2 = 0.988, Fig. 1d), and (iii) that based on whole-genome sequencing (WGS) data (R2 = 0.988, Fig. 1e), and they all showed extremely high correlations. These results indicate that our HRD assay can quantify the HRD status of tumor samples accurately.

a A diagram showing the components of the Precision Human HRD Assay, created with BioRender.com. b Distribution of HRD-score probes by chromosomes. The correlation of the HRD score with those from c myChoice-Plus, d WES+Backbone, and e WGS. The WGS samples were a subset of WES+Backbone samples, and these samples are highlighted in red in (d, e).
Tumor characteristics of EOC patients
To evaluate the impact of HRD status on the treatment of Chinese EOC patients, we retrospectively surveyed the medical records of patients from two leading hospitals in China: Peking Union Medical College Hospital (Beijing, China) and West China Hospital of Sichuan University (Chengdu, China). Among the eligible participants who underwent first-line adjuvant chemotherapy, we successfully genotyped 380 patients using our HRD assay and defined them as the FACT cohort (Supplementary Table 2). Among the eligible participants who underwent first-line PARPi maintenance therapy, we successfully genotyped 83 patients and defined them as the FPMT cohort (Supplementary Table 3). Thus, our study included 463 participants in total (Supplementary Fig. 1).
From 36 DDR genes covered by our HRD assay, we further defined several HRR gene subsets, including HRR12, HRR14, HRR26, and HRR28 (Supplementary Table 1). We next examined the mutation status of these genes or subsets in our cohorts. Among all the participants, 172/463 (37.1%) were BRCA1/2 mutated, and 164/463 (35.4%) had BRCA1/2 bi-allelic loss-of-function (BILOF). Among 380 participants in the FACT cohort, 133/380 (35%) were BRCA1/2 mutated, and 130/380 (34.2%) had BRCA1/2 BILOF. Among 83 participants in the FPMT cohort, 39/83 (47%) were BRCA1/2 mutated, and 34/83 (41%) had BRCA1/2 BILOF. In the FACT cohort, 9.2% of the participants were BRCA1/2 wild-type with at least one of the HRR26 genes mutated; therefore, a total of 44.2% of the participants had at least one of the HRR28 genes mutated, and 55.8% participants had all HRR28 genes as wild-type (Fig. 2a, b). Among all the mutations observed on HRR28 genes, BRCA1 (58%) and BRCA2 (13.3%) were the most frequent ones, followed by RAD51D (5.9%), RAD51C (3.7%), PTEN (2.7%), and PALB2 (2.1%). In the FPMT cohort, we observed a higher frequency of BRCA1/2 mutations, likely due to the treatment strategy (Fig. 2c, d).

HRR gene mutation rates in the FACT cohort are summarized a by patients and b by HRR gene mutations. HRR gene mutation rates in the FPMT cohort are summarized c by patients and d by HRR gene mutations. Mutation rates in each pie chart sum to 100%. HRR26 is a gene list composed of 26 HRR genes, as defined in Supplementary Table 1. A patient is defined as HRR26-mutated if having at least one of the HRR26 genes mutated. HRD score distribution stratified by BRCA1/2 BILOF is illustrated e by a histogram and f by a violin plot. On each violin plot, the lower whisker, lower bound of box, center line, upper bound of box, and upper whisker represent the minimum, lower quartile, median, upper quartile, and maximum, respectively, with the curve representing kernel density plot. MUT mutated, WT wild-type.
Bi-allelic alterations in HRR genes are necessary for loss of function according to the two-hit hypothesis. We tested whether the bi-allelic inactivation of HRR genes led to genomic scarring consistent with the underlying DNA-repair deficiency. We observed strong positive associations between BILOF of HRR genes and HRD score for HRR28 (47.7% increase in median HRD score relative to the wild-type; P < 0.001), HRR14 (47.7% increase; P < 0.001), and BRCA1/2 (44.4% increase; P < 0.001) in the FACT cohort (Supplementary Fig. 2a). We further analyzed each DDR gene and observed strong positive associations for BRCA1 (40.8% increase; P < 0.001), RAD51D (28.2% increase; P = 0.038), and TP53 (31.3% increase; P < 0.001) (Supplementary Fig. 2b). We finally examined the association between BRCA1 promoter methylation and HRD score in the FACT cohort and found a positive association (Supplementary Fig. 3). We observed a clear distinction in HRD score distribution when stratified by BRCA1 promoter methylation status (BRCA1 promoter methylation score ≥0.5). These results indicate a good performance of our HRD assay.
