
Abstract
Limited efforts have been made in assessing the effect of genome-wide profiling of RNA splicing-related variation on lung cancer risk. In the present study, we first identified RNA splicing-related genetic variants linked to lung cancer in a genome-wide profiling analysis and then conducted a two-stage (discovery and replication) association study in populations of European ancestry. Discovery and validation were conducted sequentially with a total of 29,266 cases and 56,450 controls from both the Transdisciplinary Research in Cancer of the Lung and the International Lung Cancer Consortium as well as the OncoArray database. For those variants identified as significant in the two datasets, we further performed stratified analyses by smoking status and histological type and investigated their effects on gene expression and potential regulatory mechanisms. We identified three genetic variants significantly associated with lung cancer risk: rs329118 in JADE2 (P = 8.80E−09), rs2285521 in GGA2 (P = 4.43E−08), and rs198459 in MYRF (P = 1.60E−06). The combined effects of all three SNPs were more evident in lung squamous cell carcinomas (P = 1.81E−08, P = 6.21E−08, and P = 7.93E−04, respectively) than in lung adenocarcinomas and in ever smokers (P = 9.80E−05, P = 2.70E−04, and P = 2.90E−05, respectively) than in never smokers. Gene expression quantitative trait analysis suggested a role for the SNPs in regulating transcriptional expression of the corresponding target genes. In conclusion, we report that three RNA splicing-related genetic variants contribute to lung cancer susceptibility in European populations. However, additional validation is needed, and specific splicing mechanisms of the target genes underlying the observed associations also warrants further exploration.
Similar content being viewed by others


Introduction
RNA splicing is a process in which an mRNA precursor can produce multiple mRNA isoforms that dramatically diversify the transcriptome and the proteome in eukaryotic cells1,2,3. In humans, it is estimated that up to 94% of the genes are differentially spliced, and the resulting protein isoforms can contribute to proteome complexity and have diverse or even opposite biological functions, with profound consequences on cellular processes and phenotypes4. Several studies have described aberrant or dysregulated RNA splicing events as well as alternative RNA splicing events, which can frequently be affected by genetic variants and mutations, that contribute to cancer cell phenotypes by directly or indirectly regulating the genome, epigenome, transcriptome, and proteome5,6,7,8,9. Aberrant RNA splicing has been implicated in the ancestry-related biology of cancer disparities and as a potential source of novel targets for precision oncology10. The role of aberrant splicing as a primary cause of Mendelian diseases has been widely accepted by decades of related studies11,12,13. However, much less has been reported and appreciated regarding the extent of physiological RNA splicing variation among human populations and the phenotypic variability and disease susceptibility affected by them in humans5.
Lung cancer is the most common malignancy in humans, leading the causes of cancer death worldwide14. Non-small cell lung cancer (NSCLC) is the most prevalent and heterogeneous subtype of lung cancer, including lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC)15. Although a large number of germline mutations within cancer susceptibility genes have been reported, genetic etiology of lung cancer is still not fully understood16,17. In addition, until recently, despite the important role of RNA splicing in cancer, limited efforts have been made in the genome-wide profiling of RNA splicing-related variation in cancer patients, including in NSCLC15.
We have previously reported single nucleotide polymorphisms (SNPs) in RNA splicing-related regulatory sequences in prostate cancer stemness-associated genes that are associated with race and/or survival18,19. To conduct a comprehensive characterization of a genome-wide profile of common lung cancer genetic susceptibility loci associated with RNA splicing, we first performed a meta-analysis to discover potential RNA splicing-related SNPs using summary statistics from eight published lung cancer genome-wide association studies (GWASs) from the Transdisciplinary Research in Cancer of the Lung (TRICL) and the International Lung Cancer Consortium (ILCCO)20. Those significant SNPs discovered were then validated using data from the OncoArray platform that provides an unprecedented opportunity for additional de novo discovery as well as for fine mapping of lung cancer susceptibility16,21. For those identified SNPs that were found to be significant in both discovery and validation datasets, we further performed stratified analyses by smoking status and histological type and investigated their effects on gene expression and potential regulatory mechanisms in cell lines and tissues by using the available genomic and genetic data from multiple public databases.
Results
Meta-analysis of the main effects
We focused on the joint analyses of both TRICL-ILLCO and OncoArray sets, representing the largest sample size of European ancestry to date. The study populations (29,266 lung cancer cases and 56,450 non-cancer controls) for the eight GWASs from TRICL-ILLCO consortia (i.e., ICR, MDACC, IARC, NCI, Toronto, GLC, Harvard, and deCODE GLC) and OncoArray dataset and the demographic characteristics of the final dataset are summarized in Supplementary Table 1, and the work workflow is depicted in Fig. 1.

CEU Caucasian, MAF minor allele frequency, FDR false discovery rate, eQTL expression quantitative trait loci.
The overview of overall association results is shown in the Manhattan plot (Fig. 2a). We found that there were 295 SNPs related to RNA splicing with a nominal P < 0.05, of which 14 SNPs remained with a false discovery rate (FDR) < 0.20 for multiple testing correction. There was no heterogeneity observed for the effect estimates of these 14 SNPs from the eight GWASs (Supplementary Table 2).

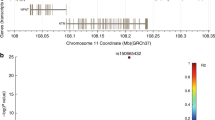
a Manhattan plot of the overall results. There were 295 SNPs related to RNA splicing with a nominal P < 0.05, 14 of which remained with FDR < 0.20. The x-axis indicates the chromosome number and the y-axis shows the association P values with lung cancer risk (as −log10 P values). The horizontal blue line represents P values of 0.05, while the red line indicated the FDR threshold 0.20. Regional association plot, which shows the LD between the top SNP rs329118 on JADE2 (b), rs2285521 on GGA2 (c), and rs198459 on MYRF (d), and other SNPs in the region of 500 kb up- or downstream of the top SNP. Locations, functional prediction, and position weight matrix based Sequence Logo of three SNPs. JADE2 rs329118 (e) and GGA2 rs2285521 (f) are located within one CpG island and presented strong signals of active enhancer and promoter functions (indicated by H3K4 methylation, histone modification H3K27 acetylation, and DNase hypersensitivity, respectively). MYRF rs198459 (g) is located within one CpG island and presented strong signals of active enhancer and promoter functions (indicated by H3K4 methylation and DNase hypersensitivity, respectively). The panels were adapted from the UCSC Genome Browser. Three SNPs are located on the AP2B motif (e), the MYOD1 motif (f), and the ELK3 motif (g), respectively.
