
Abstract
Patients with metastatic NSCLC bearing a ROS1 gene fusion usually experience prolonged disease control with ROS1-targeting tyrosine kinase inhibitors (TKI), but significant clinical heterogeneity exists in part due to the presence of co-occurring genomic alterations. Here, we report on a patient with metastatic NSCLC with a concurrent ROS1 fusion and KRAS p.G12C mutation at diagnosis who experienced a short duration of disease control on entrectinib, a ROS1 TKI. At progression, the patient continued entrectinib and started sotorasib, a small molecule inhibitor of KRAS p.G12C. A patient-derived cell line generated at progression on entrectinib demonstrated improved TKI responsiveness when treated with entrectinib and sotorasib. Cell-line growth dependence on both ROS1 and KRAS p.G12C was further reflected in the distinct downstream signaling pathways activated by each driver. Clinical benefit was not observed with combined therapy of entrectinib and sotorasib possibly related to an evolving KRAS p.G12C amplification identified on repeated molecular testing. This case supports the need for broad molecular profiling in patients with metastatic NSCLC for potential therapeutic and prognostic information.
Similar content being viewed by others

Introduction
Lung cancer remains the leading cause of cancer-related deaths for men and women in the United States1. For patients newly diagnosed with metastatic non-small cell lung cancer (NSCLC), treatment decisions are based on broad molecular profiling of the tumor including tissue sequencing and oncogenic fusion and amplification detection2. In a subset of these patients, molecular testing will identify targetable alterations such as an epidermal growth factor receptor (EGFR) mutation or ROS proto-oncogene 1 (ROS1) gene fusion. Patients with NSCLC bearing a targetable alteration, such as a ROS1 fusion, can experience durable, prolonged disease control with molecularly targeted, tyrosine kinase inhibitors.
For the 1–2% of patients diagnosed with NSCLC positive for a ROS1 fusion (ROS1+), a current first-line therapy option, entrectinib, results in a disease response in the majority of patients with expected survival measured in years3. Despite effective TKI therapy, nearly all ROS1+ NSCLC develop treatment resistance and disease progression occurs. At time of disease progression, repeat tissue or cell free DNA molecular testing are frequently pursued as some mechanisms of TKI resistance can be inhibited with other clinically available therapies4. Acquired mechanisms of resistance to ROS1 TKIs include ROS1-dependent mechanisms, such as ROS1 kinase domain mutations, or ROS1-independent mechanisms including bypass signaling through the mitogen-activated protein kinase (MAPK) pathway. Acquired Kristen rat sarcoma virus (KRAS) mutations or amplifications have been reported in a subset of patients with ROS1+ NSCLC5. Resistance arising through KRAS mutations, specifically KRAS p.G12C mutations, are of particular interest given the development and clinical availability of KRAS G12C inhibitors6. These concurrent alterations in other targetable drivers are primarily noted at the time of TKI resistance, and rarely at diagnosis7. Here we report on a patient with metastatic lung adenocarcinoma bearing both a ROS1 fusion and KRAS p.G12C mutation at diagnosis. Despite molecularly targeted therapies for both oncogenic alterations, the patient did not experience clinical benefit from therapy likely due to evolving outgrowth of a KRAS p.G12C bearing clone and eventual KRAS p.G12C amplification. This case underscores the importance of broad molecular profiling in advanced NSCLC both at the time of diagnosis and at treatment milestones such as progression to personalize therapy and outcome expectations for patients.
Results
Clinical history
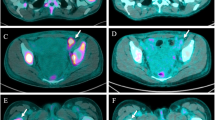
A 57-year-old man with a smoking history of less than 5 pack-years presented with bilateral leg pain due to deep venous thrombosis in both legs. A computed tomography (CT) scan of the chest demonstrated multiple, bilateral acute and subacute pulmonary emboli and a left hilar nodal mass with left-sided mediastinal lymphadenopathy. A bronchoscopy with endobronchial ultrasound guided biopsies demonstrated adenocarcinoma consistent with a lung primary at station 11L (Fig. 1a). While holding his anticoagulation after the procedure, the patient presented with expressive aphasia and was found to have an acute middle cerebral artery infarct. During his recovery, molecular testing on the biopsy reported a ROS1 fusion identified by fluorescence in situ hybridization (FISH), and next-generation sequencing (NGS) of tumor DNA identified a KRAS p.G12C mutation and a telomerase reverse transcriptase (TERT) promoter mutation (c.-124C>T). At diagnosis variant allele frequency (VAF) determined was 3.2% for KRAS p.G12C and 12.9% TERT mutation. Positron emission tomography (PET) demonstrated FDG avid left hilar mass and extensive adenopathy throughout the neck and mediastinum (Fig. 1b). Magnetic resonance imaging (MRI) of the brain showed no evidence of intracranial metastases. The patient was started on first line entrectinib.