To make a binary call on the HRD status of a tumor (i.e., HRD-positive vs. HRD-negative), we aimed to identify a threshold of HRD score that can effectively separate BRCA1/2 BILOF from the others. The HRD score threshold training set contained 199 BILOF participants in total, including 130 BRCA1/2 BILOF participants from the FACT cohort, 34 BRCA1/2 BILOF participants from the FPMT cohort, and 35 BRCA1/2 BILOF participants from our routine genetic testing services who met the shared eligibility criteria of FACT and FPMT cohort. Within the HRD score threshold training set, the 5th percentile of the HRD score was 41.9, for which the sensitivity in predicting BRCA1/2 BILOF was 95% (Fig. 2e). Thus, a tumor was defined as HRD-positive if the HRD score was ≥42 or BRCA1/2 was mutated (Fig. 2f).
Effects of HRD status on patient survival time in the FACT cohort
To validate the clinical value of HRD status in EOC patients, we first examined the correlations of HRD status with both chemotherapy-related progression-free survival (PFS) and overall survival (OS) in the FACT cohort. For the PFS analysis stratified by HRD status, the median follow-up time was 35.7 months (IQR 27.3–44.6). Figure 3 shows PFS and OS analyses stratified by BRCA1/2 mutation and HRD status. The median PFS was 17.7 months for participants with BRCA1/2 mutation versus 12.1 months for participants with wild-type BRCA1/2 (HR, 0.629; 95% CI, 0.487–0.814; P = 0.003) (Fig. 3a). When the combination of HRD score and BRCA1/2 mutation status was used to obtain the HRD status, the median PFS was 15.6 months in HRD-positive participants versus 9.4 months in the HRD-negative participants (HR, 0.688; 95% CI, 0.526–0.899; P = 0.003) (Fig. 3b). Even in the BRCA1/2 wild-type subgroup, there was an improved PFS in participants with a positive HRD status (median, 13 months versus 9.4 months; HR, 0.769; 95% CI, 0.578–1.023; P = 0.071) (Fig. 3c) with marginal significance. We observed similar improvements in OS for participants who were BRCA1/2 mutated, HRD-positive, and HRD-positive with wild-type BRCA1/2. The magnitude of the OS benefit was most prominent in BRCA1/2 mutated participants (median, 90.9 months versus 70.5 months; HR, 0.6; 95% CI, 0.39–0.922; P = 0.004) (Fig. 3d), followed by the HRD-positive (median, 89.5 months versus 60.9 months; HR, 0.636; 95% CI, 0.423–0.955; P = 0.008) (Fig. 3e), and the least in BRCA1/2 wild-type participants with a positive HRD status, showing no statistical significance (median, 74.9 months versus 60.9 months; HR, 0.724; 95% CI, 0.468–1.122; P = 0.147) (Fig. 3f).

PFS analyses in the FACT cohort stratified by a BRCA1/2 mutation, b HRD status, and stratified by c HRD status in BRCA1/2 wild-type participants are presented. OS analyses in the FACT cohort stratified by d BRCA1/2 mutation, e HRD status, and stratified by f HRD status in BRCA1/2 wild-type participants are presented. WT wild-type.
We next performed PFS and OS analyses on HRD-positive participants stratified by different causes of HRD. Participants whose tumors were HRD-positive due to genetic changes (BRCA1/2 mutation) outperformed the other two groups in both PFS and OS (Supplementary Fig. 4). Participants whose tumors were HRD-positive due to epigenetic changes (BRCA1/2 wild-type and BRCA1 promoter methylation status high) showed similar PFS and OS compared with those whose tumors were HRD-positive due to unknown reasons (BRCA1/2 wild-type, BRCA1 promoter methylation status low, and HRD status positive).
Finally, we investigated whether HRD status could predict the platinum sensitivity status in the FACT cohort. BRCA1/2 mutations were strongly associated with being platinum-sensitive: 87.2% of participants with BRCA1/2 mutations were platinum-sensitive, whereas only 72.9% of participants without BRCA1/2 mutations were platinum-sensitive (P = 0.001) (Supplementary Fig. 5a). HRD status also increased the platinum sensitivity rate (80.6% versus 68.6%, P = 0.026) (Supplementary Fig. 5b). HRD status achieved a smaller increase in platinum sensitivity rate in the BRCA1/2 wild-type participants (75.2% versus 68.6%, P = 0.295) (Supplementary Fig. 5c), with no statistical significance.