Among the 14 SNPs identified, six were unreported, which were then validated using the OncoArray dataset (14,803 cases and 12,262 controls), and three SNPs reached a nominal P < 0.05 (Table 1). All three SNPs were imputed with the overall imputation information/ R-squared in the eight TRICL-ILLCO GWAS presented in Supplementary Table 3. After a final combined analysis, as shown in Table 1, JADE2 rs329118 T>C was found to be associated with a significantly decreased risk of lung cancer [odds ratio (OR) = 0.94, 95% confidence interval (CI) = 0.92–0.96, P = 8.80 × 10−9], while two other SNPs were associated with a significantly increased risk of lung cancer (GGA2 rs2285521 C>T: OR = 1.08, 95% CI = 1.05–1.11, P = 4.43 × 10−8 and MYRF rs198459 G>A: OR = 1.07, 95% CI = 1.04–1.11, P = 1.60 × 10−6). No heterogeneity was observed for the effect estimates of these three SNPs from the eight GWASs and the OncoArray dataset (Supplementary Fig. 1). The regional association plots of these three SNPs are shown in Fig. 2b–d.
We then performed functional prediction for these three significant SNPs by using three bioinformatics tools (SNPinfo, regulomDB, and HaploReg) to predict their potential effects on gene expression or biological functions for further analysis (Supplementary Table 4).
Stratified analyses
To assess whether histological types of lung cancer may be impacted by different genetic factors, we performed stratified analyses by LUAD and LUSC. By using 11,273 LUAD and 7,426 LUSC from both the TRICL-ILLCO and OncoArray datasets, we found that the effects of all three SNPs (JADE2 rs329118, GGA2 rs2285521, and MYRF rs198459) were more evident in LUSC (OR = 0.91, 95% CI = 0.88–0.95; OR = 1.13, 95% CI = 1.08–1.19; OR = 1.08, 95% CI = 1.03–1.13, respectively) than in LUAD (OR = 0.95, 95% CI = 0.91–0.98; OR = 1.04, 95% CI = 0.99–1.09; OR = 1.05, 95% CI = 1.00–1.12, respectively)). However, no significant heterogeneity was found for either of the two histological strata for these three SNPs (Table 2 and Supplementary Fig. 1).
One of the major risk factors for lung cancer is cigarette smoking, which may interact with genetic factors. According to available smoking data, study subjects were divided into two groups as ever smokers and never smokers in stratified analyses. We found that rs329118, rs2285521, and rs198459 all had a significant risk effect in ever smokers (OR = 0.94, 95% CI = 0.91–0.97; OR = 1.07, 95% CI = 1.03–1.12; OR = 1.02, 95% CI = 1.06–1.18, respectively) (Table 2 and Supplementary Fig. 1), while no significant association was observed in never smokers for all three SNPs. The forest plots of the overall and stratification results for these three SNPs are shown in Supplementary Fig. 1a–c.
In silico analyses
The three SNPs were not only predicted to potentially affect RNA splicing but also were predicted to potentially affect mRNA transcription (Supplementary Table 4). According to experiment-based data (e.g., histone modification, DNase cluster, transcription factor (TF) binding, RNAseq) from the ENCODE project (Fig. 2e, f), we found that two SNPs (JADE2 rs329118 and GGA2 rs2285521) are located within one CpG island with strong signals for active enhancer and promoter functions (indicated by H3K4 tri-methylation, histone modification H3K27 acetylation, and DNase hyper-sensitivity). MYRF rs198459 was also predicted to be located within a regulatory region with evidence for H3K4 mono-methylation and DNase cluster (Fig. 2g). Further TF binding analysis (using ChIP-seq data) showed that rs329118, rs2285521, and rs198459 are located within the AP2B, MYOD-1, and ELK3 motifs, respectively, as shown by the position weight matrix (PWM)-based Sequence Logo (Fig. 2e–g), which suggest that the allele difference might influence the binding activity of the TF.
In the following analyses using three mRNA expression and genotype databases, rs2285521 in GGA2 was assessed directly or indirectly from the lymphoblastoid cell lines, normal lung tissue, and lung cancer tissue, while the other two variants rs329118 and rs198459 only had available data either from lymphoblastoid cell lines or whole blood cells. The rational for using different tissues was to test the potential tissue-specific regulation of these SNPs.
Expression quantitative trait loci (eQTL) analysis was first performed to assess the association between each SNP and its target gene mRNA expression level in the lymphoblastoid cell lines from 373 subjects of European ancestry in the 1000 Genomes project. From this analysis, we found that the JADE2 rs329118 T allele was significantly associated with increased expression levels of JADE2 in a recessive model (P = 0.027), but not in an additive or dominant model (Fig. 3a–c). The GGA2 rs2285521 C allele was significantly related to decreased mRNA expression levels in all additive (P = 5.30 × 10−4), dominant (P = 1.30 × 10−3), and recessive (P = 0.034) models (Fig. 3d–f).

Correlation between JADE2 rs329118 and JADE2 mRNA expression levels in additive (a), dominant (b), and recessive (c) models in 373 blood cells from 373 Europeans individuals in 1000 genomes project (P = 0.094, 0.487 and 0.027, respectively). Correlation between GGA2 rs2285521 and GGA2 mRNA expression levels in additive (d), dominant (e), and recessive (f) models in 373 blood cells from 373 Europeans individuals in 1000 genomes project (P = 5.30 × 10−4, 0.0013 and 0.034, respectively). Correlation between GGA2 rs2285521 (g) and MYRF rs198459 (h), and mRNA expression levels in normal lung tissues or whole blood cells of GTEx project (P = 0.014 and P = 6.20 × 10−10, respectively). i, j Correlation between EARS2 rs6497670 in additive (i) and dominant (j) models in lung cancer tissues of TCGA project (P = 4.85 × 10−3 and P = 8.29 × 10−3, respectively). k Pair-wise LD plot between GGA2 rs2285521 (T>C) and EARS2 rs6497670 (C>T). a–j P value was calculated from linear regression. Center-line indicates the median expression level across all participants in that group, and the hinges represent the lower (Q1) and upper (Q3) quartile, with lower whisker indicating the smallest value within 1.5 interquartile range (IQR) below Q1 and upper whisker indicating the largest value within 1.5 IQR above Q3.
