
Abstract
Recent studies show that rare, deleterious variants (RDVs) in certain genes are critical determinants of heritable cancer risk. To more comprehensively understand RDVs, we performed the largest-to-date germline variant calling analysis in a case-control setting for a multi-cancer association study from whole-exome sequencing data of 20,789 participants, split into discovery and validation cohorts. We confirm and extend known associations between cancer risk and germline RDVs in specific gene-sets, including DNA repair (OR = 1.50; p-value = 8.30e-07; 95% CI: 1.28–1.77), cancer predisposition (OR = 1.51; p-value = 4.58e-08; 95% CI: 1.30–1.75), and somatic cancer drivers (OR = 1.46; p-value = 4.04e-06; 95% CI: 1.24–1.72). Furthermore, personal RDV load in these gene-sets associated with increased risk, younger age of onset, increased M1 macrophages in tumor and, increased tumor mutational burden in specific cancers. Our findings can be used towards identifying high-risk individuals, who can then benefit from increased surveillance, earlier screening, and treatments that exploit their tumor characteristics, improving prognosis.
Similar content being viewed by others


Introduction
Inherited genetic variants play an important role in cancer susceptibility. These variants are associated with disease risk in a spectrum, from common variants that tend to have weak effects, to rare variants (<0.5% minor allele frequency, MAF) with often large effects1,2. In fact, some studies suggest that greater than 95% of variants predicted to be functionally important are rare3,4. In addition to their high penetrance, rare variants are also abundant3. Studies in cancer prone families were the first to identify rare, deleterious variants (RDVs) with statistically significantly elevated cancer risk. Well-known examples include BRCA1/2 in inherited breast and ovarian cancer syndrome5, DNA mismatch repair genes in Lynch syndrome6, TP53 in Li-Fraumeni syndrome7, and APC in familial adenomatous polyposis8. Genetic screening for these inherited cancer syndromes has constituted one of the first applications of genomics in precision medicine, as it allows tailored cancer screening, prevention, and in certain cases, therapies5,6,7,8. While the vast majority of genes identified this way and currently screened for in the cancer clinic are in DNA damage repair (DDR) genes, RDVs in genes for which cancer risks are less well-characterized present a challenge in the clinic9.
While genome-wide association studies (GWAS) have identified multiple susceptibility loci as determinants of increased cancer risk in relatively large cohorts, these studies examine associations with common variants only, which typically have modest effects that explain only a small fraction of heritability. RDVs can have larger effect sizes than common variants, yet large cohorts are needed to obtain statistical power for conclusive analyses on their cancer risk, which are often prohibitive. More recently, next generation sequencing studies by large consortia have produced and aggregated data from thousands of germlines and matched tumors. These studies have revealed many germline risk variants10,11,12,13,14, and provide a rich resource for investigating the association of RDVs with cancer risk11,12,15,16. Our team has previously utilized such resources in focused studies on lung cancer, which revealed that RDVs in ATM increase risk for lung adenocarcinomas16 and in Fanconi Anemia (FA) genes for lung squamous cell carcinomas15. In these studies, we addressed the issue of low power observed in single, recurrent RDV studies by conducting case-control analyses for RDVs that may affect risk of cancer cumulatively as part of a gene or gene group. We reason that a similar approach focused on RDVs will provide novel insights into risk across cancers, as it is unlikely to miss weaker associations. To better characterize the roles of RDVs in cancer risk and other cancer-related outcomes, it is paramount to comprehensively assess RDVs in such cancer germline datasets, including analysis of the effects of RDVs aggregated per each individual. This will enable the development of new predictive tools and precision preventive strategies for the clinic.
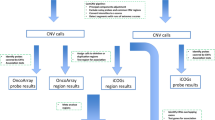
Towards this end, we have performed the largest-to-date germline sequencing data analysis in a cancer case-control setting, aggregating existing whole-exome sequencing (WES) datasets on 20,789 participants, split into discovery and validation cohorts. In discovery analysis, we processed germline WES datasets on 15,709 participants (13,018 total post-Quality Control, with 6371 cases and 6647 controls) where the cases span 24 different cancers from The Cancer Genome Atlas, TCGA17. We then replicated our discovery cohort findings in an independent validation cohort, by using WES data from 7771 participants (1571 cases, 6200 controls) from the Icahn School of Medicine at Mount Sinai (ISMMS) BioMe Biobank. In these studies, as the low frequencies of RDVs make genome-wide discovery difficult without large cohorts, we investigated their roles by collapsing RDVs at gene and gene-set levels for cohort-level RDV burden (RDVs in cases vs. controls). Furthermore, unlike previous studies that compared RDVs in cases only to population-level databases for controls, our variant calling in a case-control setting enabled us to identify the number of RDVs, or RDV load for each control as well as case participant, and thereby to study the association of personal RDV load with cancer risk (>1 RDV vs. 1 RDV vs. 0 RDV). In Fig. 1, we graphically represent the RDV burden and RDV load concepts. Finally, we studied the association of personal RDV load in cancer individuals with their age of disease onset, tumor mutation burden and cellular composition of tumor immune microenvironment.

Flowchart of the study pipeline to identify rare deleterious variants (RDVs) and to perform RDV burden and RDV load analyses.
RDV burden analysis results revealed that individuals with RDVs in specific cancer genes and gene-sets are at increased risk for cancers. While TCGA germline RDVs have been categorized previously12, this study reports result where germline variants are processed in a case-control setting, which enabled us to study associations between personal RDV load and cancer risk. Importantly, this study also replicates germline discovery case-control cohort findings in an independent case-control validation cohort.
Overall, our multi-scale analysis results first confirm known associations in TCGA12 and then provide novel observations on the associations of germline RDVs in specific gene-sets with cancer risk. Furthermore, we show that having a germline RDV load in certain gene-sets (i.e. RDVs in more than one gene in the same pathway) is a potential biomarker for younger age of disease onset, tumor mutational burden (TMB), and characteristics of the tumor immune microenvironment (TME) in a dose-dependent manner (i.e. >1 RDV vs. 1 RDV vs. 0 RDV).
Results
Germline variant calling in a case-control setting identified sites of rare, deleterious variants (RDVs) and their genotypes across the cohort
In the discovery cohort, to enable case-control analyses (Fig. 1) while avoiding biases potentially introduced by different calling algorithms, we first realigned and called variants in the germline WES data in a case-control setting. These included 8321 TCGA cases (Supplementary Table 1), and 7388 controls from dbGaP (Supplementary Table 2), for a total of 15,709 participants. To reduce confounding due to population stratification, we focused on those participants in the discovery cohort who comprised the largest group by ancestry, which clustered via principal component analysis (PCA) with individuals of known European ancestry (Fig. 2). After sample and variant QC (see methods), we observed 941,609 variants (Supplementary Table 3) in 17,507 genes across the autosomes and X chromosomes of 13,018 participants (6371 cases and 6647 controls) (clinical characteristics in Table 1). We note that recently an independent multi-cancer analysis of TCGA cases12 (‘case-only analysis’) called each case sample separately (rather than calling variants across all samples in a case-control setting) and used the union of several calling software packages for variant identification. Unlike that case-only approach, our calling approach in a case-control setting can distinguish between instances where a variant site is wild type and where it does not have enough sequence coverage to make a call.

PCA based on common SNPs (MAF ≥ 0.05) showing the top two principal components of (a) the study cohort together with 1000 Genomes and The Ashkenazi Genome Consortium (TAGC) samples and of (b) the gated samples from the study cohort with European ancestry (6371 cases and 6647 controls).