Effects of HRD status on patient survival time in the FPMT cohort
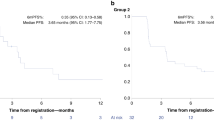
To further assess whether HRD status could stratify Chinese EOC patients for PARPi (Niraparib or Olaparib) maintenance therapy in the first-line setting, we analyzed PFS and OS in FPMT cohort stratified by BRCA1/2 mutation and HRD status (Fig. 4). For the PFS analysis stratified by HRD status, the median follow-up time was 22.1 months (IQR 10.9–29.3). BRCA1/2 mutation was a strong predictor for PFS (median, NA vs. 20 months; HR, 0.423; 95% CI, 0.198–0.906; P = 0.023) (Fig. 4a) but not as much for OS (median, NA vs. NA; HR, 0.592; 95% CI, 0.141–2.477; P = 0.467) (Fig. 4d). There was a significantly improved PFS in HRD-positive participants (median, NA vs. 12 months; HR, 0.438; 95% CI, 0.201–0.957; P = 0.033) (Fig. 4b), and HRD status also showed an obvious benefit in OS (median, NA vs. NA; HR, 0.12; 95% CI, 0.029– 0.505; P = 0.001) (Fig. 4e). Furthermore, in the BRCA1/2 wild-type subgroup, HRD status also demonstrated a significant benefit in OS (Fig. 4f).

PFS analyses in the FPMT cohort stratified by a BRCA1/2 mutation, b HRD status, and stratified by c HRD status in BRCA1/2 wild-type participants are presented. OS analyses in the FPMT cohort stratified by d BRCA1/2 mutation, e HRD status, and stratified by f HRD status in BRCA1/2 wild-type participants are presented. WT wild-type.
HRD status versus HRR genes in BRCA1/2 wild-type participants
We investigated potential predictors for efficacy in BRCA1/2 wild-type participants (Fig. 5). For the PFS analyses in the FACT cohort, HRD status, HRR28, and HRR14 were all satisfactory predictors but did not reach statistical significance (Fig. 5a). For OS analyses in the FACT cohort, HRR28 and HRR14 performed slightly worse compared to HRD status, and none of them reached statistical significance (Fig. 5b). For both PFS and OS analyses in the FPMT cohort, HRR14 and HRR28 were poor predictors with no statistical significance (Fig. 5c, d). In the FPMT cohort, HRD status was a strong predictor for both PFS and OS, especially for OS (statistically significant) (Fig. 5c, d). Collectively, these results suggest that HRD status was a better predictor for efficacy than the mutational status of HRR genes in BRCA1/2 wild-type participants.

Forest plots illustrate whether HRD status, HRR28, and HRR14 were strong predictors for efficacy in BRCA1/2 wild-type participants. Explorations were split by a PFS in the FACT cohort, b OS in the FACT cohort, c PFS in the FPMT cohort, and d OS in the FPMT cohort.
We further explored whether alterations of each gene were associated with HRD status in BRCA1/2 wild-type participants. In the combined dataset (FACT and FPMT cohorts combined), TP53 BILOF was significantly enriched in HRD-positive participants in the BRCA1/2 wild-type subgroup (P < 0.001, Supplementary Fig. 6). In contrast, the frequency of TP53 mono-allelic loss-of-function (MOLOF) was significantly higher in the HRD-negative group than the HRD-positive group (P = 0.016), but in the context of co-occurrence with BRCA1-LOH, the difference between the two groups became insignificant (P = 0.854, Supplementary Fig. 6). TP53 BILOF did not show any correlation with efficacy in the FACT or FPMT cohort (Supplementary Fig. 7).
Discussion
Here we developed a rigorous HRD assay and assessed the impact of HRD status on the efficacy of first-line chemotherapy and PARPi maintenance therapy in Chinese patients with primary EOC. Our HRD scoring system demonstrates its utility in predicting therapy efficacy in Chinese EOC patients. Furthermore, consistent with the results in the PRIMA trial8 and ATHENA-MONO23, even in BRCA wild-type patients, HRD status in our study demonstrated survival benefits in both PFS and OS when appropriate HRD score threshold was selected. Despite some fundamental disparities between the FACT and FPMT cohorts, including different mechanisms of drug resistance and patient pharmacogenomic features (e.g., intratumor heterogeneity and HRR gene mutation rate)24,25,26,27,28, our HRD assay demonstrates robust correlations with clinical outcomes.