SNP rs2285521 was also significantly correlated with mRNA expression levels of GGA2 in normal lung tissues based on the Genotypes-Tissue Expression (GTEx) database (P = 0.014, Fig. 3g), which is consistent with the results in the lymphoblastoid cell lines. Similar results were found for MYRF rs198459 in whole blood cells (P = 6.20 × 10−10) (Fig. 3h). Using both genotyping and gene expression data for lung cancer in the Cancer Genome Atlas (TCGA) database, we performed SNP-mRNA correlation analyses for the three SNPs. However, we were only able to retrieve the genotype data for rs6497670 in EASR2 (Fig. 3i, j), a SNP in complete linkage disequilibrium (LD) (r2 = 1) with rs2285521 in GGA2, with available data after performing imputation for these top three SNPs (Fig. 3k). Once again, SNP rs2285521 showed a significant correlation with decreased mRNA expression levels of GGA2 in lung cancer tissues, specifically in both additive (P = 4.85 × 10−3, Fig. 3i) and recessive (P = 8.29 × 10−3, Fig. 3j) models for LUSC, but not for LUAD (Supplementary Fig. 2e, f).
Results from other studies collected in the cancer microarray database Oncomine also provided evidence for a higher expression level of JADE2 in NSCLC tissue (both LUAD and LUSC) than in normal lung tissue. Both GGA2 and MYRF were found to exhibit lower expression in NSCLC (both LUAD and LUSC) tissue than in normal lung tissue (Supplementary Fig. 3a–f).
For GGA2, we compared two GGA2 transcripts (GenBank number: NC_000016) (Fig. 4a–d), which were designated as GGA2-X1 and GGA2-X2 in the GenBank of National Center of Biotechnology Information. We analyzed the amino acid (aa) sequence and secondary structure of the putative protein isoforms of GGA2-X1 and GGA2-X2 with ExPASy and SWISS-MODEL, respectively. The aa sequence (Fig. 4a) and secondary structure differed between the two isoforms (Fig. 4b). The GGA2-X1 isoform contained 576 aa, while the GGA2-X2 isoform contained 526 aa. There is complete homology between GGA2-X1 and GGA2-X2 in the last nine exons of C-terminal sequences, but GGA2-X2 lacks 50 aa (aa1-aa50) in the N-terminal sequences, compared with GGA2-X1, as shown in Fig. 4c, d.

a Results of SNP rs2285521 sequencing in the 53 bp sequence of 5’ UTR region in Exon 1 of the GGA2: Genomic structure of the GGA2-X1 and GGA2-X2. Splicing pattern of the GGA2 variant with SNP rs2285521 T>C. The position of the A nucleotide in the start codon (ATG) is defined as +1. b Part of RNA secondary structure of the GGA2 rs2285521 U and GGA2 rs2285521 C. c The aa sequence of GGA2-X1 and GGA2-X2. The GGA2-X1 isoform encodes a 576 aa protein and GGA2-X2 encodes a 526 aa protein. The aa sequence of red is where GGA2-X2 starts. d Part of the secondary structures and domains of putative isoforms of GGA2-X1 and GGA2-X2. aa1-aa50 circled by red was the part two isoforms differed.
Discussion
To our knowledge, we are the first to report a genome-wide profiling analysis of RNA splicing-related genetic variants in lung cancer, using a total of 29,266 cases and 56,450 non-cancer controls of European ancestry. We identified three genetic variants (rs329118, rs198459, and rs2285521) to be associated with lung cancer risk, residing in a gene for apoptosis and differentiation in epithelia 2 (JADE family PHD zinc finger 2; JADE2; PHF16) at 5q31.1, myelin regulatory factor gene (MYRF) at 11q12.2, and golgi-localized, γ-adaptin ear-containing, ADP ribosylation factor-binding protein 2 (GGA2) at 16p12.2, respectively.
Among the three identified susceptibility genes in the present study, GGA2 belongs to the GGA family, which consists of three isoforms in vivo, namely GGA1, GGA2, and GGA322. These isoforms have been reported to have transport functions in cells, with GGA2 predominantly in the trans-Golgi network (TGN) and endoplasmic reticulum, participating in the separation of the TGN and polyvesicles22,23. GGA2 cooperates with mannose 6-phosphate receptor and adaptor protein-1 in regulating protein sorting, showing significant co-localization with transporter glucose transporter 4 (GLUT4) recovery pool24. It also stimulates the activation of EGFR signal transduction and promotes the occurrence and development of several malignancies22,25. SNP rs2285521 T>C is located in the first exon of GGA2 on chr16, which lies upstream of the coding sequence, 48 bp away from the translation initiation site, belonging to the 5 ‘untranslated region (UTR). It appears that rs2285521 T>C does not directly change the sequence or the amino acid types of GGA2, but the mRNA structure analysis suggests that the conversion of rs2285521 allele T to C changes a loop-stem-loop secondary structure. In the GenBank of National Center of Biotechnology Information, there are two isoforms of GGA2: GGA2-X1 and GGA2-X2, generated by an exon skipping/inclusion event. Compared with GGA2-X1, GGA2-X2 lacks 50 amino acids (aa1–aa50) in the N-terminal sequences. The exon skipping/inclusion event leads to a difference in the N-terminal sequences between GGA2-X1 and GGA2-X2, while keeping the complete homology of the two isoforms in the remaining nine exons of C-terminal sequences. Whether rs2285521 is involved in the regulation of this exon skipping/inclusion event remains to be determined. Meanwhile, both the eQTL data from lymphocytes and GTEx data from lung tissue suggest that rs2285521 is involved in transcriptional regulation possibly by reducing the mRNA expression of GGA2. In addition, TCGA data showed that the substitution of the T allele with the C allele would lead to a decrease in GGA2 mRNA expression in LUSC. The in silico results of these three analyses supported the observed genetic associations. These data also suggest that different histological subtypes of lung cancer may have different genetic etiology, with GGA2 rs2285521 more likely having significance in LUSC.