a Timeline of the patient’s diagnosis, treatment, and molecular testing results. b Radiographic changes associated with patient’s clinical course.
Approximately 3 months later, the patient developed a left sided pleural effusion and a thoracentesis was positive for metastatic lung adenocarcinoma. Molecular testing again demonstrated a ROS1 fusion by FISH and a KRAS p.G12C mutation. Allele frequency of KRAS p.G12C was 62% compared to 40% for the TERT mutation suggesting outgrowth of clones bearing a KRAS p.G12C mutation. Subsequent tissue sequencing by NGS at our institution did not detect a ROS1 kinase domain mutation and the fusion event was determined to be TPM3(ex8)-ROS1(ex35). PET imaging revealed near complete resolution of the previously FDG avid left hilar mass and neck and mediastinal adenopathy but a new FDG avid posterior left lower lobe nodule and left pleural thickening was noted (Fig. 1b). At an outside institution, the patient was started on pembrolizumab along with continued entrectinib.
One month later, the patient presented to our institution with progressive, expressive aphasia. MRI of the brain demonstrated new multifocal supratentorial lesions predominantly within the left frontal and left parietal lobes (Fig. 1b). The patient received radiation therapy to the brain lesions. Approximately 4 weeks later, the patient started sotorasib plus entrectinib and pembrolizumab was discontinued. One day after initiation of dual targeted therapies, the patient required hospitalization for progressive hypoxia due to bilateral pulmonary emboli and new community acquired pneumonia which were not thought due to his previous immunotherapy or his dual targeted therapies. During his treatment course on both targeted therapies, he was frequently evaluated by his primary oncologist and clinical team. The patient did not experience adverse events that could be considered related to combination therapy. Specifically the patient did not require antiemetic therapy and had stable weights during his evaluations. No liver function test or electrolyte abnormalities were noted and creatinine was normal. Approximately 2 months later, the patient returned with progressive worsening of his baseline aphasia along with 1 week of right arm weakness and paresthesias. MRI imaging of the brain showed interval progression in 3 hemorrhagic lesions within the left cerebral hemisphere (Fig. 1b). CT imaging of the chest, abdomen and pelvis showed progression on the left pleural disease with new abdominal lymphadenopathy. During the hospitalization, a biopsy of a left pleural mass was performed and molecular testing demonstrated a KRAS p.G12C VAF of 98% compared to 23% for the TERT mutation. While hospitalized, the patient was experiencing a clinical recovery of his speech and right arm function. Anticoagulation was restarted with argatroban due to a 40% decrease in platelet counts over 5 days with recent low molecular weight heparin exposure. The day of argatroban initiation, the patient experienced acute clinical deterioration while ambulating was noted to have fixed and dilated pupils, and died likely due to an acute intracranial hemorrhage.
Cell line characterization
Prior to initiation of sotorasib, pleural fluid was collected for cell line generation (Fig. 1a). The cell line that grew out was designated CUTO64 (TPM3-ROS1) and demonstrated resistance to first line ROS1 targeted therapies entrectinib and crizotinib, with half-maximal inhibitory concentrations (IC50) > 2000 nM for both agents in contrast to a ROS1 TKI responsive cell line derived from a different patient, CUTO37 (CD74-ROS1) that has an IC50 of 56 nM with crizotinib and an IC50 of 39 nM with entrectinib (Fig. 2a). CUTO64 was similarly resistant to ceritinib in addition to second-line lorlatinib with an IC50 > 2000 nM for both. (Fig. 2a). CUTO28 (TPM3-ROS1), a cell line derived from a different patient, exhibits sensitivity to entrectinib with an IC50 of 8.3 nM (Supplementary Fig. 2) suggesting the ROS1 TKI resistance exhibited by CUTO64 is not due to the ROS1 fusion partner.