To compare these two approaches in more detail, we focused on data from 6275 TCGA participants that were analyzed by both methods. From our case-control analysis, after QC we identified 588,287 variants for which at least one alternate allele was called. Of these, 460,373 (78%) were also called by the case-only analysis, while 22% were unique to our calling. Conversely, the case-only analysis call set, which used several genotype callers and took the union of calls, identified 1,681,769 variants, of which 27% were also called by our approach (Supplementary Fig. 1). To estimate the fraction of variants unique to each of these call sets that represent true variant sites versus spurious false positives, we used the imputation panel from the TOPMed consortium18 as an external reference. This panel was generated from a large set of participants of diverse ancestry for whom whole genome sequencing (WGS) data are available. Importantly, TCGA participants were not included in this panel. As this panel has been shown to enable imputation of variants at low MAF (0.01%)19, we reasoned that it would allow us to assess the quality of many of the low-MAF calls in the call sets. Of the 460,373 germline variants that were called in both the case-control and the case-only analyses, 67% were on the TOPMed panel (Supplementary Table 4). Most importantly, at the variant level, a greater fraction of the alternate alleles at sites found uniquely in our case-control data were concordant with TOPMed compared to the case-only approach (Supplementary Table 5).
Based on the comparison results between the two calling approaches, we proceeded with our case-control analysis approach to identify germline RDVs that associate with cancer risk relative to controls. Overall, we identified 7241 RDVs (MAF < 2% in cases or controls, MAF ≤ 1% in ExAC non-TCGA Non-Finnish European population and MAF ≤ 1% gnomAD Ashkenazi Jewish population) across 1787 genes in the discovery cohort. Similarly, focusing on 1571 cases and 6200 control participants of European ancestry in the validation cohort, we identified 5766 RDVs in 1814 genes.
Gene-set level burden analysis revealed that RDVs in cancer predisposition, DNA damage repair, Fanconi Anemia and somatic cancer driver gene-sets are associated with cancer risk
Though we performed tests at the levels of individual genes, we did not find any significant (p ≤ 0.05), replicable associations beyond the well-known association of BRCA1 (discovery cohort: OR = 2.91; p-value = 1.14e-04; 95% CI: 1.67–5.32, validation cohort: OR = 2.91; p-value = 1.06e-03; 95% CI: 1.55–5.35) and BRCA2 (discovery cohort: OR = 3.04; p-value = 9.35e-04; 95% CI: 1.55–6.46, validation cohort: OR = 2.50; p-value = 1.04e-03; 95% CI: 1.46–4.18) with breast and ovarian cancer risk (Supplementary Data 1). Instead, we hypothesized that collapsing RDVs at the gene-set level would be more powerful and enable better understanding of risk-associated biological processes. Therefore, we compared the RDV burden (Table 2, Fig. 3) in a priori defined gene groups (Supplementary Table 6). We first tested the set of 94 genes in the TruSight Cancer Gene panel (Illumina https://www.illumina.com/products/by-type/clinical-research-products/trusight-cancer.html), which is often used in genetic testing clinics, and observed statistically significant RDV burden (p ≤ 0.05) across cancers in the discovery cohort (OR = 1.51; p-value = 4.58e-08; 95% CI: 1.30–1.75), which replicated in the validation cohort (OR = 1.24; p-value = 0.04; 95% CI: 1.01–1.51). Similarly, when we focused on 95 DNA repair genes involved in known functional DDR pathways20, which included those known to associate with autosomal dominant cancer predisposition (CPD) syndromes (20 genes)21 (Supplementary Table 6), we observed a statistically significant enrichment of RDVs in cases compared to controls in both discovery (OR = 1.50; p-value = 8.30e-07; 95% CI: 1.28–1.77) and validation cohorts (OR = 1.59; p-value = 1.17e-04; 95% CI: 1.26–2.00). Next, we hypothesized that germline mutations in genes with known somatic mutations may increase cancer risk, and tested the germline RDV burden on 299 known somatic cancer driver (SCD) genes22. We again observed a statistically significant burden of RDVs across cancers in cases vs. controls in the discovery cohort (OR = 1.46; p-value = 4.04e-06; 95% CI: 1.24–1.72), which replicated in the validation cohort (OR = 1.72; p-value = 2.00e-06; 95% CI: 1.38–2.14).

The whiskers span the 95% confidence interval for OR values (penalized logistic regression). The black circle outline indicates significant burden p ≤ 0.05.
Based on our earlier findings15 that RDVs in Fanconi Anemia (FA) genes increased risk for squamous lung cancer (LUSC), we also investigated the burden of FA (subset of 22 DDR genes) RDVs in other tissues and multi-cancer. Consistent with our LUSC findings, we observed an increased RDV burden cross-cancers in cases vs. controls in both discovery (OR = 2.05; p-value = 6.14e-08; 95% CI: 1.58–2.70) and validation (OR = 2.01; p-value = 1.07e-04; 95% CI: 1.42–2.80) cohorts. In the discovery cohort, this signal was significantly driven by cancers of the breast, bladder, stomach and ovary (Supplementary Data 2). Furthermore, the 9 FA core complex genes and 11 FA genes involved in DNA repair both had a statistically significant signal cross-cancers in the discovery cohort (OR = 1.93; p-value = 0.02; 95% CI: 1.10–3.49 and OR = 2.12; p-value = 2.15e-06; 95% CI: 1.54–2.93, respectively). The finding on 11 FA genes involved in DNA repair further replicated in the validation cohort (OR = 2.48; p-value = 3.14e-06; 95% CI: 1.71–3.56), while on 9 FA core complex genes trended in the expected direction (OR = 1.18; p-value = 0.74; 95% CI: 0.41–2.88). Next, to make sure the signals we observed in these gene-sets (CPD, DDR, SCD and FA) were not solely driven by BRCA1/2, as a sensitivity analysis we removed BRCA1/2 and repeated the burden analyses, and observed that these gene-sets were still significant (Supplemental Table 7, Supplementary Fig. 2). Furthermore, our analysis of the MSigDB gene-sets (below) supported our findings on the association of the FA gene-set with cancer risk. Note that while we further tested for the confounding effects of gender for all gene-sets multi-cancer, we did not observe a noticeable difference in the OR results for any gene-set (Supplementary Fig. 3). We provide the gender distribution in Supplementary Table 8.
Finally, we performed a similar gene-set level burden analysis (Supplementary Data 3) for sets of cancers grouped by histological or anatomical relation, including pan-gastrointestinal23, pan-kidney24, pan-gynecological25, and pan-squamous26. Pan-gastrointestinal and pan-gynecological cohorts were associated with significant (p ≤ 0.05) cancer risk with RDVs in all four gene-sets (CPD, DDR, SCD and FA). Furthermore, in pan-kidney and pan-squamous analyses, we observed significant burden of RDVs in cases compared to controls in CPD and FA gene-sets, respectively (Supplementary Data 3).