Two other variants, rs329118 and rs198459 are located in the introns of JADE2 and MYRF, respectively. JADE2 is a member of the small JADE family that also includes JADE1 (PHF17) and JADE3 (PHF15) paralogs26. All three JADE proteins bear in tandem two Plant Homeo-domains (PHD), which are zinc finger domains26. Jade-1 was reported to inhibit Wnt signaling through its E3 ubiquitin ligase activity of β-catenin and was therefore defined as a PHD-finger-type E3 ubiquitin ligase27. JADE2 rs329118 was recently shown to be associated with age of initiation of regular smoking, but no relation to lung cancer was reported28. So far, it is known that JADE1 mRNA encodes two protein products: the full length isoform JADE1L and the truncated isoform JADE1S as a result of alternative RNA splicing26, but the RNA splicing pattern of JADE2, as well as its molecular function, remain unclear.
MYRF encodes an endoplasmic reticulum membrane protein that undergoes auto-processing to release its N-terminal fragment, which enters the nucleus, forms a homo-trimer, and functions as a TF29. There is an increasing evidence that MYRF may play a critical role in the development of various organs, including the heart, lungs, diaphragm, and genitourinary tract. For example, particular missense or frameshift variants in MYRF have been identified to be linked to mild encephalitis/encephalopathy or congenital heart defects, and/or congenital diaphragmatic hernia30. Interestingly, members of our team have recently reported that MYRF is a differentially spliced gene in LUSC between patients of West African and European ancestry who are smokers31.
In summary, the present study, with the largest lung cancer study population of European ancestry ever reported, identified three genetic variants in genome-wide profiling RNA splicing-related genes to be associated with lung cancer risk. We believed that all susceptibility alleles, if biologically meaningful, should be correlated with expression levels of the corresponding genes in normal lymphocytes and/or lung tissues. Our results suggest that the combination of genetics and in silico analyses helps identify and emphasize potential functional importance of RNA splicing-related loci in lung cancer susceptibility, providing insights into the etiology of this complex disease. However, replication of the results is also required in different populations as well in larger prospective studies. Because the underlying molecular splicing mechanisms of the RNA splicing-related genetic variants in lung cancer are not completely understood, further biological validation both in vitro and in vivo are warranted in the future to better understand the role of these three SNPs.
Methods
Study design and subjects
The present study adopted a two-stage design with discovery and replication datasets. The discovery set was comprised of 14,463 lung cancer cases and 44,188 non-cancer controls of European ancestry from eight centers. The replication series was comprised of 14,803 cases and non-cancer 12,262 controls of European ancestry from 31 sites, of which some centers (with no overlapping study subjects) also participated in the discovery phase (Supplementary Table 1).
Discovery set
The study populations of the discovery set have been described in previous publications from TRICL-ILCCO20,32. Briefly, eight published lung cancer GWASs were from the TRICL-ILCCO consortia, which consists of 14,463 lung cancer cases and 44,188 controls of European ancestry. The GWAS participants included Institute of Cancer Research (ICR), The University of Texas MD Anderson Cancer Center (MDACC), International Agency for Research on Cancer (IARC), National Cancer Institute (NCI), Lunenfeld-Tanenbaum Research Institute study (Toronto), German Lung Cancer Study (GLC), the Harvard Lung Cancer Study, and Icelandic Lung Cancer Study (deCODE)20,33. (Supplementary Table 1)
Replication set
The replication series was comprised of 14,803 cases and 12,262 controls from 31 study sites, of which some centers (but not study subjects) also participated in the discovery phase. Comprehensive details of each series have been previously reported16,21,34,35,36. After excluding samples genetically identified as overlapping between the OncoArray and the TRICL-ILCCO, 14,463 cases and 44,188 controls from the discovery set and 14,803 cases and 12,262 controls from the OncoArray were included in the final analyses. Most of the lung cancer cases had been histologically confirmed to be lung adenocarcinoma, followed by LUSC, and lung small cell carcinoma. Given distinct differences in smoking status and histological subtypes, the subgroup analyses, including ever and never-smokers as well as lung adenocarcinomas and LUSC, were performed. All ever smokers in the present study were defined as individuals having smoked at least 100 cigarettes in their lifetime and never-smokers defined as individuals who had smoked less than 100 cigarettes during their lifetime. A written informed consent was obtained from each participant of each dataset. The present study was approved by the Duke University Health System Institutional Review Board, and all methods performed in the present study were in accordance with the relevant guidelines and regulations.
Genotyping and quality control
For all of the GWAS datasets in TRICL-ILCCO, multiple genotyping platforms were applied, including Illumina HumanHap 317, 317 + 240S, 370Duo, 550, 610, or 1M arrays37. For the meta-analyses, imputation was performed based on the reference data from the 1000 Genomes Project (phase I integrated release 3, March 2012) by using both IMPUTE2 v2.1.138 and MaCH v1.0 + minimac (version 2012.10.3) softwares39. Only SNPs with an information score ≥0.40 in IMPUTE2 or an r2 ≥ 0.30 in MaCH were included in the final analyses. Standard quality control on samples was performed on all scans, excluding individuals with a low call rate (<90%), extremely high or low heterozygosity (P < 1.0 × 10−4), and non-European ancestry (using the HapMap phase II CEU, JPT/CHB, and YRI populations as reference).
The OncoArray consortium genotyping was completed at the Center for Inherited Disease Research (CIDR), the Helmholtz Center Munich (HMGU), Copenhagen University Hospital, and the University of Cambridge. The quality control procedures for the OncoArray dataset were identical and are reported elsewhere16,21,36. Briefly, genotype definition was undertaken using Genome Studio and jointly clustered data from 57,775 individuals and 533,631 SNPs. This included 44,591 samples associated with this study of lung cancer, 12,901 individuals from other unrelated OncoArray studies, and 283 HapMap control individuals of European, African, Chinese, and Japanese origin. Among 44,591 OncoArray lung cancer samples, 17,526 samples, including 1193 QC duplicate samples, 7633 samples overlapped with the discovery sets, 1708 with low call rate less than 95%, 1280 samples with PI-HAT greater than 0.95 or between 0.45 and 0.95 in IBD analysis, 306 with sex inconsistency information, and 5407 individuals with non-Caucasian ancestry were removed from the OncoArray when performing the validation OncoArray analysis and the joint analysis of the discovery and OncoArray sets. A total of 27,065 OncoArray samples were included in final association analysis including 14,803 lung cancer cases and 12,262 controls. Additionally, 4348 samples genotyped on the OncoArray and in a prior study including 1926 from MDACC, 2422 from IARC, and 9,811 samples without linked disease information used for genotype clustering were removed from the OncoArray set in the joint analysis of the discovery and OncoArray sets. Finally, 25,978 samples remained. The OncoArray genotyping platform queried 533,631 SNPs for fine mapping of lung cancer susceptibility loci as well as for additional de novo discovery. We used OncoArray samples for a validation of six top variants from TRICL-ILCCO samples, and we performed a meta-analysis of the discovery set and the validation set.