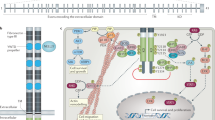
a CUTO64 is resistant to clinically available 1st line ROS1 TKIs (ceritinib, crizotinib, entrectinib) and 2nd line lorlatinib, in comparison to CUTO37 a patient derived cell line responsive to ROS1 TKIs. Inhibition of KRAS G12C with clinically available sotorasib or adagrasib increases CUTO64 responsiveness to ROS1 inhibition with b entrectinib or c crizotinib. d Inhibition of ROS1 and the MAPK pathway with trametinib increases CUTO64 responsiveness to TKI. Showing the mean ± SD, n = 3 biological replicates. e Treatment of CUTO64 with entrectinib resulted in pROS1 inhibition and a reduction in downstream Akt activity but had limited impact on ERK signaling. f Single agent sotorasib reduced ERK signaling but had no impact on pROS1 or pAkt. The combination of entrectinib plus sotorasib resulted in inhibition of both the MAPK and Akt pathway.
As the patient’s clinical testing also demonstrated a KRAS p.G12C mutation, we used the combination of entrectinib plus KRAS G12C inhibitors sotorasib or adagrasib which reduced the cell viability compared to either agent alone (Fig. 2b). Entrectinib plus sotorasib resulted in an IC50 of 5.8 nM while entrectinib plus adagrasib resulted in an IC50 of 7.8 nM compared to single agent IC50 of >1900 nM, >2600 nM, and >1000 nM for entrectinib, sotorasib, and adagrasib, respectively. Treatment of CUTO64 with entrectinib and a KRAS G12C inhibitor was synergistic with combination indices (CI) of 0.12 and 0.17 for the combination with sotorasib or adagrasib, respectively. A similar pattern was observed with crizotinib plus sotorasib or adagrasib (Fig. 2c) with an IC50 > 1000 nM for each drug individually. Combination of crizotinib and a KRAS G12C inhibitor was synergistic with a CI of 0.16 for crizotinib plus sotorasib and 0.21 for crizotinib plus adagrasib. Inhibition of the MAPK signaling pathway with trametinib, a mitogen-activated protein kinase 1 and 2 (MEK1/2) inhibitor, reduced CUTO64 viability when combined with entrectinib or crizotinib (Fig. 2d).
As concurrent inhibition of ROS1 and KRAS G12C increased cell sensitivity to targeted therapy, we evaluated the signaling pathways downstream of each driver. Treatment of CUTO64 with entrectinib resulted in pROS1 inhibition suggesting resistance was not due to a kinase domain mutation commonly observed in approximately 40% of patients at time of progression on single agent TKI therapy5. ROS1 inhibition lead to a dose dependent reduction in downstream protein kinase B (Akt) activity but had limited impact on extracellular-signal-regulated kinase (ERK) signaling at higher drug concentrations (Fig. 2e). Single agent sotorasib reduced ERK signaling but had no impact on pROS1 or pAkt (Fig. 2f). The combination of entrectinib plus sotorasib resulted in potent inhibition of both the MAPK and Akt pathways (Fig. 2f).
Identification of KRAS amplification through NGS and FISH
In previous studies, KRAS amplification has been reported as an acquired mechanism of resistance in patients with progression on ROS1 targeted therapies, in KRAS p.G12C positive preclinical models testing sotorasib and in patients receiving other KRAS G12C inhibitors5,8,9,10. Clinical testing on biopsy specimens from the patient demonstrated an increase in KRAS p.G12C variant allele frequency in comparison to a passenger mutation in TERT over time, suggesting evolving selection of KRAS p.G12C co-mutation and amplification as an acquired mechanism of resistance through the patient’s course of therapy (Fig. 1a). Expanded testing with the research-based 498-gene NGS-based approach did not detect KRAS p.G12C amplification at diagnosis through our bioinformatics pipeline that includes assessment for copy number alterations (Fig. 3a). After therapy initiation, KRAS p.G12C amplification was not detected at time of progression on entrectinib and was readily detectable at time of disease progression on entrectinib and sotorasib. We further verified this amplification with the tissue sample at time of progression on entrectinib and sotorasib by FISH which demonstrated a KRAS/CEP12 ratio of 11.5 and uniform ROS1 positivity across tumor cells (Fig. 3b).

a KRAS copy number alterations identified on sequencing of clinical biopsies at time of diagnosis (left), progression on entrectinib (middle), and progression on entrectinib and sotorasib (right). b ROS1 and KRAS FISH on patient biopsy after progression on entrectinib and sotorasib.