Our study shows a moderately high percentage of being HRD-positive in the BRCA1/2 wild-type participants, which is similar to the findings from Japan29,30 and another study in China31 but higher than those reported in previous studies8,17. Sinha et al.32 demonstrated higher genomic instability, HRD, and chromothripsis among tumors from African Americans across many cancer types (including ovarian cancer) compared with European Americans in The Cancer Genome Atlas. Hsiao et al.33 reported that genome-wide HRD scores showed racial differences between the Caucasian population and Asian population across cancer types, with a significantly higher score observed in the Asian population. Furthermore, BRCA1/2 mutation rates reached 35% in our study, which was slightly higher than other reports from China34,35 and higher than other countries36,37. Ethnic-specific BRCA1/2 variation in Asian populations may be explained as follows:38 low overlapping between Asian and non-Asian BRCA1/2 variants, low overlapping within different Asian populations, different variation spectrum and variation types, and differences in BRCA1/2 founder mutations. BRCA1 c.5470_5477del, which is a reported Chinese BRCA1 founder mutation39, has a high occurrence in patients from our routine genetic testing services and from patients in this study. Taken together, our results suggest that genetic ancestry is a major factor for the higher rate of HRD positivity observed in this study.
Consistent with a previous report30, we show that among the HRD-positive participants, positive HRD status caused by BRCA1/2 mutations demonstrated a better prognosis than those caused by epigenetic changes or unknown reasons. These findings suggest that BRCA1/2 alterations contributed greatly to the HRD status. Contrary to BRCA1/2 mutations, mutations in other HRR genes had no inherent association with survival outcomes in the FACT cohort. These findings are compatible with previous studies and current recommendations5,40,41. We found that a higher BRCA1 methylation score was associated with higher HRD scores. However, in this and previous studies30, epigenetic changes in BRCA1/2 genes were less associated with survival outcomes compared with genetic HRD, but this difference did not reach statistical significance. Furthermore, we found that TP53-mutated participants had higher median HRD scores than the TP53 wild-type, especially for TP53 BILOF, which was significantly enriched in high HRD score participants in the BRCA1/2 wild-type subgroup. These findings are consistent with previous observations in prostate cancer42 and pan-cancer analysis43. Loss of TP53 may lead to chromosomal instability and aneuploidy44, which is associated with a higher HRD score.
In conclusion, we developed an effective HRD assay for Chinese EOC patients, which provides precise predictions for first-line chemotherapy and PARPi efficacy, including PFS and OS. The HRD status shows high accordance with BRCA1/2 mutations, BILOF of HRR genes, and BRCA1 promoter methylation scores. These findings should be further validated in future cohorts or RCT trials with larger sample sizes and universal treatment protocols.
Methods
HRD assay development
The Precision Human HRD Assay contains two sets of probes, HRD-score probes (~50 K) and DDR-gene probes, which were used to evaluate HRD score and genotype 36 DDR genes (Supplementary Table 1), respectively. To validate the analytical performance, the HRD score was compared with the scores of myChoice-Plus, WES+Backbone, and WGS. The WES+Backbone (IDT xGen Exome Research Panel v1.0, IDT xGen Human CNV Backbone Hybridization Panel) and WGS data were sequenced on the NovaSeq platform in PE150 mode by a mean depth of 150× (tumor) and 50× (control). The WES+Backbone and WGS data were analyzed using FACETS45 and scarHRD46.
The SNPs targeted by the Precision Human HRD Assay were selected based on the following criteria: SNPs on the Y chromosome were removed; mitochondrial SNPs were removed; SNPs with minor allele-frequency (MAF) less than 1% in European or West African population were removed; SNPs with MAF less than 5% in East Asian population were removed; SNPs significantly deviated from Hardy–Weinberg Equilibrium (HWE) in any of the three populations mentioned above were removed; SNPs with Fst (fixation indices) <0.05 in East Asian population were removed; SNPs with CG-content <40% or >60% were removed; SNPs located on Short Tandem Repeats (STRs) were removed; SNPs evenly covered the human genome.