SNP selection
All SNPs were selected from both the 1000 Genomes project40 and GTEx Portal project (383 normal lung tissues)41. By using the genotyping data from the 1000 Genomes project, we filtered out SNPs with a minor allele frequency (MAF) < 0.01 in populations of European ancestry. After that, there remained 9,739,729 SNPs with MAF ≥ 0.01 in Caucasian populations. We then performed functional prediction for these SNPs using SNPinfo software42 and the Ensembl Variant Effect Predictor (VEP) software43, which integrated the RNA splicing prediction algorithms of FAS–ESS, RESCUE ESE, ESEfinder, MaxEntScan, Ada Boost, and Random Forest in dbscSNV44. The algorithm of SNPinfo uses GWAS SNP P-value data and finds all SNPs in high LD with GWAS SNPs, so that selection is from a much larger set of SNPs than the GWAS itself41. The Ensembl VEP software provides tools and methods for a systematic approach to annotate and prioritize variants in both large-scale sequencing projects and smaller analysis studies42. By using SNPinfo, we found 36,142 predicted RNA splicing-related SNPs in Caucasian populations. Through the Ensembl VEP, we identified 16,306 potential RNA splicing-related SNPs. After combining all SNPs, there were 48,305 non-redundant SNPs in Caucasian populations. Meanwhile, 92,213 and 837,755 potential RNA splicing-related SNPs with MAF ≥ 0.01 were predicted from the GTEx Portal project by sQTLseekeR package and Altrans method, respectively45,46. sQTLseekeR is an R package to identify splicing quantitative trait loci (sQTL) in transcriptome population studies. It can be downloaded from http://big.crg.cat/computational_biology_of_rna_processing/sqtlseeker. sQTLseekeR could be directly employed for joint analysis of gene expression across tissues. it could also be used to identify SNPs affecting expression networks, where the multivariate phenotype is the relative expression of gene compared with the total expression output of the network44. Altrans is another method for discovery of alternative sQTLs45. In searching for alternative sQTLs, nearly all methods have to infer quantifications of transcripts or splice junctions, and each method has its relative merits. However, Altrans is capable of identifying thousands of sQTLs, many of which are missed by other methods. It is very sensitive and performs comparably to other methods45. Because each of these tools has its own unique algorithm and merits, we expected some variation in the final results generated by these tools.
After comparing the final SNP sets from the two projects mentioned above, a total of 5,182 mutual SNPs with P-value < 0.05 were retained. The P-values were extracted from the sQTL results of lung tissues of the GTEx project. More details about the sQTL analysis could be found in the previous GTEx publication40. Briefly, two complementary approaches (sQTLseekeR44 and Altrans45) were used to quantify the relative expression of splicing isoforms. For sQTLseekeR, the association between SNPs with ±5 kb of the corresponding gene and the splicing ratios of gene transcript isoforms by using a non-parametrical approach: a F score was calculated by comparing the variability of splicing ratios within genotypes with the variability between genotypes, after which permutation was performed to estimate the significance of the score and calculate P value. For Altrans, the association between SNPs within ±1 Mb region around transcription start sites and the expression levels of exon junctions were tested by using the Spearman’s rank correlation test with adjustment for the first three principal components. There were finally a total of 4482 SNPs extracted in the TRICL-ILLCO dataset. Additional details are shown in Fig. 1.
In silico functional analyses
Based on the association results of genetic variants and lung cancer risk, bioinformatic analyses for functional prediction were performed on the top three significant SNPs, in particular their potential ability to affect mRNA splicing function and transcription. Four in silico tools: F-SNP47, SNPinfo Web Server42, RegulomeDB48, and HaploReg49 were selected for further predictive functional analyses of the significant SNPs. We then performed an eQTL analysis to assess the association between SNPs and mRNA expression level of the corresponding gene using the mRNA expression data from the lymphoblastoid cell lines of 373 subjects of European ancestry available in the 1000 Genomes Project40 and the eQTL results from the GTEx project41 as well as 127 NSCLC tissues in TCGA50,51. If no direct genotyping data for the three SNPs could be retrieved in the databases, then all other SNPs, which were in complete LD (r2 = 1) with the corresponding SNP generated by the imputation were also checked. The available data of those SNPs would then be retrieved and analyzed. In addition, we compared the mRNA expression levels of target genes between NSCLC and adjacent normal tissues available in the OncomineTM database52. To explore the potential regulation mechanism of the aberrant splicing, possible allelic effects of these variants on TF-binding motifs were determined using PrEdict Regulatory Functional Effect of SNPs by Approximate P value Estimation (PERFECTOS-APE; http://opera.autosome.ru/perfectosape/), which determines the probability of a TF motif (using position weight matrices, from HOCOMOCO-10, JASPAR, HTSELEX, SwissRegulon, and HOMER databases) in the DNA sequence overlapping each variant. The fold change in the probability of a TF binding site present for each allele of a variant was then calculated53.
Analysis and prediction of GGA2 splicing variants
The secondary RNA structure of human GGA2 splicing variants was predicted with RNA-Folding-Form54. The secondary protein structure of human GGA2 splicing variants was predicted with SWISS-MODEL55.