Data-driven analysis of 17,810 gene-sets in MSigDB again identified RDV burden in Fanconi Anemia genes associated with increased cancer risk
Next, to ensure we did not miss any additional gene-sets with significant RDV burden in cases vs. controls, we performed a data-driven exome-wide gene-set analysis. For this purpose, we tested all gene-sets (17,810) contained in the Molecular Signatures Database (MsigDB)27 (Supplementary Data 4). Consistent with our observations on pre-selected gene-sets, the strongest signal was in Fanconi Anemia pathway genes from REACTOME (p = 1.4e-11; OR = 2.54; 95% CI = 1.92–3.40) in the discovery cohort, which was further replicated in the validation cohort (p = 1.2e-05; OR = 2.21; 95% CI = 1.56–3.09). Other cancer-related pathways, for the majority including the strongest DDR genes, were found statistically significant after adjusting for multiple comparisons. Of note in the discovery cohort, we observed significant risk association for RDV burden in the set of genes targeted by the eukaryotic translation initiation factors EIF4EBP1 and EIF4EBP2 (OR = 1.43; p-value = 4.4e-05; 95% CI: 1.20–1.69). While we did not observe significant association in the validation cohort, we still observed higher frequency with the same direction of effect in cases with RDVs compared to controls (OR = 1.14; p-value = 0.39; 95% CI: 0.85–1.51).
Germline RDV load in key cancer genes is a potential marker for increased cancer risk
RDVs in cancer are usually considered in a binary context: a patient either has an RDV in a gene of interest or does not (similar to our RDV burden analysis). Given that even when the penetrance of RDVs in cancer risk is high, it is not absolute (i.e., some individuals with RDVs in known cancer predisposition genes will never develop cancer), we hypothesized that the accumulation of RDVs within a set of related genes in an individual could increase their cancer risk. We refer to this concept as personal “germline RDV load”. We hypothesized that an increased personal germline RDV load in particular gene-sets is associated with an increased risk of cancer. The premise of this hypothesis is that the RDVs damage, but do not fully destroy a particular pathway; therefore, additional RDVs in the same pathway add to the damage and increase cancer risk further. To test this hypothesis, we evaluated the associations between the participant-level RDV load within gene-sets (i.e., CPD, DDR, SCD and FA) (Supplementary Table 6) and cancer risk (Fig. 4 and Supplementary Table 9). For each gene-set, we divided the participants into three participant groups: (i) participants with no RDVs; (ii) participants where RDVs occurred in only one gene; and (iii) participants with RDVs in two or more genes (Supplementary Table 9), recognizing that the small number of participants with RDVs in >1 gene makes precise estimation of the effect of RDV load difficult. Consistent with our hypothesis on RDV load, for each studied gene-set, we observed a greater association with cancer risk for participants that had higher number of genes with germline RDVs, as shown in Fig. 4 and Supplementary Table 9 (except for FA due to the limited number of genes in this gene-set). Notably, all our RDV load findings replicated in the validation cohort. Note that in SCD and FA gene-sets, we did not observe significant cancer risk association (p-value ≤ 0.05) with individuals with RDVs in more than one gene.

a Discovery cohort; b Validation cohort. The whiskers span the 95% confidence interval for OR values (penalized logistic regression). The black circle outline indicates significant burden p ≤ 0.05.
We further hypothesized that a higher RDV load in these gene-sets is associated with a younger age of diagnosis. To test this hypothesis, we used the detailed clinical information available on the discovery cohort of TCGA participants. We evaluated the associations between each participant’s germline RDV load with their age at cancer diagnosis, and observed that those participants with a germline RDV indeed exhibited a statistically significant early age of diagnosis than those individuals without RDVs (Fig. 5) in CPD, DDR and FA gene-sets. Additionally, we observed that the individuals with high RDV load (>1 RDV) in CPD and SCD gene-sets had significantly younger age of diagnosis than those individuals without RDV.

a Cancer predisposition genes b DNA damage repair genes c Somatic cancer driver genes d Fanconi Anemia genes. p-values are calculated based on Mann–Whitney U-test. Boxplot elements: center line indicates median; box limits represent lower (25th percentile) and upper (75th percentile) quartiles; whiskers extend to 1.5 times the interquartile range.
Germline RDV load is associated with altered tumor immune microenvironment and increased tumor mutation burden
Next, in light of recent findings on the impact of germline variants on tumor immune microenvironment (TME) in various cancers28,29,30, we tested the association of increased germline RDV load in CPD, DDR, SCD and FA gene-sets with TME immune cell fractions. Briefly, to avoid any potential biases from B-cell immune signatures in blood cancers, we focused on 6277 participants with solid tumors in TCGA by excluding 94 TCGA participants with hematological malignancies. We then utilized previously reported annotations on the 22 infiltrating immune cell types on solid tumors of TCGA participants31, based on analysis by the CIBERSORT tool that used their tumor RNA-sequencing data32. Next, for each participant, we tested the association of tumor immune cell fraction with their germline RDV load in the CPD, DDR, SCD and FA gene-sets (Supplementary Fig. 4, Supplementary Data 5). Remarkably, the germline RDV load in CPD, DDR or FA genes exhibited the strongest association with increased levels of M1 macrophages in the TME. Specifically, we observed that participants with a germline RDV in DDR or FA gene-sets developed tumors with a statistically significantly higher fraction of M1 macrophages (Fig. 6), compared to participants with no RDV. Additionally, we observed that the individuals with high RDV load (>1 RDV) in CPD and DDR gene-sets had higher fraction of M1 macrophages than those individuals without RDV although it was not statistically significant. This signal was mostly driven by the increased levels of the chemokine ligands CXCL10/11. We provide the complete set of results in Supplementary Data 6. We next asked whether increased M1 macrophages in TME of participants with solid tumors associated with survival (Supplementary Table 10). We observed that those participants with increased M1 macrophages exhibited worse survival (p ≤ 0.05 without correction for multiple testing) in brain lower grade glioma, kidney renal clear cell carcinoma and kidney renal papillary cell carcinoma.

a Cancer predisposition genes b DNA damage repair genes c Somatic cancer driver genes d Fanconi Anemia genes. The marker in the violin plot indicates mean.
Finally, since tumor mutation burden (TMB) is currently being investigated as a biomarker for durable response to life-extending cancer immunotherapies33, we also tested the association of germline RDV load in CPD, DDR, SCD and FA gene-sets with TMB for each cancer type with solid tumors (Supplementary Data 7). We observed the most significant TMB association with germline RDV load in FA genes in breast cancer patients (p = 0.00052). Overall, germline RDVs associated with TMB in: (i) DDR, CPD and SCD genes in colon cancer; (ii) DDR genes in kidney clear cell and cervical cancers; (iii) CPD genes in kidney papillary cell cancer; (iv) SCD genes in stomach and ovarian cancers; and (v) FA genes in breast, cervical, ovarian and stomach cancers. This signal for FA genes was mostly driven by BRCA1/2; when we removed BRCA1/2 and reperformed the analysis, we still observed associations of germline RDV load in FA genes and TMB for stomach cancer. Details are in Supplementary Data 7.