Discussion
This case report highlights the critical importance of broad molecular profiling in patients with metastatic lung cancer as co-occurring alterations may inform prognostic discussions and additional therapeutic options11. To the best of our knowledge, this case is also the first report of a lung adenocarcinoma bearing both a ROS1 fusion and KRAS p.G12C mutation at diagnosis and the concurrent use of targeted therapies for both drivers. KRAS-mediated resistance to ROS1 inhibitors was predicted in preclinical models using crizotinib where a KRAS p.G12C mutation was identified in the HCC78 (SLC34A2-ROS1) ROS1+ cell line12. This clinical case is also consistent with previous work by our group showing primary resistance to TKI therapy in a patient with concurrent ALK gene fusion and KRAS p.G12C mutation13,14. Bypass signaling through the MAPK pathway is a known driver of resistance to ROS1 TKI therapy and ongoing clinical trials are exploring the utility of combining ROS1 TKIs with MEK inhibition12,15,16,17. However, as illustrated by this case, precision therapies targeting driver alterations can be rapidly surmounted by adaptive resistance. Clinical management of systemic progression on a ROS1 fusion targeting TKI can be informed by subsequent molecular testing at time of progression18. For this patient, management options included a combination strategy for the known oncogenic drivers or chemotherapy with or without the continuation of targeted therapy. While KRAS G12C inhibition represents a new therapeutic approach for patients with lung cancer, prospective data report response rates of 40.7% in the subsequent line setting and durability of response of approximately 1 year19. Retrospective series on pemetrexed-based chemotherapy in patients with ROS1 fusion positive NSCLC reports a range in disease response from 23.8% to 54.5%20,21. Historically, continuing TKI therapy while receiving subsequent line chemotherapy did not improve outcomes but this remains an active area of investigation especially in the era of CNS-penetrant TKIs22,23,24.
Notably even in the presence of a KRAS p.G12C mutation, initial therapy with entrectinib resulted in clinical improvement in the patient’s mediastinal tumor burden that persisted until the patient’s death. Studies with CUTO64, derived at time of entrectinib progression, suggests the tumor remained partially dependent on ROS1 as maximal reduction in cell viability was achieved when ROS1 inhibition was paired with MAPK pathway inhibition. Similarly, single agent KRAS G12C inhibitors had limited impact on CUTO64 viability in the absence of ROS1 inhibition. CUTO64 dependence on both ROS1 and KRAS G12C was further reflected in the distinct downstream signaling pathways activated by each driver. However, clinical benefit was not observed with combined therapy of entrectinib and sotorasib likely related to the KRAS amplification observed on molecular testing. Clinically, the patient did not have access to KRAS G12C inhibitors until later in the disease course when subsequent line sotorasib was approved by the Food and Drug Administration (FDA). Based on the partial dependence of CUTO64 on both drivers, clinical benefit may have been derived if combined entrectinib and sotorasib were started at diagnosis. Due to the initial response to therapy and clinical considerations, tumor biopsies were not obtained from the same site and the spatial heterogeneity of the obtained samples is a limitation of our study25. Molecular testing of samples at times progression may reflect tissue related factors shaping the genomic landscape of the cancer cells in addition to cancer-cell intrinsic mechanisms of therapeutic resistance25.
The presence of co-occurring, actionable drivers in NSCLC is uncommon, but the application of NGS testing to clinical care has improved the recognition of this event. In a retrospective series of 3,077 patients diagnosed with NSCLC, NGS testing identified co-occurring, potentially targetable oncogenic drivers in 46 patients, including 16 patients identified prior to therapy initiation26. Patients with NSCLC bearing multiple targetable alterations have been reported to experience sustained disease control either through alternating oncogene targeting therapies or combination approaches26,27,28. These reported cases frequently involve EGFR mutations alongside ALK fusions or MET amplifications. Fewer studies have described ROS1 positive NSCLC with concomitant driver oncogenes, and there has been some discordance in the reported frequency of co-occurring drivers likely due to the method of ROS1 fusion detection7,29,30. NGS testing on 166 ROS1+ NSCLC without associated clinical information identified 1 patient with ROS1+ NSCLC with a concurrent EGFR L858R mutation and 3 patients with ROS1+ NSCLC and a KRAS activating mutation (Q61R, G12R, G12C)7. In the setting of progression on a ROS1 TKI, bypass activation of the MAPK signaling pathway, including KRAS activating mutations and amplifications, have also been reported as acquired changes5. Pre-clinical studies associated entrectinib resistance with the acquisition of a KRAS p.G12C mutation and amplification of both KRAS and FGFR3 in HCC78, an NSCLC cell line bearing a ROS1 fusion8. In this model, entrectinib resistance was associated sustained pERK activation and resensitization to ROS1 inhibition was achieved with the addition of the MEK1/2 inhibitor selumetinib. KRAS amplification is also a reported mechanism of resistance to sotorasib in a patient with KRAS p.G12C mutant NSCLC and a patient with KRAS p.G12C mutant colon cancer10. One potential approach to overcoming KRAS amplification is to target upstream or downstream components of the KRAS signaling pathway as is being explored in early clinical trials with SHP2 inhibitors and ERK1/2 inhibitors31.