Statistical methods
We performed an unconditional logistic regression to estimate ORs and 95% CIs per effect allele for each GWAS dataset using R (v2.6), Stata (v10, State College, TX, USA), and PLINK (v1.06) software with adjustment for the top significant principal components32. We used a fixed effects model to perform meta-analysis by the inverse variance method56. If the Cochran’s Q test P-value ≤ 0.100 or the heterogeneity statistic (I2) ≥ 25%, a random-effects model was employed. We used the linear step-up method of Benjamini and Hochberg to calculate FDR with a cut-off value of 0.20 to correct for multiple comparisons57 and used linear regression for the eQTL analysis and paired t-test for the gene differential expression analysis between tumor and adjacent normal tissues. Based on the 1000 Genomes Phase I integrated variant set (release 201203), we used Haploview v4.258 to construct the LD plots, respectively. SNP pruning was applied, and SNPs with pair-wise r2 < 0.30 were considered as independent. All other analyses were conducted with SAS (version 9.4; SAS Institute, Cary, NC, USA), if not specified otherwise.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Selected data that support the findings of this study are available from the corresponding author upon reasonable request and ccorrespondence should be addressed to S.R.P. or Q.W. The primary and processed data used to generate the analyses presented here are available in the following locations: The phase1 release of variant calls of the 1000 Genomes Project39 was downloaded from https://mathgen.stats.ox.ac.uk/impute/impute_v2.html. The data deposited in and available from the GTEx Portal40 can be downloaded from www.gtexportal.org. Some other data that support the findings of this study are available via the dbGaP (www.ncbi.nlm.nih.gov/gap, database of genotypes and phenotypes) repository, among which the data are controlled-access, so interested readers will need to request access. The information on how to do so can be found on pages linked to below. The access numbers are https://identifiers.org/dbgap:phs000876.v2.p159 for TRICL study, https://identifiers.org/dbgap:phs001273.v3.p260 for the OncoArray study, and https://identifiers.org/dbgap:phs000178.v11.p861 for TCGA study.
Code availability
We used SAS (version 9.4; SAS Institute, Cary, NC, USA), R (v2.6), Stata (v10, State College, TX, USA), and PLINK (v1.06) softwares for data analysis. Only standard scripts were used for this study. No custom codes or mathematical algorithms were applied.
References
Oltean, S. & Bates, D. O. Hallmarks of alternative splicing in cancer. Oncogene 33, 5311–5318 (2014).
Wang, E. T. et al. Alternative isoform regulation in human tissue transcriptomes. Nature 456, 470–476 (2008).
Nilsen, T. W. & Graveley, B. R. Expansion of the eukaryotic proteome by alternative splicing. Nature 463, 457–463 (2010).
Biamonti, G., Infantino, L., Gaglio, D. & Amato, A. An intricate connection between alternative splicing and phenotypic plasticity in development and cancer. Cells 9, 34 (2019).
Park, E., Pan, Z., Zhang, Z., Lin, L. & Xing, Y. The expanding landscape of alternative splicing variation in human populations. Am. J. Hum. Genet. 102, 11–26 (2018).
Zhang, Y. et al. Pan-cancer analysis of clinical relevance of alternative splicing events in 31 human cancers. Oncogene 38, 6678–6695 (2019).
El Marabti, E. & Younis, I. The cancer spliceome: Reprograming of alternative splicing in cancer. Front. Mol. Biosci. 5, 80 (2018).
Escobar-Hoyos, L., Knorr, K. & Abdel-Wahab, O. Aberrant RNA splicing in cancer. Annu. Rev. Cancer Biol. 3, 167–185 (2019).
Pagani, F. & Baralle, F. E. Genomic variants in exons and introns: Identifying the splicing spoilers. Nat. Rev. Genet. 5, 389–396 (2004).
Al Abo, M. et al. Differential alternative RNA splicing and transcription events between tumors from African American and White patients in The Cancer Genome Atlas. Genomics 113, 1234–1246 (2021).
Dong, X. & Chen, R. Understanding aberrant RNA splicing to facilitate cancer diagnosis and therapy. Oncogene 39, 2231–2242 (2020).
Scotti, M. M. & Swanson, M. S. RNA mis-splicing in disease. Nat. Rev. Genet. 17, 19–32 (2016).
Boussaad, I. et al. A patient-based model of RNA mis-splicing uncovers treatment targets in Parkinson’s disease. Sci. Transl. Med. 12, eaau3960 (2020).
Bray, F. et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Ca. Cancer J. Clin. 68, 394–424 (2018).
Li, Y. et al. Prognostic alternative mRNA splicing signature in non-small cell lung cancer. Cancer Lett. 393, 40–51 (2017).
McKay, J. D. et al. Large-scale association analysis identifies new lung cancer susceptibility loci and heterogeneity in genetic susceptibility across histological subtypes. Nat. Genet. 49, 1126–1132 (2017).
Li, T., Kung, H. J., Mack, P. C. & Gandara, D. R. Genotyping and genomic profiling of non-small-cell lung cancer: implications for current and future therapies. J. Clin. Oncol. 31, 1039–1049 (2013).
Freedman, J. A. et al. Single-nucleotide polymorphisms of stemness genes predicted to regulate RNA splicing, microRNA, and oncogenic signaling are associated with prostate cancer survival. Carcinogenesis 39, 879–888 (2018).
Wang, Y. et al. Associations between RNA splicing regulatory variants of stemness-related genes and racial disparities in susceptibility to prostate cancer. Int. J. Cancer 141, 731–743 (2017).
Liu, H. et al. Functional variants in DCAF4 associated with lung cancer risk in European populations. Carcinogenesis 38, 541–551 (2017).
Amos, C. I. et al. The OncoArray Consortium: A network for understanding the genetic architecture of common cancers. Cancer Epidemiol. Biomark. Prev. 26, 126–135 (2017).
Uemura, T., Kametaka, S. & Waguri, S. GGA2 interacts with EGFR cytoplasmic domain to stabilize the receptor expression and promote cell growth. Sci. Rep. 8, 1368 (2018).
Deng, Y. et al. Gga2 mediates sequential ubiquitin-independent and ubiquitin-dependent steps in the trafficking of ARN1 from the trans-Golgi network to the vacuole. J. Biol. Chem. 284, 23830–23841 (2009).
Daboussi, L., Costaguta, G., Ghukasyan, R. & Payne, G. S. Conserved role for Gga proteins in phosphatidylinositol 4-kinase localization to the trans-Golgi network. Proc. Natl Acad. Sci. USA 114, 3433–3438 (2017).
O’Farrell, H. et al. Integrative genomic analyses identifies GGA2 as a cooperative driver of EGFR-mediated lung tumorigenesis. J. Thorac. Oncol. 14, 656–671 (2018).
Panchenko, M. V. Structure, function and regulation of jade family PHD finger 1 (JADE1). Gene 589, 1–11 (2016).