We developed an in-house HRD score algorithm to assess genomic instability and calculate an overall HRD score through four steps. The first step was to split the genome into segments. Sequencing depth and allele frequency for each SNP site were calculated based on the alignment of the sequencing data to the human reference genome (GRCh37/UCSC hg19), and SNPs were then grouped into different segments. Only heterozygous SNPs were used during the computation. The second step was to estimate the parameters in each segment. Based on the sequencing depth and allele frequency in each segment, four key parameters were estimated using maximum likelihood estimation, including major allele count per segment, minor allele count per segment, tumor purity, and tumor ploidy. The third step was to calculate a score for each of the three features based on the estimated major allele count per segment and minor allele count per segment, similar to that described in previous studies: LOH47, TAI48, and LST49. Finally, the HRD score was the sum of LOH, TAI, and LST scores. For genes included in the panel, a custom bioinformatic analysis pipeline was used to detect single nucleotide variants (SNVs) and small insertions and deletions (indels) in protein-coding regions and intron/exon boundaries of the 36 genes. Variants were classified according to the recommendations of the American College of Medical Genetics and Genomics (ACMG) for standards in the interpretation of sequence variations50. Clinically significant variants were classified as “class 5: pathogenic” or “class 4: likely pathogenic.”
EOC participants
The study was approved by the Ethics Committee of Peking Union Medical College Hospital, Beijing (project ID: JS–1932). Medical records of EOC patients from Peking Union Medical College Hospital and West China Hospital of Sichuan University were surveyed retrospectively. Eligibility criteria included being at least 18 years old at diagnosis; a histological/pathologic diagnosis of EOC (ovarian cancer, fallopian tube cancer, or primary peritoneal cancer); high-grade serous or grade-3 endometroid histological subtype; FIGO stage II, III, or IV; first-line surgery performed; administered at least five rounds of first-line chemotherapy. Eligibility criteria unique to the FACT cohort included no maintenance therapy of any kind, no PARPi administered during first-line therapies for any purpose, and the date of the last dose of first-line adjuvant chemotherapy between 2009/12/01 and 2020/05/01. The FACT cohort patients were collected from two different investigators. Eligibility criteria unique to FPMT included achieving complete or partial response after first-line adjuvant chemotherapy and PARPi administered as first-line maintenance therapy. FPMT cohort patients were collected from two different investigators.
Molecular analyses of patient samples
For each participant, at least 10 slides of 5μm formalin-fixed paraffin-embedded (FFPE) tumor samples were collected with the patient’s informed consent (both written informed consent for this study and informed consent in the medical record). The first slide was stained with hematoxylin and eosin (H&E). The H&E slide was reviewed by two independent pathologists to determine the histological type and neoplastic cellularity (30% minimum). Genomic deoxyribonucleic acid (gDNA) was extracted and quantified from the patient’s specimen(s) using a standardized methodology. Tumor-only genetic testing was conducted with the Precision Human HRD Assay. Among the 36 DDR genes, we further defined several HRR gene lists (Supplementary Table 1). If at least one gene in a gene list was mutated (class 5 or 4), then the gene list was considered mutated. An additional 100 ng genomic DNA was used to evaluate BRCA1 promoter methylation status via bisulfite sequencing. We defined the average methylation ratio across CpG sites (i.e., BRCA1 promoter methylation score) ≥0.5 as high methylation status.
Endpoints
For the FACT cohort, the primary endpoint was PFS, defined as the time from the last dose of chemotherapy to disease progression or death, whichever occurred first. Participants with uncontrolled disease, defined as the participants whose disease progressed during first-line adjuvant chemotherapy, had PFS time set as zero and censoring status set as the event occurred. Secondary endpoints included OS, defined as the time from the last dose of chemotherapy to death, and platinum sensitivity status (PSS), defined as whether progression-free survival was longer than 6 months. For the FPMT cohort, the primary endpoint was PFS, defined as the time from the first dose of PARPi to disease progression or death, whichever occurred first. The secondary endpoint was OS, defined as the time from the first dose of PARPi to death. The cutoff date for assessing disease progression and survival of participants was February 23, 2022.