Our data along with other reports highlights the importance of evaluating for alterations in the MAPK pathway as part of a broad assessment at time of ROS1 TKI resistance. In addition to identifying a likely mechanism of resistance, the therapeutic landscape for targeting MAPK pathway alterations continues to expand including clinically available KRAS G12C inhibitors, and ongoing clinical trials testing inhibition of non-G12C KRAS mutations as well as downstream targets such as MEK and ERK32,33. In conclusion, this case report underscores the need for comprehensive molecular testing of patients with metastatic lung cancer at the time of diagnosis as well as at progression on targeted therapy, as multiple, targetable oncogene drivers may influence treatment choice and expectations for disease control.
Methods
Clinical testing
Initial clinical testing was performed through the NeoTYPE™ Analysis Lung Profile (Neogenomics) which analyzes 49 biomarkers through a combination of next-generation sequencing (NGS), fluorescence in situ hybridization, and immunohistochemistry34.
Mutational analysis of clinical samples was also performed using a research-based 498-gene NGS-based approach at the University of Colorado (Colorado Molecular Correlates Laboratory). Briefly, total nucleic acid was extracted and mechanically sheared. Sheared nucleic acid was then submitted to library preparation using the KAPA Hyper Prep kit (Roche, Basel, Switzerland). Target enrichment of the library was performed using xGen Lockdown Hybridization probes and kit (IDT, Coralville, IA). Enriched library was then sequenced on an Illumina NextSeq instrument (Illumina, San Diego CA). Raw sequence data was analyzed for mutations and insertions/deletions with a custom bioinformatic pipeline that utilized Novosort for read alignment, GATK/Mutect2 and VarScan2 for variant calling, and Ensenbl Variant Effect Predictor for variant annotation. Copy number alterations were determined using CNVkit35.
Dual color FISH assays were performed on 4 µm sections of formalin fixed paraffin embedded (FFPE) tumor specimen with ROS1 break-apart (Vysis, Abbott molecular, and Agilent) and KRAS/12 centromere enumeration (Empire Genomics) probes, using proteinase K enzymatic digestion, or on patient-derived cultured cells. Fluorescent probe signals were evaluated in minimum 50 tumor nuclei per specimen. Specimens were counted as positive for ROS1 rearrangement if 15% or more of cells displayed a split 5’/3’ and/or single 3’ signal pattern. Separation between 5’ and 3’ ROS1 signals of 1 or more signal diameter was required to be considered as a split. KRAS amplification was defined as ratio of KRAS probe to 12 centromere enumeration probe of >2.0.
Cell line derivation and cell culture and reagents
CUTO64 and CUTO37 cell lines were derived as previously described with patient consent under COMIRB-approved protocol 11–1621 with written consent36. In brief, the CUTO64 line was derived from a sample of the patient’s pleural effusion which was applied onto the Ficoll density gradient medium (Histopaque-1077, Sigma-Aldrich # 10771-100 ML) and underwent centrifugation as recommended by the manufacturer. The resulting cell pellet was resuspended in RPMI 1640 media (Gibco # 11875093) supplemented with 10% fetal bovine serum (Atlas Biologicals # F-0500-A) and plated out onto a 25 cm flask. Based on visual inspection of cell morphology, when adherent tumor cells became the predominately established cell type in the culture flask, the culture was subjected to differential trypsinization and mechanical dissociation in order to dislodge the remaining minor population of stromal cells. Cell lines were cultured in RPMI 1640 with 10% FBS and incubated at 37 °C in 5% CO2.
MTS
CUTO64 and CUTO37 cells were plated at a density of 1500 to 2000 cells per well in a 96-well plate and treated with designated drug 24 h post plating. After 72 h exposed to drug, media was changed to remove cell debris and MTS reagent was added to the manufacturer’s recommendations (Promega, G7570). Absorbance was measured on a microplate reader (BioTek) at 490 nm. Non-linear regression curves and IC50 were calculated using GraphPad Prism v9.3.1 (GraphPad Software) and CompuSyn v1.0 (www.combosyn.com). The combination index was calculated using the Chou-Talay method with CompuSyn v1.037. Adagrasib, sotorasib, trametinib, ceritinib, crizotinib, entrectinib, and lorlatinib were all purchased from ChemieTek.