Liu, M. Z. et al. Association studies of up to 1.2 million individuals yield new insights into the genetic etiology of tobacco and alcohol use. Nat. Genet. 51, 237–244 (2019).
Han, X. et al. Destabilizing LSD1 by Jade-2 promotes neurogenesis: An antibraking system in neural development. Mol. Cell. 55, 482–494 (2014).
Hornig, J. et al. The transcription factors Sox10 and Myrf define an essential regulatory network module in differentiating oligodendrocytes. PLoS Genet. 9, e1003907 (2013).
Rossetti, L. Z. et al. Review of the phenotypic spectrum associated with haploinsufficiency of MYRF. Am. J. Med. Genet. A 179, 1376–1382 (2019).
Deveaux, A. E. et al. RNA splicing and aggregate gene expression differences in lung squamous cell carcinoma between patients of West African and European ancestry. Lung Cancer 153, 90–98 (2021).
Wang, Y. et al. Rare variants of large effect in BRCA2 and CHEK2 affect risk of lung cancer. Nat. Genet. 46, 736–741 (2014).
Thorgeirsson, T. E. et al. A variant associated with nicotine dependence, lung cancer, and peripheral arterial disease. Nature 452, 638–642 (2008).
Wang, T. et al. Pleiotropy of genetic variants on obesity and smoking phenotypes: Results from the Oncoarray Project of The International Lung Cancer Consortium. PLoS One 12, e0185660 (2017).
Li, Y. et al. Genetic variants and risk of lung cancer in never smokers: A genome-wide association study. Lancet Oncol. 11, 321–330 (2010).
Hung, R. J. et al. Lung cancer risk in never-smokers of European Descent is associated with genetic variation in the 5p15.33 TERT-CLPTM1Ll region. J. Thorac. Oncol. 14, 1360–1369 (2019).
Wang, Y. et al. Deciphering associations for lung cancer risk through imputation and analysis of 12,316 cases and 16,831 controls. Eur. J. Hum. Genet. 23, 1723–1728 (2015).
Howie, B. N., Donnelly, P. & Marchini, J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 5, e1000529 (2009).
Roshyara, N. R. et al. Comparing performance of modern genotype imputation methods in different ethnicities. Sci. Rep. 6, 34386 (2016).
Lappalainen, T. et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature 501, 506–511 (2013).
Consortium, G. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science 348, 648–660 (2015).
Xu, Z. & Taylor, J. A. SNPinfo: Integrating GWAS and candidate gene information into functional SNP selection for genetic association studies. Nucleic Acids Res. 37, W600–W605 (2009).
McLaren, W. et al. The ensembl variant effect predictor. Genome Biol. 17, 122 (2016).
Jian, X., Boerwinkle, E. & Liu, X. In silico prediction of splice-altering single nucleotide variants in the human genome. Nucleic Acids Res. 42, 13534–13544 (2014).
Monlong, J., Calvo, M., Ferreira, P. G. & Guigo, R. Identification of genetic variants associated with alternative splicing using sQTLseekeR. Nat. Commun. 5, 4698 (2014).
Ongen, H. & Dermitzakis, E. T. Alternative splicing QTLs in European and African populations. J. Am. Hum. Genet. 97, 567–575 (2015).
Lee, P. H. & Shatkay, H. F-SNP: Computationally predicted functional SNPs for disease association studies. Nucleic Acids Res. 36, D820–D824 (2008).
Boyle, A. P. et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 22, 1790–1797 (2012).
Ward, L. D. & Kellis, M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 40, D930–D934 (2012).
Cancer Genome Atlas Research, N. Comprehensive molecular profiling of lung adenocarcinoma. Nature 511, 543–550 (2014).
Cancer Genome Atlas Research, N. Comprehensive genomic characterization of squamous cell lung cancers. Nature 489, 519–525 (2012).
Rhodes, D. R. et al. ONCOMINE: A cancer microarray database and integrated data-mining platform. Neoplasia 6, 1–6 (2004).
Kulakovskiy, I. V., Vorontsov, I. E. & Makeev, V. PERFECTOS-APE-predicting regulatory functional effect of SNPs by approximate P-value estimation. In Conference: 6th International Conference on Bioinformatics Models, Methods and Algorithms, BIOINFORMATICS Lisbon, Portugal Vol. 1, 102–108 (2015).
Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 31, 3406–3415 (2003).
Waterhouse, A. et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–W303 (2018).
Begum, F., Ghosh, D., Tseng, G. C. & Feingold, E. Comprehensive literature review and statistical considerations for GWAS meta-analysis. Nucleic Acids Res. 40, 3777–3784 (2012).
Grant, G. R., Liu, J. & Stoeckert, C. J. Jr. A practical false discovery rate approach to identifying patterns of differential expression in microarray data. Bioinformatics 21, 2684–2690 (2005).
Barrett, J. C., Fry, B., Maller, J. & Daly, M. J. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21, 263–265 (2005).
Transdisciplinary Research Into Cancer of the Lung (TRICL)-Meta Analysis. dbGaP https://identifiers.org/dbgap:phs000876.v2.p1 (2017).
Oncoarray Consortium-Lung Cancer Studies. dbGaP https://identifiers.org/dbgap:phs001273.v3.p2 (2017).
National Institutes of Health The Cancer Genome Atlas (TCGA). dbGaP https://identifiers.org/dbgap:phs000178.v11.p8 (2017).