Discussion
Here, we have performed the largest-to-date case-control WES study to identify germline rare, deleterious variants (RDVs) in specific genes and gene-sets that associate with multi-cancer risk and to further investigate whether this risk increases with the number of RDVs an individual has. The study findings were made possible by performing variant calling analysis in a case-control setting by including thousands of controls, which allowed the examination of combinations of variants for individual control participants. We then used an independent pan-cancer cohort from the ISMMS BioMe Biobank for validation. To examine the cumulative effects of RDVs in functionally related gene groups, we used a gene-set based variant collapsing approach. Specifically, we focused both on a few cancer-associated gene groups, as well as in a function-agnostic manner on all 17,810 gene-sets in MSigDB27. For cancer-associated gene groups, mindful of the fact that many risk variants have been shown to exhibit tissue specificity34, we tested RDVs in cancer predisposition (CPD) and DNA damage repair (DDR) genes, which we hypothesized would pleiotropically associate with risk across cancer sites, due to the accumulation of mutations that fail to be properly repaired35. In addition, we tested whether germline RDVs in genes with known somatic mutations (somatic cancer drivers, SCDs) increase cancer risk22. Finally, based on our earlier findings15 that RDVs in Fanconi Anemia (FA) genes are associated with lung squamous cancer risk, we tested RDVs in the FA genes. Remarkably, we observed increased cancer risk associated with RDVs in all four tested gene-sets (CPD, DDR, SCD, and FA) in both discovery and validation cohorts. Furthermore, our analysis of the MSigDB gene-sets supported our findings on the association of the FA gene-set with cancer risk.
Our results demonstrate the value of variant calling in a case-control setting in germline risk variant discovery. Notably, unlike prior works12 that have simply compared germline variant frequencies in TCGA cancer individuals to those in databases such as ExAC36 non-TCGA or gnomAD37 as controls, we performed germline variant calling in a case-control setting. Doing so enabled us to identify control participants with more than one RDV in a given gene or gene-set and to examine the role of personal RDV load in cancer risk (see Fig. 1). This is a significant finding, which prior works12 were unable to examine. While multiple studies have focused on the somatic tumor mutation burden (TMB), our study highlights the critical importance of germline RDV load in key cancer gene-sets. The association of increased RDV load in CPD, DDR, FA and SCD gene-sets with increased personalized cancer risk in both discovery and validation cohorts have important implications for our understanding of how germline genetic factors govern cancer risk, and can impact the clinical management of cancer patients with one or more germline RDVs, and their families.
We further observed that personal RDV load in specific gene-sets associated with age of disease onset, tumor immune microenvironment (TME) and TMB in TCGA matching tumor data. That individuals who have RDVs in CPD, DDR or FA genes have statistically significantly earlier age of diagnosis than those without warrants research in whether these individuals should start screening and surveillance efforts at younger ages than the currently recommended guidelines for the general public.
TME and TMB are two tumor characteristics important in immunotherapy response and prognosis. Recent studies show that germline variants can shape certain immune features within the TME of solid tumors28,29,30. Consistent with and complementary to these observations, our results show that the personal germline RDV load of a cancer patient is associated with their TME (see Supplementary Fig. 4, Supplementary Data 5). Interestingly, the germline RDV load in CPD, DDR or FA genes exhibited the strongest association with increased levels of M1 macrophages in the TME (Fig. 6). Unlike T-cells which may or may not be in the TME38, macrophages are often present in tumors as the most dominant cells, including tissue resident cells and infiltrating cells, with relative proportions varying in different patients. However, the potential impact of TME macrophage elevation is currently somewhat controversial, as the historical dichotomy between good (M1) vs. bad (M2) subsets is being challenged by more granular data on macrophage subsets and lineage based on single-cell transcriptomics39. While increased levels of M2 macrophages in tumors generally associate with poor survival40,41, the levels of M1 macrophages have so far not been associated with poor outcome. M1 macrophages have been shown to likely contribute to a more immunogenic tumor environment by providing activating signals, including Th1 polarizing cytokines or chemokines42. They also are likely to active via their phagocytic function, either directly through eating up tumor cells and contributing to antigen presentation and priming of T cells as well, or via antibody dependent cell-mediated cytotoxicity43. As TME can impact response to standard-of-care therapies44, including chemotherapy, radiation and angiogenic inhibitors, and since clinical trials that examine the combinations of these treatments are currently on-going, our results warrant further investigations into identifying specific macrophage markers that may correlate with immunotherapy treatment efficacy and prognosis of individuals with germline RDV load in CPD, DDR or FA genes in solid tumors.
We further observed statistically significant association of RDV load with TMB in different gene-sets for different cancer types (see Supplementary Data 7). Since TMB has been suggested as a biomarker for durable response to cancer immunotherapies33,45, these results further support our findings on the potential importance of RDV load in specific gene-sets in shaping tumor immune characteristics (Supplementary Data 5). For example, in colon cancer RDV load in most gene-sets we tested associated with increased TMB.
Finally, we asked whether RDV load is associated with survival. While we had limited statistical power to answer this question directly, we did observe that increased M1 macrophages (which correlated with RDV load) associated with worse survival (p ≤ 0.05 without correction for multiple testing, Supplementary Table 10) in specific cancers (brain lower grade glioma, kidney renal clear cell carcinoma and kidney renal papillary cell carcinoma). The functionality of macrophages in these histologies compared to other tumor types in relation to antigen presentation capacity or inflammatory potential remains to be further explored. Future studies in larger, independent cohorts are needed for validation of this finding and will help better understand the nature of interactions between germline RDV load, tumor characteristics and survival.
This study should be considered in the context of its limitations. First, the multi-cancer analysis needs to consider the overrepresentation of rarer cancers in the TCGA data. Thus, strong association signals specific to these less common cancers in the TCGA data may be weaker when studied in a cohort reflective of the population incidences of cancers of various sites. Similarly, inclusion of some cancer types with small sample size increased the heterogeneity of our study, risking a diluted signal for associations with more common cancers. Second, while focusing on variants annotated as pathogenic in ClinVar46 was necessary to ensure the clinical reliability of our results, it also restricts our analysis to those genes previously known to have a clinical impact. There are variants, such as rs11571833 (p.Lys3326Ter) in BRCA2, for which research studies strongly support a role in cancer risk15,47 but not annotated as pathogenic in ClinVar. Alternative approaches to identifying pathogenic variants will be needed to address these issues. Third, while we had well-annotated gender data available to study its potential confounding effects, information on other potential confounders such as age and smoking was not available for all controls in the dbGaP studies. Thus, we were unable to investigate their potential confounding effects and cannot distinguish direct genetic effects on cancer risk versus genetic effects on risk factors for cancer. Fourth, our results do not explore the interplay between specific RDVs, RDV load, and environmental and clinical exposures. Future efforts that link genetic information with epidemiological exposure and clinical information from electronic health records (EHR) (e.g. blood measurements; smoking and alcohol history; viral infections, etc.) will be needed to understand such interactions. This work also did not consider the role of common genetic polymorphisms in cancer risk, such as that captured by polygenic risk scores48,49. Future efforts towards understanding if and how the penetrance of RDV load (and visa versa) will be impacted with polygenic background could be quite informative50,51. We did not include cancer types for which data are currently available in TCGA, but were not available when we first began this project. Finally, the statistical power of our validation was somewhat limited. As data from larger population- or hospital-based BioBanks (e.g. UKBiobank) become available, larger studies to interrogate the role of RDV load in multi-cancer and tissue-specific cancer risk will become possible.
Methods
Data sources
For discovery, we used case data from TCGA (Supplementary Table 1) and control data from twelve population-based studies (Supplementary Table 2) in the database of Genotypes and Phenotypes (dbGaP) (http://www.ncbi.nlm.nih.gov/gap, RRID:SCR_002709). Briefly, we downloaded TCGA germline WES bam files from National Cancer Institute Cancer Genomics Hub (cgHub), a predecessor to the Genomic Data Commons which is no longer online. We extracted control fastq files from the NCBI Short Read Archive (SRA) for dbGaP studies listed in Supplementary Table 2. For replication, we used the exome calls from BioMe Biobank52 of Icahn School of Medicine at Mount Sinai (ISMMS). The relevant data access committees at NIH under project #8668 approved TCGA and dbGaP data usage. The usage of BioME data in our study (HS# 19-01088) was approved by the Program for the Protection of Human Subjects (PPHS) office at ISMMS as exempt human research (IRB-19-02546).