Western blot
Cells were plated in a 6 well plate, cultured to 80–100% confluency and were then treated with the indicated drug(s) and dose(s) for 3 h and processed in parallel. Cells were then lysed with 150 μL of T-PER Tissue protein Extracting Reagent (ThermoFisher) with 1× Halt Protease Inhibitor (ThermoFisher) for 20 min at 4 °C. The lysing solution was collected and centrifuged. The supernatant was collected and stored at −80 °C. The lysate was then diluted 3:1 with 4× Protein Loading Buffer (LI-COR) with 0.1 mM DTT (ThermoFisher). The samples were loaded onto a 10% APS Gel with Chameleon Duo Ladder (LI-COR) and Page Ruler Prestained Plus Protein Ladder (ThermoFisher) and run. Gels were transferred onto a nitrocellulose membrane (LI-COR) and transferred using the Thermofisher PierceG2 Fast Blotter as per the manufacturer’s recommendations. Membranes were blocked in 1:1 1× TBS (Research Products International) and Intercept Blocking Buffer (LI-COR) and then tagged with indicated antibody. Membranes were then rinsed and tagged with Goat Anti-Mouse Antibody IRDye 800 CW (LI-COR P/N: 926–32210) and Goat Anti-Rabbit IRDye 680 LT Antibody 690 (LI-COR P/N: 926–68021). All secondary antibodies were diluted 1:10,000. The membranes were visualized using a LI-COR Odyssey Machine. Western data processing is done with LI-COR Image Studio software and GraphPad Prism.
Antibodies used were ROS1 (69D6) Mouse mAb (3266S), p-ROS1 (Y2274) Rabbit Ab (3078S), ERK1/2 p44/p42 MAPK (L34F12) Mouse mAb (4696S), p-ERK1/2 T202/Y204 (197G2) Rabbit mAb (4337S), Akt (pan) (40D4) Mouse mAb (2920S), p-Akt S473 (D9E) Rabbit mAb (4060S) from Cell Signaling Technologies, and Mouse mAb GAPDH (65C) from Millipore (CB1001). pROS1 and ROS1 antibodies were diluted to 1:1000 and all other Cell Signaling Technology antibodies were diluted to 1:2000. GAPDH was diluted to 1:105.
Uncropped and unprocessed scans of the manuscript western blots are shown in Supplementary Fig. 1. All blots were derived from the same experiment and were processed in parallel.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All the data and resources generated for this study are available in the article or from the corresponding author upon request. The data have been deposited with links to BioProject accession number PRJNA909827 in the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/). BioSample metadata are available in the NCBI BioSample database (http://www.ncbi.nlm.nih.gov/biosample/) under accession numbers SAMN32100022, SAMN32100023, and SAMN32100024.
Code availability
Bioinformatics pipeline code is available upon request.
References
Siegel, R. L. et al. Cancer statistics, 2022. CA Cancer J. Clin. 72, 7–33 (2022).
Lindeman, N. I. et al. Updated molecular testing guideline for the selection of lung cancer patients for treatment with targeted tyrosine kinase inhibitors: guideline from the College of American Pathologists, the International Association for the Study of Lung Cancer, and the Association for Molecular Pathology. Arch. Pathol. Lab. Med. 142, 321–346 (2018).
Dziadziuszko, R. et al. Updated integrated analysis of the efficacy and safety of entrectinib in locally advanced or metastatic ROS1 fusion-positive non-small-cell lung cancer. J. Clin. Oncol. 39, 1253–1263 (2021).
Lin, J. J. & Shaw, A. T. Resisting resistance: targeted therapies in lung cancer. Trends Cancer 2, 350–364 (2016).
Lin, J. J. et al. Spectrum of mechanisms of resistance to crizotinib and lorlatinib in ROS1 fusion-positive lung cancer. Clin. Cancer Res. 27, 2899–2909 (2021).
Skoulidis, F. et al. Sotorasib for lung cancers with KRAS p.G12C mutation. N. Engl. J. Med. 384, 2371–2381 (2021).
Lin, J. J. et al. ROS1 fusions rarely overlap with other oncogenic drivers in non-small cell lung cancer. J. Thorac. Oncol. 12, 872–877 (2017).
Ku, B. M. et al. P2.14-61 acquired resistance to entrectinib associated with activation of RAS signaling pathway in ROS1-rearranged non-small cell lung cancer. J. Thorac. Oncol. 14, S855 (2019).