HRD score threshold training
The HRD score threshold training set was composed of three parts: BRCA1/2 BILOF participants from the FACT cohort, BRCA1/2 BILOF participants from the FPMT cohort, and selective BRCA1/2 BILOF participants from our routine genetic testing services who met the shared eligibility criteria of FACT and FPMT cohort. The third part of the HRD score threshold training set was added to further expand the number of BRCA1/2 BILOF participants since a larger training set produced a more robust HRD score threshold. The HRD score threshold was trained by reaching 95% sensitivity in predicting BRCA1/2 BILOF, i.e., the HRD score threshold was the 5th percentile of HRD score in BRCA1/2 BILOF participants. BRCA1/2 BILOF was defined as meeting one of the following three criteria: (i) one allele as class 4/5 mutated and the other allele as LOH, (ii) two class 4/5 mutations in the same gene, or (iii) one allele as methylation status high and the other allele as LOH.
Statistical analyses
Statistical analyses were conducted using R 4.2.1. PFS and OS were estimated using the Kaplan–Meier method. Treatment effect differences were assessed using the log-rank test. In the FACT cohort, HR and associated 95% CI were calculated using the CoxPH model, adjusted for age, FIGO stage, surgery residual, surgery type, pre-treatment CA125, concurrent use of bevacizumab, and the round of chemotherapy. For analyses in the FPMT cohort and subgroup analyses in the FACT cohort, HR and associated 95% CI were calculated using the unadjusted CoxPH model. If one arm in PFS or OS analysis had zero events, Firth’s penalized maximum likelihood bias reduction method for the CoxPH model was applied. Fisher’s exact test was used to assess whether the proportion of platinum-sensitive participants was significantly different between strata. Two-sided statistical tests were used.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Raw sequencing data files of patients cannot be publicly shared under the obtained institutional review board approval, as patients did not consent to share raw sequencing data beyond the research and clinical terms. However, the datasets generated and analyzed during the current study are available from the corresponding author on reasonable request and upon a data usage agreement.
Code availability
Code used for all processing and analysis is available upon reasonable request.
References
Howlader, N. et al. (eds). SEER Cancer Statistics Review, 1975–2018, National Cancer Institute. Bethesda, MD, https://seer.cancer.gov/csr/1975_2018/, based on November 2020 SEER data submission, posted to the SEER web site (2021).
Sung, H. et al. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71, 209–249 (2021).
Zheng, R. et al. Cancer incidence and mortality in China, 2016. J. Natl Cancer Cent. 2, 1–9 (2022).
Samimi, G. et al. Traceback: a proposed framework to increase identification and genetic counseling of BRCA1 and BRCA2 mutation carriers through family-based outreach. J. Clin. Oncol. 35, 2329–2337 (2017).
Konstantinopoulos, P. A. et al. Germline and somatic tumor testing in epithelial ovarian cancer: ASCO guideline. J. Clin. Oncol. 38, 1222–1245 (2020).
Coleman, R. L. et al. Veliparib with first-line chemotherapy and as maintenance therapy in ovarian cancer. N. Engl. J. Med. 381, 2403–2415 (2019).
Enomoto, T. et al. The first Japanese nationwide multicenter study of BRCA mutation testing in ovarian cancer: CHARacterizing the cross-sectionaL approach to Ovarian cancer geneTic TEsting of BRCA (CHARLOTTE). Int. J. Gynecol. Cancer 29, 1043–1049 (2019).
González-Martín, A. et al. Niraparib in patients with newly diagnosed advanced ovarian cancer. N. Engl. J. Med. 381, 2391–2402 (2019).
Coleman, R. L. et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 390, 1949–1961 (2017).
Ledermann, J. et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer. Obstet. Gynecol. Surv. 69, 594–596 (2014).
Ledermann, J. et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 15, 852–861 (2014).
Ledermann, J. A. et al. Overall survival in patients with platinum-sensitive recurrent serous ovarian cancer receiving olaparib maintenance monotherapy: an updated analysis from a randomised, placebo-controlled, double-blind, phase 2 trial. Lancet Oncol. 17, 1579–1589 (2016).
Matulonis, U. A. et al. Olaparib maintenance therapy in patients with platinum-sensitive, relapsed serous ovarian cancer and a BRCA mutation: overall survival adjusted for postprogression poly(adenosine diphosphate ribose) polymerase inhibitor therapy. Cancer 122, 1844–1852 (2016).
Mirza, M. R. et al. Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. N. Engl. J. Med. 375, 2154–2164 (2016).
Pujade-Lauraine, E. et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 18, 1274–1284 (2017).
Lee, A. Fuzuloparib: first approval. Drugs 81, 1221–1226 (2021).