Study cohorts
For the discovery cohort, we realigned and called germline variants in a case-control cohort setting for 8321 TCGA cases and 7388 dbGaP controls. Cases included participants with 24 different cancer types of different sample sizes, as listed in Supplementary Table 1. For the control samples, when the underlying dbGaP study was a case-control study focused on a disease, we only included control individuals who did not have that disease in question.
For the validation cohort, we utilized available datasets from ISMMS BioMe52, which is an electronic health record (EHR) linked biobank. BioMe encompasses a wide array of phenotypic and genetic data from the diverse population of ISMMS patients with a vast spectrum of medical disorders. WES data (Illumina v4 HiSeq 2500) already exist for 30,813 BioMe participants52. Focusing on BioMe participants with existing WES data, we identified cancer patients and healthy controls based on their International Classification of Diseases (ICD)-9 and ICD10 codes53, which led to 1,571 cases and 6,200 controls of European ancestry (filtering details below). The clinical characteristics of the cohorts are listed in Table 1. The study design is provided in Fig. 1.
Discovery cohort variant discovery
We first realigned and called germline variants from WES datasets in a case-control setting using GVCF-based best practices for the genome analysis toolkit (GATK, https://www.broadinstitute.org/gatk/, RRID:SCR_001876) as implemented in a custom pipeline at the Icahn School of Medicine at Mount Sinai (ISMMS)54 and previously15,55,56. Briefly, we independently aligned all samples to human genome build GRCh37 with BWA57 (RRID:SCR_010910), performed indel realignment, duplicate marking and base quality score recalibration using GATK and Picard (RRID:SCR_006525), and finally called to a GVCF file with HaplotypeCaller. The case-control germline variant calling step consisted of calling variants from GVCF files and variant quality score calibration with GATK, where we only included samples for which over 75% of the exome was callable (depth ≥ 20, mapping quality ≥ 10, base quality ≥ 20) and for which there was no evidence of contamination (VerifyBamID < 3%).
Discovery cohort sample QC
We first removed samples with 15% or more missing genotype data. To filter samples that are duplicates or from first or second degree relative pairs, we performed relatedness analysis with KING software58 (RRID:SCR_009251) and then removed a sample from each such pair that had the highest fraction of missing data. To remove any bias that may arise due to systematic ancestry-based variations in allele frequency differences between cases and controls (i.e. population stratification) we used Principal Component Analysis (PCA). Briefly, we first removed indels and rare variants (defined by <5% of minor allele frequency, MAF), using 1000 Genomes dataset59 (RRID:SCR_008801) and The Ashkenazi Genome Consortium (TAGC, https://ashkenazigenome.org) as reference. For the remaining variants, we performed linkage disequilibrium (LD) pruning, filtered for a call rate of at least 0.99, and performed PCA with smartpca using EIGENSOFT 5.0.1 software (RRID:SCR_004965). We filtered for the least ancestry-based variation by focusing our downstream analyses on the largest set of case-control individuals clustered within the PCA plot by examining the PCA plot and selecting thresholds on PC1 and PC2 to corresponded to individuals of European ancestry (EA); analogous to flow cytometry we call this approach “gating”. The PCA plots along with the gated region are shown in Fig. 2. To adjust for population-level differences, we used the first two principal components from PCA of the gated individuals as covariates in the burden analyses. After sample QC, 6371 cases and 6647 controls remained.
Discovery cohort variant-level QC
For participants that passed PCA gating, we focused on ensuring high-quality genotype/variant calls for analysis. For this purpose, we filtered for variants with: read genotype quality ≥20; read depth ≥10; allelic depth of alternate allele ≥4; sites with: quality score ≥50; quality by depth score ≥2; mapping quality ≥40; read position rank sum > –3; mapping quality rank sum > –10 and variant tranche <99%. For heterozygous genotypes, we filtered for alternative allele ratio between 0.30 and 0.70. To reduce any differences between samples in cases and controls, we kept sites that have differential missing variant fraction ≤ 0.05 between the cases and controls. Finally, we kept sites with ≥88% of data (in both cases and controls).
Validation cohort participant selection
Next, to replicate the discovery cohort findings within an ancestry-matched validation cohort, we focused on those of EA in BioMe. Briefly, we used the PCA performed on the common variants60 and gated for EA individuals based on the first two principal components which captured the majority of the variance, resulting in 10,784 BioMe participants of (Supplementary Fig. 5). Next, we also ensured we used data from unrelated participants up to second degree. Finally, we identified participants with cancer based on their ICD9 and ICD10 codes available within BioMe52. Specifically, to avoid any false positives, we filtered for participants who had ICD9/10 codes related to cancer at least twice in their diagnosis files on separate dates. To avoid any conflicts in categorizations, we also removed participants with the following diagnoses (or diagnoses of similar nature): benign neoplasms, neoplasm of uncertain behavior and genetic susceptibility to malignant neoplasm. The complete list of ICD9/10 codes used for classification and elimination of cancer diagnosis are listed in Supplementary Data 8. We considered all other participants who were unaccounted for in the above categories as controls. These led to 1571 cases and 6200 control participants of EA in the BioMe cohort.
Validation cohort data generation and variant QC
BioMe WES data generation and QC steps have been discussed in detail previously52. Briefly, we filtered out sites with missingness >0.02 and biallelic sites with allele balance (<0.3 or >0.8). Additionally, to be consistent with the discovery cohort variant QC, we filtered for variants with: read genotype quality ≥20; read depth ≥10; allelic depth of alternate allele ≥4; sites with: quality score ≥50; quality by depth score ≥2; mapping quality ≥40; read position rank sum > –3; mapping quality rank sum > –10; differential missing variant fraction ≤ 0.05 between the cases and controls and site missingness <12% (in both cases and controls).
Variant filtering (both cohorts)
After sample and variant QC, we focused on rare, deleterious variants (RDVs) with known pathogenicity. To filter out common polymorphisms, we removed any variant present in both case and control cohorts at MAF > 2% or in Exome Aggregation Consortium (ExAC)36 non-TCGA Non-Finnish European population at MAF > 1% or in Genome Aggregation Database (gnomAD)37 Ashkenazi Jewish population at MAF > 1%. We considered variants that pass these filters to be rare. We then filtered the remaining variants for functional impact based on those present in the ClinVar database46 (RRID:SCR_006169) using the Annovar tool (http://annovar.openbioinformatics.org, RRID:SCR_012821). We considered a variant to be deleterious if: (i) it is listed as pathogenic/likely pathogenic in ClinVar; or (ii) it is a frameshift or stopgain variant located 5′ of a variant described to be a pathogenic LOF variant in ClinVar (nonsense and frameshift). We also performed a sensitivity analysis for our MAF cutoff by removing any variant present in both case and control cohorts at MAF > 1% or in ExAC non-TCGA Non-Finnish European population at MAF > 1% or in gnomAD Ashkenazi Jewish population at MAF > 1% and did not observe a difference in the top genes or gene-set level burden analyses (data not shown).