Zhao, Y. et al. Diverse alterations associated with resistance to KRAS(G12C) inhibition. Nature 599, 679–683 (2021).
Awad, M. M. et al. Acquired resistance to KRAS(G12C) inhibition in cancer. N. Engl. J. Med. 384, 2382–2393 (2021).
Skoulidis, F. & Heymach, J. V. Co-occurring genomic alterations in non-small-cell lung cancer biology and therapy. Nat. Rev. Cancer 19, 495–509 (2019).
Cargnelutti, M. et al. Activation of RAS family members confers resistance to ROS1 targeting drugs. Oncotarget 6, 5182–5194 (2015).
Doebele, R. C. et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non–small cell lung cancer. Clin. Cancer Res. 18, 1472–1482 (2012).
McCoach, C. E. et al. Resistance mechanisms to targeted therapies in ROS1+ and ALK+ non–small cell lung cancer. Clin. Cancer Res. 24, 3334–3347 (2018).
Sato, H. et al. MAPK pathway alterations correlate with poor survival and drive resistance to therapy in patients with lung cancers driven by ROS1 fusions. Clin. Cancer Res. 26, 2932–2945 (2020).
Massachusetts General Hospital, Array Biopharma & Pfizer. Lorlatinib combinations in lung cancer. https://clinicaltrials.gov/ct2/show/NCT04292119 (2022).
University of California, San Francisco, Takeda & Array Biopharma. Brigatinib and binimetinib in treating patients with stage IIIB-IV ALK or ROS1-rearranged non-small cell lung cancer. https://clinicaltrials.gov/ct2/show/NCT04005144 (2022).
Almquist, D. & Ernani, V. The road less traveled: a guide to metastatic ROS1-rearranged non–small-cell lung cancer. JCO Oncol. Pract. 17, 7–14 (2021).
Dy, G. K. et al. Long-Term Outcomes with Sotorasib in Pretreated KRASp.G12C-Mutated NSCLC: 2-Year Analysis of CodeBreaK100 (American Association for Cancer Research, 2022).
Zhang, L. et al. Efficacy of crizotinib and pemetrexed-based chemotherapy in Chinese NSCLC patients with ROS1 rearrangement. Oncotarget 7, 75145–75154 (2016).
Chen, Y.-F. et al. Efficacy of pemetrexed-based chemotherapy in patients with ROS1 fusion–positive lung adenocarcinoma compared with in patients harboring other driver mutations in East Asian populations. J. Thorac. Oncol. 11, 1140–1152 (2016).
Soria, J.-C. et al. Gefitinib plus chemotherapy versus placebo plus chemotherapy in EGFR-mutation-positive non-small-cell lung cancer after progression on first-line gefitinib (IMPRESS): a phase 3 randomised trial. Lancet Oncol. 16, 990–998 (2015).
AstraZeneca & Parexel. A study to evaluate chemotherapy plus osimertinib against chemotherapy plus placebo in patients with non-small cell lung cancer (NSCLC). https://clinicaltrials.gov/ct2/show/NCT04765059 (2022).
Loxo Oncology, Inc. & Eli Lilly and Company. A study of selpercatinib (LY3527723) in participants with advanced or metastatic RET fusion-positive non-small cell lung cancer. https://clinicaltrials.gov/ct2/show/NCT04194944 (2023).
Dagogo-Jack, I. & Shaw, A. T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 15, 81–94 (2018).
Zhao, Y. et al. Co-occurring potentially actionable oncogenic drivers in non-small cell lung cancer. Front. Oncol. 11, 665484 (2021).
Baldi, L. et al. Concomitant EGFR mutation and ALK rearrangement in lung adenocarcinoma is more frequent than expected: report of a case and review of the literature with demonstration of genes alteration into the same tumor cells. Lung Cancer 86, 291–295 (2014).
Peters, T. L. et al. Evolution of MET and NRAS gene amplification as acquired resistance mechanisms in EGFR mutant NSCLC. NPJ Precis. Oncol. 5, 91 (2021).
Warth, A. et al. ROS1 expression and translocations in non-small-cell lung cancer: clinicopathological analysis of 1478 cases. Histopathology 65, 187–194 (2014).
Wiesweg, M. et al. High prevalence of concomitant oncogene mutations in prospectively identified patients with ROS1-positive metastatic lung cancer. J. Thorac. Oncol. 12, 54–64 (2017).
Nagasaka, M. et al. KRAS inhibitors– yes but what next? Direct targeting of KRAS– vaccines, adoptive T cell therapy and beyond. Cancer Treat. Rev. 101, 102309 (2021).