Ray-Coquard, I. et al. Olaparib plus bevacizumab as first-line maintenance in ovarian cancer. N. Engl. J. Med. 381, 2416–2428 (2019).
Wu, X. H. et al. Niraparib maintenance therapy in patients with platinum-sensitive recurrent ovarian cancer using an individualized starting dose (NORA): a randomized, double-blind, placebo-controlled phase III trial☆. Ann. Oncol. 32, 512–521 (2021).
Liu, J. et al. Olaparib maintenance monotherapy in Chinese patients with platinum-sensitive relapsed ovarian cancer: China cohort from the phase III SOLO2 trial. Asia. Pac. J. Clin. Oncol. 13753, https://doi.org/10.1111/ajco.13753 (2022).
Li, N. et al. An open-label, multicenter, single-arm, phase II study of fluzoparib in patients with germline BRCA1/2 mutation and platinum-sensitive recurrent ovarian cancer. Clin. Cancer Res. 27, 2452–2458 (2021).
Li, N. et al. Fuzuloparib maintenance therapy in patients with platinum-sensitive, recurrent ovarian carcinoma (FZOCUS-2): a multicenter, randomized, double-blind, placebo-controlled, phase III trial. J. Clin. Oncol. 10–12 https://doi.org/10.1200/jco.21.01511 (2022).
Wu, L. et al. Olaparib maintenance therapy in patients with newly diagnosed advanced ovarian cancer and a BRCA1 and/or BRCA2 mutation: SOLO1 China cohort. Gynecol. Oncol. 160, 175–181 (2021).
Monk, B. J. et al. A randomized, phase III trial to evaluate rucaparib monotherapy as maintenance treatment in patients with newly diagnosed ovarian cancer (ATHENA–MONO/GOG-3020/ENGOT-ov45). J. Clin. Oncol. 40, 3952–3964 (2022).
Takaya, H. et al. Intratumor heterogeneity and homologous recombination deficiency of high-grade serous ovarian cancer are associated with prognosis and molecular subtype and change in treatment course. Gynecol. Oncol. 156, 415–422 (2020).
Patel, J. N. et al. Characterisation of homologous recombination deficiency in paired primary and recurrent high-grade serous ovarian cancer. Br. J. Cancer 119, 1060–1066 (2018).
Vanderstichele, A., Busschaert, P., Olbrecht, S., Lambrechts, D. & Vergote, I. Genomic signatures as predictive biomarkers of homologous recombination deficiency in ovarian cancer. Eur. J. Cancer 86, 5–14 (2017).
Maxwell, K. N. et al. BRCA locus-specific loss of heterozygosity in germline BRCA1 and BRCA2 carriers. Nat. Commun. 8, 319 (2017).
Pellegrino, B. et al. Preclinical in vivo validation of the RAD51 test for identification of homologous recombination-deficient tumors and patient stratification. Cancer Res. 82, 1646–1657 (2022).
Fujiwara, K. et al. Olaparib plus bevacizumab as maintenance therapy in patients with newly diagnosed, advanced ovarian cancer: Japan subset from the Paola-1/engot-ov25 trial. J. Gynecol. Oncol. 32, e82 (2021).
Takaya, H., Nakai, H., Takamatsu, S., Mandai, M. & Matsumura, N. Homologous recombination deficiency status-based classification of high-grade serous ovarian carcinoma. Sci. Rep. 10, 1–8 (2020).
Lai, C. H. et al. Maintenance of pegylated liposomal doxorubicin/carboplatin in patients with advanced ovarian cancer: Randomized study of an Asian gynecologic oncology group. J. Gynecol. Oncol. 31, 1–14 (2020).
Sinha, S. et al. Higher prevalence of homologous recombination deficiency in tumors from African Americans versus European Americans. Nat. Cancer 1, 112–121 (2020).
Hsiao, Y. W. & Lu, T. P. Race-specific genetic profiles of homologous recombination deficiency in multiple cancers. J. Pers. Med. 11, 1287 (2021).
You, Y. et al. Germline and somatic BRCA1/2 mutations in 172 Chinese women with epithelial ovarian cancer. Front. Oncol. 10, 295 (2020).
Li, W. et al. Germline and somatic mutations of multi-gene panel in Chinese patients with epithelial ovarian cancer: a prospective cohort study. J. Ovarian Res. 12, 1–9 (2019).
Pennington, K. P. et al. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin. Cancer Res. 20, 764–775 (2014).