Statistical analysis
Background variation correction
To test for possible background variation between cases and controls, we calculated the tally of rare autosomal synonymous variants per each study participant. We defined synonymous variants as rare at Exac MAF ≤ 0.005% and cohort MAF ≤ 0.05%. Supplementary Fig. 6 provides the distribution and background variation statistics of genes with rare synonymous variants between the cases and controls in both cohorts. We accounted for differences in background variation by using the number of genes with rare synonymous variants of each individual as a covariate during the burden analyses.
Gene level RDV burden analyses
Next, to evaluate the cumulative effects of multiple RDVs in each gene, and thereby increase the statistical power to identify cancer risk genes, we performed gene level RDV burden tests. Briefly, to accommodate for data sparsity, we performed aggregate RDV burden for each gene using penalized logistic regression analysis (PLRA), using the logistf package in R (https://cran.r-project.org/web/packages/logistf/index.html). To adjust for background variation, we used the number of genes with rare synonymous variants as a covariate for each individual in both cohorts. Additionally, in the discovery cohort we used the first two principal components as covariates to adjust for population difference. We deemed genes with p-value ≤ 0.05 and odds ratio >1 as statistically significant risk genes. We performed the burden analysis in pan-cancer and tissue-specific cancer types. All statistical tests were two-sided.
Gene-set level RDV burden analyses
Next, we evaluated the RDV burden of gene-sets that typically play key roles in cancer risk and progression: (i) known cancer predisposition genes (Illumina, https://www.illumina.com/products/by-type/clinical-research-products/trusight-cancer.html); (ii) DNA damage repair genes20,21; (iii) somatic cancer driver genes22 and (iv) Fanconi Anemia genes61. We performed the burden analysis for (i) tissue type specific cancers; (ii) histologically or anatomically related cancer types, including pan-gastrointestinal23, pan-kidney24, pan-gynecological25, and pan-squamous26, and (iii) pan-cancer. We provide a complete list of genes in these gene-sets in Supplementary Table 6. For burden analyses, we used PLRA and considered all participants with at least one RDV in a gene within the considered gene-set. Furthermore, for an unbiased data-driven exome-wide gene-set analysis, we also tested all gene-sets (17,810) in Molecular Signatures Database (MSigDB)27 using the same RDV burden approach. We considered statistically significant burden as p-value ≤ 0.05.
Gender effects
The gender breakdown of participants for each cancer type in TCGA is provided in Supplementary Table 8. To study the effect of gender on burden analyses, we first removed samples with missing gender data (Supplementary Table 8) and then used PLRA with gender as an additional covariate. Resulting ORs and p-values of all gene-sets are in Supplementary Fig. 3. Please note that we did include gender-biased cancer subtypes breast, prostate, ovarian, cervical and uterine (endometrial and sarcoma) in the multi-cancer analysis of gender effects.
Cancer type grouping
While cancer type is traditionally defined based on tissue of origin and histologic type, recently62, histologically or anatomically related cancer types have been studied together, including pan-gastrointestinal23, pan-kidney24, pan-gynecological25, and pan-squamous analyses26. In light of these studies, we performed gene-set level burden analyses in these four sub-groups. We included 634 patients with stomach, colon or rectum adenocarcinoma for pan-gastrointestinal; 528 patients with clear cell, papillary or chromophobe renal cell carcinoma for pan-kidney; 1392 patients with ovarian serous cystadenocarcinoma, uterine corpus endometrial carcinoma, cervical squamous cell carcinoma and endocervical adenocarcinoma, uterine carcinosarcoma or breast invasive carcinoma for pan-gynecological; and 1038 patients with lung squamous cell carcinoma, head and neck squamous cell carcinoma, or cervical and bladder cancers with squamous differentiation26 for pan-squamous analyses. For all of these groupings, we compared the cases with the full set of controls.
Germline RDV load effects
Next, we asked whether the accumulation of personal germline RDVs, or “RDV load” of an individual impacts their personal cancer risk. Towards this end, we first divided the discovery cohort participants based on their RDV loads into three groups: participants with (i) no RDVs; (ii) one RDV; and (iii) more than one RDV. We then tested and compared the association of germline RDV load with age of diagnosis between these groups using the Mann–Whitney U test. Next, we asked whether the germline RDV load is associated with tumor immune microenvironment (TME). For this purpose, we used existing datasets31 on the relative fraction of 22 different immune cell types within TME across TCGA cancers, as estimated by the CIBERSORT tool32 (RRID:SCR_016955), which included 5917 cases. To obtain the total cell fraction in tissue, we multiplied the relative immune cell fractions with leukocyte fraction31. We then compared the immune cell fractions between the groups using the Kruskal-Wallis test. Next, to study the effect of M1 macrophages on survival, we used Cox proportional hazards regression model. Finally, to study the effect of germline RDVs on tumor mutation burden (TMB) between the groups, we used Mann–Whitney U test. To calculate the TMB, we used the publicly available TCGA somatic mutations MAF file (mc3.v0.2.8.PUBLIC.maf.gz) which included 6225 cases. We used the TMB definition as the total number of somatic, missense, nonsense, frameshift/inframe mutations per megabase (Mb) of genome examined, with 38 Mb as an estimate of exome size.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The results here are in part based upon data generated by the TCGA Research Network (https://www.cancer.gov/tcga) and datasets from dbGaP (http://www.ncbi.nlm.nih.gov/gap) through dbGaP accession numbers phs000209, phs000276, phs000296, phs000298, phs000424, phs000654, phs000687, phs000806, phs000876, phs000971, phs001000, phs001101. BioMe BioBank (http://icahn.mssm.edu/research/ipm/programs/biome-biobank) WES data from Mount Sinai Health system requires PPHS/IRB approval.
Code availability
All analyses were performed utilizing standard publicly available software. Any specific analysis code details are available from the authors upon request.
References
Stadler, Z. K. et al. Genome-wide association studies of cancer. J. Clin. Oncol. 28, 4255–4267 (2010).
Li, X. et al. The impact of rare variation on gene expression across tissues. Nature 550, 239–243 (2017).
Tennessen, J. A. et al. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science 337, 64–69 (2012).
Nelson, M. R. et al. An abundance of rare functional variants in 202 drug target genes sequenced in 14,002 people. Science. 337, 100–104 (2012).
Robson, M. & Offit, K. Clinical practice. Management of an inherited predisposition to breast cancer. N. Engl. J. Med. 357, 154–162 (2007).
Lindor, N. M. et al. Recommendations for the care of individuals with an inherited predisposition to Lynch syndrome: a systematic review. JAMA 296, 1507–1517 (2006).
Villani, A. et al. Biochemical and imaging surveillance in germline TP53 mutation carriers with Li-Fraumeni syndrome: a prospective observational study. Lancet Oncol. 12, 559–567 (2011).
Galiatsatos, P. & Foulkes, W. D. Familial adenomatous polyposis. Am. J. Gastroenterol. 101, 385–398 (2006).
Hall, E. T. et al. Pathogenic variants in less familiar cancer susceptibility genes: What happens after genetic testing? JCO Precis. Oncol. https://doi.org/10.1200/PO.18.00167 (2018).
Zhang, J. et al. Germline mutations in predisposition genes in pediatric cancer. N. Engl. J. Med. 373, 2336–2346 (2015).
Lu, C. et al. Patterns and functional implications of rare germline variants across 12 cancer types. Nat. Commun. 6, 10086 (2015).
Huang, K. et al. Pathogenic germline variants in 10,389 adult cancers. Cell 173, 355–370.e14 (2018).