Drosten, M. & Barbacid, M. Targeting the MAPK pathway in KRAS-driven tumors. Cancer Cell 37, 543–550 (2020).
Lindsay, C. R. et al. On target: rational approaches to KRAS inhibition for treatment of non-small cell lung carcinoma. Lung Cancer 160, 152–165 (2021).
NeoGenomics. NeoTYPE® lung tumor profile. https://neogenomics.com/test-menu/neotyper-lung-tumor-profile (2022).
Talevich, E. et al. CNVkit: genome-wide copy number detection and visualization from targeted DNA sequencing. PLOS Comput. Biol. 12, e1004873 (2016).
Davies, K. D. et al. Resistance to ROS1 inhibition mediated by EGFR pathway activation in non-small cell lung cancer. PLoS ONE 8, e82236 (2013).
Chou, T.-C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 70, 440–446 (2010).
Acknowledgements
E.L.S. is supported by NIH grant K12 CA086913 and LUNGevity Career Development Award #25B1267. This study was supported in part by the NIH P30CA06934 Pathology Shared Resource – Cytogenetic Section RRID:SCR_021991 and Molecular Pathology Section RRID:SCR_021995. We thank the Colorado Genetics Laboratory, University of Colorado, Anschutz Medical Campus, Aurora, Colorado, for their assistance with clinical sample analysis. We would like to thank the ROS1ders patient advocacy group (ros1cancer.com) for their support of ROS1 research and their donation of tissue samples for the creation of novel ROS1 cell lines and the Addario Lung Cancer Medical Institute for research funding.
Author information
Authors and Affiliations
Contributions
K.P. contributed to the design, acquisition, and interpretation of the data, and drafting and revising of the manuscript; A.L., A.G., G.B.R., and M.M. contributed to the acquisition of the data; H.N. contributed to the design, acquisition, and interpretation of the data, and revising of the manuscript; K.D.D. contributed to the design, acquisition, and interpretation of the data, and revising of the manuscript; C.L. contributed to the design, acquisition, and interpretation of the data; E.D. contributed to the design, acquisition, and interpretation of the data; R.C.D. contributed to the acquisition and interpretation of the data and revising of the manuscript; L.B. contributed to the acquisition and interpretation of the data and revising of the manuscript; D.L.A. contributed to the design, acquisition, and interpretation of the data, and drafting and revising of the manuscript; E.L.S. contributed to the overall study conception, design, acquisition, and interpretation of the data, and drafting and revising of the manuscript.
Corresponding author
Ethics declarations
Competing interests
K.D.D. received sponsored travel from ArcherDX. R.C.D. has royalties or licenses through Rain Therapeutics, Foundation Medicine, Takeda, Pearl River, Scorpion Therapeutics, ThermoFisher, Genentech, Black Diamond, Voronoi, received consulting fees from Rain Therapeutics, Anchiano, Blueprint Medicines, Takeda, AstraZeneca, Genentech/Roche, Bayer, AstraZeneca, Green Peptide, received payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing or educational events: Medscape, Princess Margaret Hospital, Targeted Oncology, OncLive, received support for attending meetings and/or travel: Genentech/Roche, Blueprint Medicines, Rain Therapeutics, has patents planned, issued or pending: Rain Therapeutics, Abbott Molecular, has a leadership or fiduciary role with Rain Therapeutics serves on the advisory board of Rain Therapeutics. D.L.A. reports consulting fees from Takeda, Sonif-Genzyme, Loxo Oncology, Bluepring Medicines, and Genentech. E.L.S. reports speaker fees from OncLive, Physicians’ Education Resource, Takeda, Roche/Genetech, IDEO Oncology, Sanofi/Regeneron, consultant fees from Actinium, Bionest Partners, ExpertConnect, FCB Health, Guidepoint Network, the KOL Connection Ltd, Prescient Advisory, served on an advisory board for Regeneron, Janssen, and G1 therapeutics. No other authors have any competing interests to disclose.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Priest, K., Le, A., Gebregzabheir, A. et al. Evolution of acquired resistance in a ROS1+ KRAS G12C+ NSCLC through the MAPK pathway. npj Precis. Onc. 7, 9 (2023). https://doi.org/10.1038/s41698-023-00349-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41698-023-00349-0
This article is cited by
-
Screening biomarkers for predicting the efficacy of immunotherapy in patients with PD-L1 overexpression
Journal of Cancer Research and Clinical Oncology (2023)