Norquist, B. M. et al. Inherited mutations in women with ovarian carcinoma. JAMA Oncol. 2, 482–490 (2016).
Bhaskaran, S. P. et al. Ethnic-specific BRCA1/2 variation within Asia population: evidence from over 78 000 cancer and 40 000 non-cancer cases of Indian, Chinese, Korean and Japanese populations. J. Med. Genet. 58, 752–759 (2021).
Meng, H. et al. BRCA1 c.5470_5477del, a founder mutation in Chinese Han breast cancer patients. Int. J. Cancer 146, 3044–3052 (2020).
Miller, R. E. et al. ESMO recommendations on predictive biomarker testing for homologous recombination deficiency and PARP inhibitor benefit in ovarian cancer. Ann. Oncol. 31, 1606–1622 (2020).
Vergote, I. et al. European experts consensus: BRCA/homologous recombination deficiency testing in first-line ovarian cancer. Ann. Oncol. 33, 276–287 (2022).
Lotan, T. L. et al. Homologous recombination deficiency (HRD) score in germline BRCA2- versus ATM-altered prostate cancer. Mod. Pathol. 34, 1185–1193 (2021).
Sokol, E. S. et al. Pan-cancer analysis of BRCA1 and BRCA2 genomic alterations and their association with genomic instability as measured by genome-wide loss of heterozygosity. JCO Precis. Oncol. 442–465. https://doi.org/10.1200/po.19.00345 (2020).
Bronder, D. et al. TP53 loss initiates chromosomal instability in fallopian tube epithelial cells. DMM Dis. Model. Mech. 14, dmm049001 (2021).
Shen, R. & Seshan, V. E. FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res. 44, 1–9 (2016).
Sztupinszki, Z. et al. Migrating the SNP array-based homologous recombination deficiency measures to next generation sequencing data of breast cancer. npj Breast Cancer 4, 8–11 (2018).
Abkevich, V. et al. Patterns of genomic loss of heterozygosity predict homologous recombination repair defects in epithelial ovarian cancer. Br. J. Cancer 107, 1776–1782 (2012).
Birkbak, N. J. et al. Telomeric allelic imbalance indicates defective DNA repair and sensitivity to DNA-damaging agents. Cancer Discov. 2, 366–375 (2012).
Popova, T. et al. Ploidy and large-scale genomic instability consistently identify basal-like breast carcinomas with BRCA1/2 inactivation. Cancer Res. 72, 5454–5462 (2012).
Richards, S., Aziz, N., Bale, S., Bick, D. & Das, S. ACMG standards and guidelines standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 17, 405–424 (2015).
Acknowledgements
This study was supported by a grant from CAMS Innovation Fund for Medical Sciences (CIFMS-2017-I2M-1–002) and grants from the National High Level Hospital Clinical Research Funding (No. 2022-PUMCH-A-117, 2022-PUMCH-B-083, 2022-PUMCH-C-010, 2022-PUMCH-C-022, and 2022-PUMCH-D-003) and the Le Fund (No. KH-2020-LJJ-004, 034, and 035). We thank K. Mojumdar for editorial assistance.
Author information
Authors and Affiliations
Contributions
X.J., Z. Liang, Y.J., R.Y., M.W., and H.L. conceived and designed the study; L.L., Y.G., M.Z., X.S., Y.D., K.J., L.W., X.J., Y.Z., Z. Liang, Y.J., R.Y., M.W., and H.L. contributed to sample collection and result interpretation, Z. Li, X.X., T.S., and Y.D. performed data analysis; C.X., X.Z., and R.L. performed experiments; L.L., Z. Li, X.X., T.S., and H.L. wrote the manuscript with inputs from other authors; and L.L., Y.G., M.Z., X.S., and Z. Li are co-first authors.
Corresponding authors
Ethics declarations
Competing interests
Z. Li, X.X., T.S., Y.D., C.X., R.L., K.J., X.J., and Y.Z. are full-time employees, and H.L. is a shareholder and scientific advisor of Precision Scientific Ltd. The remaining authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, L., Gu, Y., Zhang, M. et al. HRD effects on first-line adjuvant chemotherapy and PARPi maintenance therapy in Chinese ovarian cancer patients. npj Precis. Onc. 7, 51 (2023). https://doi.org/10.1038/s41698-023-00402-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41698-023-00402-y