Ding, L. et al. Perspective on oncogenic processes at the end of the beginning of cancer genomics. Cell 173, 305–320.e10 (2018).
Campbell, P. J. et al. Pan-cancer analysis of whole genomes. Nature 578, 82–93 (2020).
Esai Selvan, M., Klein, R. J. & Gümüş, Z. H. Rare, pathogenic germline variants in Fanconi anemia genes increase risk for squamous lung cancer. Clin. Cancer Res. 25, 1517–1525 (2019).
Esai Selvan, M. et al. Inherited rare, deleterious variants in ATM increase lung adenocarcinoma risk. J. Thorac. Oncol. 15, 1871–1879 (2020).
Cancer Genome Atlas Research Network, J. N. et al. The cancer genome atlas pan-cancer analysis project. Nat. Genet. 45, 1113–1120 (2013).
Kowalski, M. H. et al. Use of >100,000 NHLBI Trans-Omics for Precision Medicine (TOPMed) Consortium whole genome sequences improves imputation quality and detection of rare variant associations in admixed African and Hispanic/Latino populations. PLoS Genet. 15, e1008500 (2019).
Taliun, D. et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature 590, 290–299 (2021).
Knijnenburg, T. A. et al. Genomic and molecular landscape of DNA damage repair deficiency across the cancer genome atlas. Cell Rep. 23, 239–254.e6 (2018).
Pritchard, C. C. et al. Inherited DNA-repair gene mutations in men with metastatic prostate cancer. N. Engl. J. Med. 375, 443–453 (2016).
Matthew Bailey, A. H. et al. Comprehensive characterization of cancer driver genes and mutations article comprehensive characterization of cancer driver genes and mutations. Cell 173, 371–385 (2018).
Liu, Y. et al. Comparative molecular analysis of gastrointestinal adenocarcinomas. Cancer Cell 33, 721–735.e8 (2018).
Ricketts, C. J. et al. The cancer genome atlas comprehensive molecular characterization of renal cell carcinoma. Cell Rep. 23, 313–326.e5 (2018).
Berger, A. C. et al. A comprehensive pan-cancer molecular study of gynecologic and breast cancers. Cancer Cell 33, 690–705.e9 (2018).
Campbell, J. D. et al. Genomic, pathway network, and immunologic features distinguishing squamous carcinomas. Cell Rep. 23, 194–212.e6 (2018).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005).
Shahamatdar, S. et al. Germline features associated with immune infiltration in solid tumors. Cell Rep. 30, 2900–2908.e4 (2020).
Lim, Y. W. et al. Germline genetic polymorphisms influence tumor gene expression and immune cell infiltration. Proc. Natl Acad. Sci. USA 115, E11701–E11710 (2018).
Tian, J. et al. CancerImmunityQTL: A database to systematically evaluate the impact of genetic variants on immune infiltration in human cancer. Nucleic Acids Res. 49, D1065–D1073 (2021).
Thorsson, V. et al. The immune landscape of cancer. Immunity 48, 812–830.e14 (2018).
Newman, A. M. et al. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 12, 453–457 (2015).
Chan, T. A. et al. Development of tumor mutation burden as an immunotherapy biomarker: Utility for the oncology clinic. Ann. Oncol. 30, 44–56 (2019).
Spira, A. et al. Precancer atlas to drive precision prevention trials. Cancer Res. 77, 1510–1541 (2017).
Jeggo, P. A., Pearl, L. H. & Carr, A. M. DNA repair, genome stability and cancer: a historical perspective. Nat. Rev. Cancer 16, 35–42 (2016).
Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016).
Karczewski, K. J. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020).
Chen, D. S. & Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 541, 321–330 (2017).
Reyes-Torres, I. et al. TREM2 sensing of tumor cell efferocytosis promotes a macrophage molecular state that limits NK cell antitumor immunity. SSRN Electron. J. https://doi.org/10.2139/ssrn.3900125 (2021).
Buscher, K. et al. Natural variation of macrophage activation as disease-relevant phenotype predictive of inflammation and cancer survival. Nat. Commun. 8, 16041 (2017).
S, T. et al. High numbers of macrophages, especially M2-like (CD163-positive), correlate with hyaluronan accumulation and poor outcome in breast cancer. Histopathology 66, 873–883 (2015).
Ardighieri, L. et al. Infiltration by CXCL10 secreting macrophages is associated with antitumor immunity and response to therapy in ovarian cancer subtypes. Front. Immunol. 12, 690201 (2021).
Pan, Y., Yu, Y., Wang, X. & Zhang, T. Tumor-associated macrophages in tumor immunity. Front. Immunol. 11, 583084 (2020).
Klemm, F. & Joyce, J. A. Microenvironmental regulation of therapeutic response in cancer. Trends Cell Biol. 25, 198–213 (2015).
Goodman, A. M. et al. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol. Cancer Ther. 16, 2598–2608 (2017).
Landrum, M. J. et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 42, D980–D985 (2014).
Wang, Y. et al. Rare variants of large effect in BRCA2 and CHEK2 affect risk of lung cancer. Nat. Genet. 46, 736–741 (2014).
Graff, R. E. et al. Cross-cancer evaluation of polygenic risk scores for 16 cancer types in two large cohorts. Nat. Commun. 12, 970 (2021).
Klein, R. J. & Gümüş, Z. H. Are polygenic risk scores ready for the cancer clinic?-a perspective. Transl. Lung cancer Res. 11, 910–919 (2022).
Fahed, A. C. et al. Polygenic background modifies penetrance of monogenic variants for tier 1 genomic conditions. Nat. Commun. 11, 1–9 (2020).
Torkamani, A., Wineinger, N. E. & Topol, E. J. The personal and clinical utility of polygenic risk scores. Nat. Rev. Genet. 19, 581–590 (2018).
Abul-Husn, N. S. et al. Exome sequencing reveals a high prevalence of BRCA1 and BRCA2 founder variants in a diverse population-based biobank. Genome Med. 12, 2 (2019).
SEER. Surveillance, Epidemiology, and End Results Program, National Cancer Institute https://seer.cancer.gov/ (2022).
Linderman, M. D. et al. Analytical validation of whole exome and whole genome sequencing for clinical applications. BMC Med. Genom. 7, 20 (2014).
Wei, X. et al. Germline Lysine-Specific Demethylase 1 (LSD1/KDM1A) mutations confer susceptibility to multiple myeloma. Cancer Res. 78, 2747–2759 (2018).
Waller, R. G. et al. Novel pedigree analysis implicates DNA repair and chromatin remodeling in multiple myeloma risk. PLoS Genet. 14, e1007111 (2018).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 26, 589–595 (2010).
Manichaikul, A. et al. Robust relationship inference in genome-wide association studies. Bioinformatics 26, 2867–2873 (2010).
Auton, A. et al. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Belbin, G. M. et al. Toward a fine-scale population health monitoring system. Cell 184, 2068–2083.e11 (2021).
Ceccaldi, R., Sarangi, P. & D’Andrea, A. D. The Fanconi anaemia pathway: new players and new functions. Nat. Rev. Mol. Cell Biol. 17, 337–349 (2016).
Hoadley, K. A. et al. Cell-of-origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell 173, 291–304.e6 (2018).