Acknowledgements
We would like to thank all individuals who participated in this study. We would also like to thank the contribution from Transdisciplinary Research in Cancer of the Lung (TRICL) Research Team and the Integrative Analysis of Lung Cancer Etiology and Risk (INTEGRAL). This work was supported in part by the National Natural Science Foundation of China (NSFC) (No. 82160535 and No. 81902876), Ningxia Natural Science Foundation (No. 2022AAC03544), and Hainan Province Clinical Medical Center. TRICL of the International Lung Cancer Consortium (ILCCO) was supported by (U19-CA203654, CA148127, and CA148127S1). The ILCCO data harmonization is supported by Cancer Care Ontario Research Chair of Population Studies to R. H. and Lunenfeld-Tanenbaum Research Institute, Sinai Health System. The TRICL-ILCCO OncoArray was supported by in-kind genotyping by the Center for Inherited Disease Research (26820120008i-0-26800068-1). Dr. Amos is a Research Scholar of the Cancer Prevention and Research Institute of Texas (CPRIT) and supported by RR170048. The Toronto study was supported by Canadian Cancer Society Research Institute (020214), Ontario Institute of Cancer and Cancer Care Ontario Chair Award to RH The ICR study was supported by Cancer Research UK (C1298/A8780 andC1298/A8362-Bobby Moore Fund for Cancer Research UK) and NCRN, HEAL and Sano-Aventis. Additional funding was obtained from NIH grants (5R01CA055769, 5R01CA127219, 5R01CA133996, and 5R01CA121197). The Liverpool Lung Project (LLP) was supported by The Roy Castle Lung Cancer Foundation, UK. The ICR and LLP studies made use of genotyping data from the Wellcome Trust Case Control Consortium 2 (WTCCC2); a full list of the investigators who contributed to the generation of the data is available from www.wtccc.org.uk. Sample collection for the Heidelberg lung cancer study was in part supported by a grant (70-2919) from the Deutsche Krebshilfe. The work was additionally supported by a Helmholtz-DAAD fellowship (A/07/97379 to MNT) and by the NIH (U19-CA148127). The KORA Surveys were financed by the GSF, which is funded by the German Federal Ministry of Education, Science, Research and Technology and the State of Bavaria. The Lung Cancer in the Young study (LUCY) was funded in part by the National Genome Research Network (NGFN), the DFG (BI576/2-1; BI 576/2-2), the Helmholtzgemeinschaft (HGF), and the Federal office for Radiation Protection (BfS: STSch4454). Genotyping was performed in the Genome Analysis Center (GAC) of the Helmholtz Zentrum Muenchen. Support for the Central Europe, HUNT2/Troms, and CARET genome-wide studies was provided by Institute National du Cancer, France. Support for the HUNT2/Troms genome-wide study was also provided by the European Community (Integrated Project DNA repair, LSHG-CT-2005-512113), the Norwegian Cancer Association and the Functional Genomics Program of Research Council of Norway. Support for the Central Europe study, Czech Republic, was also provided by the European Regional Development Fund and the State Budget of the Czech Republic (RECAMO, CZ.1.05/2.1.00/03.0101). Support for the CARET genome-wide study was also provided by grants from the US National Cancer Institute, NIH (R01 CA111703 and UO1 CA63673), and by funds from the Fred Hutchinson Cancer Research Center. Additional funding for study coordination, genotyping of replication studies and statistical analysis was provided by the US National Cancer Institute (R01 CA092039). The work reported in this paper was partly undertaken during the tenure of a Postdoctoral Fellowship from the IARC (for MNT). The Environment and Genetics in Lung Cancer Etiology (EAGLE), the Alpha-Tocopherol, Beta-Carotene Cancer Prevention Study (ATBC), and the Prostate, Lung, Colon, Ovary Screening Trial (PLCO)studies and the genotyping of ATBC, the Cancer Prevention Study II Nutrition Cohort (CPS-II) and part of PLCO were supported by the Intramural Research Program of NIH, NCI, Division of Cancer Epidemiology and Genetics. ATBC was also supported by US Public Health Service con- tracts (N01-CN-45165, N01-RC-45035, and N01-RC-37004) from the NCI. PLCO was also supported by individual contracts from the NCI to the University of Colorado Denver (NO1-CN-25514), Georgetown University(NO1-CN-25522), Pacific Health Research Institute (NO1-CN-25515),Henry Ford Health System (NO1-CN-25512), University of Minnesota(NO1-CN-25513), Washington University(NO1-CN-25516), University of Pittsburgh (NO1-CN-25511), University of Utah (NO1-CN-25524), Marshfield Clinic Research Foundation (NO1-CN-25518), University of Alabama at Birmingham (NO1-CN-75022, Westat, Inc. NO1-CN-25476), University of California, Los Angeles (NO1-CN-25404). The Cancer Prevention Study II Nutrition Cohort was supported by the American Cancer Society. The NIH Genes, Environment and Health Initiative (GEI) partly funded DNA extraction and statistical analyses (HG-06-033- NCI-01 andRO1HL091172-01), genotyping at the Johns Hopkins University Center for Inherited Disease Research (U01HG004438 and NIH HHSN268200782096C) and study coordination at the GENEVA Coordination Center (U01 HG004446) for EAGLE and part of PLCO studies. Funding for the MD Anderson Cancer Study was provided by NIH grants (P50 CA70907, R01CA121197, R01CA127219, U19 CA148127, R01 CA55769, and K07CA160753) and CPRIT grant (RP100443). Genotyping services were provided by the Center for Inherited Disease Research (CIDR). CIDR is funded through a federal contract from the NIH to The Johns Hopkins University (HHSN268200782096C). The Harvard Lung Cancer Study was supported by the NIH (National Cancer Institute) grants CA092824, CA090578, and CA074386. National Institutes of Health Feasibility and Planning Studies for Development of Specialized Programs of Research Excellence (SPOREs) to Investigate Cancer Health Disparities P20 Award 1P20CA251657-01 to SRP PI and JAF Co-Leader Project 1. The project was funded in part by GENADDICT: LSHMCT-2004–005166, the National Institutes of Health (R01-DA017932). The results published here are in whole or part based upon data generated by The Cancer Genome Atlas (TCGA) pilot project established by the NCI and NHGRI. Information about TCGA and the investigators and institutions who constitute TCGA Research Network can be found at ‘http://cancergenome.nih.gov’. The TCGA SNP data analyzed here are requested through dbGAP (accession#: phs000178.v1.p1).
Author information
Authors and Affiliations
Contributions
W.Y., H.L., R.Z., S.R.P., and Q.W. contributed to idea of the project, study design, data collection, and statistical analysis, and wrote the manuscript with contributions from all authors. J.A.F., Y.H., R.J.H., Y.B., J.M., P.B., H.B., A.R., R.S H., N.E.C., M.T.L., I.B., A.R., D.C.C., C.I.A., and X.C. contributed to the acquisition of data, quality control of data, algorithms, and the manuscript review. S.R.P. and Q.W. contributed equally to this work as co-senior authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yang, W., Liu, H., Zhang, R. et al. Deciphering associations between three RNA splicing-related genetic variants and lung cancer risk. npj Precis. Onc. 6, 48 (2022). https://doi.org/10.1038/s41698-022-00281-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41698-022-00281-9