Acknowledgements
This work was supported by grants to Z.H.G from LUNGevity Foundation, Uniting Against Lung Cancer Foundation and Cancer Moonshot R33 award # CA263705-01; to R.J.K. from the National Cancer Institute (R01 CA167824); to S.G. from the grants U24 CA224319 and U01 DK124165 and in part through the computational resources and staff expertise provided by Scientific Computing at the Icahn School of Medicine at Mount Sinai. We would also like to thank the ISMMS BioMe team for providing whole-exome sequencing calls, principal component analysis, and identification of unrelated individuals. We thank the participants, investigators and staff of the TCGA Research Network and dbGaP studies (accession numbers phs000209, phs000276, phs000296, phs000298, phs000424, phs000654, phs000687, phs000806, phs000876, phs000971, phs001000, phs001101). We thank Berk Turhan for help with one of the figures. We include the following acknowledgement statements for the dbGaP studies: phs000209: MESA and the MESA SHARe project are conducted and supported by the National Heart, Lung, and Blood Institute (NHLBI) in collaboration with MESA investigators. Support for MESA is provided by contracts N01-HC- 95159, N01-HC-95160, N01-HC-95161, N01-HC-95162, N01-HC-95163, N01-HC-95164, N01-HC-95165, N01-HC- 95166, N01-HC-95167, N01-HC-95168, N01-HC-95169, UL1-RR-025005, and UL1-TR-000040. This study is part of the NHLBI Grand Opportunity Exome Sequencing Project (GO-ESP). Funding for GO-ESP was provided by NHLBI grants RC2 HL103010 (HeartGO), RC2 HL102923 (LungGO) and RC2 HL102924 (WHISP). The exome sequencing was performed through NHLBI grants RC2 HL102925 (BroadGO) and RC2 HL102926 (SeattleGO). HeartGO gratefully acknowledges the following groups and individuals who provided biological samples or data for this study. DNA samples and phenotypic data were obtained from the following studies supported by the NHLBI: the Atherosclerosis Risk in Communities (ARIC) study, the Coronary Artery Risk Development in Young Adults (CARDIA) study, Cardiovascular Health Study (CHS), the Framingham Heart Study (FHS), the Jackson Heart Study (JHS) and the Multi-Ethnic Study of Atherosclerosis (MESA). phs000276: The NFBC1966 Study is conducted and supported by the National Heart, Lung, and Blood Institute (NHLBI) in collaboration with the Broad Institute, UCLA, University of Oulu, and the National Institute for Health and Welfare in Finland. This manuscript was not prepared in collaboration with investigators of the NFBC1966 Study and does not necessarily reflect the opinions or views of the NFBC1966 Study Investigators, Broad Institute, UCLA, University of Oulu, National Institute for Health and Welfare in Finland and the NHLBI. phs000296: This research used data generated by the COPDGene study, which was supported by NIH grants U01 HL089856 and U01 HL089897. The COPDGene project is also supported by the COPD Foundation through contributions made by an Industry Advisory Board comprised of Pfizer, AstraZeneca, Boehringer Ingelheim, Novartis, and Sunovion. This study is part of the NHLBI Grand Opportunity Exome Sequencing Project (GO-ESP). Funding for GO-ESP was provided by NHLBI grants RC2 HL103010 (HeartGO), RC2 HL102923 (LungGO) and RC2 HL102924 (WHISP). The exome sequencing was performed through NHLBI grants RC2 HL102925 (BroadGO) and RC2 HL102926 (SeattleGO). phs000298: The dataset(s) were deposited by the ARRA Autism Sequencing Collaborative, an ARRA funded research initiative. Support for the Autism Sequencing Collaborative was provided by grants: R01-MH089208 awarded to Dr. Mark Daly, R01-MH089175 awarded to Dr. Richard Gibbs, R01-MH089025 awarded to Joseph Buxbaum, R01-MH089004 awarded to Gerard Schellenberg, and R01-MH089482 awarded to James Sutcliffe. phs000424: The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health (commonfund.nih.gov/GTEx). Additional funds were provided by the NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. Donors were enrolled at Biospecimen Source Sites funded by NCI\Leidos Biomedical Research, Inc. subcontracts to the National Disease Research Interchange (10XS170), Roswell Park Cancer Institute (10XS171), and Science Care, Inc. (X10S172). The Laboratory, Data Analysis, and Coordinating Center (LDACC) was funded through a contract (HHSN268201000029C) to The Broad Institute, Inc. Biorepository operations were funded through a Leidos Biomedical Research, Inc. subcontract to Van Andel Research Institute (10ST1035). Additional data repository and project management were provided by Leidos Biomedical Research, Inc.(HHSN261200800001E). The Brain Bank was supported supplements to University of Miami grant DA006227. Statistical Methods development grants were made to the University of Geneva (MH090941 & MH101814), the University of Chicago (MH090951,MH090937, MH101825, & MH101820), the University of North Carolina - Chapel Hill (MH090936), North Carolina State University (MH101819),Harvard University (MH090948), Stanford University (MH101782), Washington University (MH101810), and to the University of Pennsylvania (MH101822). phs000654: The sequencing was performed through Epi4K Gene Discovery in Epilepsy study (NINDS U01-NS077303) and the Epilepsy Genome/Phenome Project (EPGP—NINDS U01-NS053998). phs000687: The Bulgarian Trio Sequencing study is an accumulation of exome sequencing performed and/or funded by the Broad Institute, Cardiff University, Icahn School of Medicine at Mount Sinai, and the Wellcome Trust Sanger Institute. Work at the Broad Institute was funded by Fidelity Foundations, the Sylvan Herman Foundation and philanthropic gifts from Kent and Liz Dauten, Ted and Vada Stanley, and an anonymous donor to the Stanley Center for Psychiatric Research. Work at Cardiff was supported by Medical Research Council (MRC) Centre (G0800509) and Program Grants (G0801418), the European Community’s Seventh Framework Programme (HEALTH-F2-2010-241909 (Project EU-GEI)). Work at the Icahn School of Medicine at Mount Sinai was supported by the Friedman Brain Institute, the Institute for Genomics and Multiscale Biology and National Institutes of Health grants R01HG005827 (SMP) and R01MH071681 (PS). Work at the Wellcome Trust Sanger Institute was supported by The Wellcome Trust (WT089062 and WT098051). The recruitment of the trios in Bulgaria was funded by the Janssen Research Foundation. phs000876: This research was supported in part by NCI grants U19CA148127 and P30CA023108, as well as Canadian Cancer Society Research Institute (no. 020214) and the International Agency for Research on Cancer. phs000971: This publication includes data from the ClinSeq™ study that was supported by the National Human Genome Research Institute Intramural Research Program. phs000806, phs001000 and phs001101: We thank the Broad Institute for generating high-quality sequence data supported by NHGRI funds (grant # U54 HG003067) with Eric Lander as PI.
Author information
Authors and Affiliations
Contributions
R.J.K. and Z.H.G. conceived and designed the study. Z.H.G. supervised the research. M.E.S. and R.J.K performed data analysis. M.E.S., R.J.K. and Z.H.G. wrote and reviewed the manuscript with critical feedback and support from K.O. and S.G. All authors approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
S.G. reports consultancy and/or advisory roles for Merck and OncoMed and research funding from Bristol-Myers Squibb, Genentech, Celgene, Janssen R&D, Takeda, and Regeneron. Other authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Esai Selvan, M., Onel, K., Gnjatic, S. et al. Germline rare deleterious variant load alters cancer risk, age of onset and tumor characteristics. npj Precis. Onc. 7, 13 (2023). https://doi.org/10.1038/s41698-023-00354-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41698-023-00354-3
