
Abstract
Precision medicine is associated with favorable outcomes in selected patients with cancer. Herein, we report an interim analysis of IMPACT2, an ongoing randomized study evaluating genomic profiling and targeted agents in metastatic cancer. Patients with metastatic cancer underwent tumor genomic profiling (ClinialTrials.gov: NCT02152254), and 69 patients met the criteria for randomization. Tumor board and multidisciplinary review of molecular alterations optimized treatment selection. From 5/2014 to 4/2017, 320 patients (median age, 63 years; men, 47%) had tumor molecular aberrations, and 213 (66.56%) received anticancer therapy. The most frequently mutated genes were TP53 (42%), KRAS (16%), PIK3CA (12%), and CDKN2A (11%). The median OS was 10.9 months (95% CI, 8.8–12.9). OS was shorter in patients with higher tumor mutational burden. Independent factors associated with shorter OS were age ≥60 years, liver metastases, low albumin levels, high LDH levels, and KRAS and TP53 mutations. Outcomes for randomized patients will be reported after completion of the study.
Similar content being viewed by others
Introduction
In 2007, we initiated the first personalized medicine oncology program across tumor types, Initiative for Molecular Profiling and Advanced Cancer Therapy (IMPACT), to explore whether in patients with advanced metastatic cancer, the use of investigational agents matched with patient tumor molecular alterations would improve treatment outcomes compared to investigational agents not matched with patient tumor alterations. In three separate cohorts of patients with advanced cancer treated from 2007 to 2013 in our Phase I Program, the overall response rates (ORRs) in the matched targeted therapy (MTT) groups ranged from 11 to 27% compared to 5% in patients treated with non-matched therapy (NMT). The median progression-free survival (PFS) or time to treatment failure ranged from 3.4 to 5.2 months in the MTT groups and from 2.2 to 2.9 months in the NMT groups, and the median overall survival (OS) ranged from 8.4 to 13.4 months in the MTT and from 7.3 to 9 months in the NMT groups1,2,3. Thus, our collective experience with the personalized medicine approach was encouraging. Of 3487 patients who underwent molecular testing, 711 received MTT and 596 received NMT. The respective ORRs were 16.4 and 5.4%; the ORR plus stable disease ≥ 6 months rates were 35.3 and 20.3%; the median PFS durations were 4.0 and 2.8 months; and the median OS durations were 9.3 and 7.3 months. As this was the first large precision medicine study across tumor types, it has the longest follow up. The 10-year OS rates were 6% vs. 1%, respectively, for the MTT and NMT groups (HR = 0.72; p < 0.001), and matched targeted therapy was an independent factor predicting longer OS4.
To overcome the limitations of IMPACT associated with the retrospective analysis of outcomes of patients who were prospectively profiled, the small number of alterations tested, and the subjective treatment assignment (selected by the treating physician), we initiated IMPACT2, a prospective randomized study in personalized medicine. The primary objective of the study is to determine whether patients treated with a matched targeted therapy selected on the basis of genomic alteration analysis of the tumor have longer PFS from the time of randomization than those whose treatment is not selected on the basis of alteration analysis. In this preliminary analysis, we describe the results of molecular profiling of 320 patients who participated in the first part of the study and assessed the association between OS and patient characteristics and molecular alterations. This analysis provides insights that have implications for the development of cancer genome-based medicine.
Results
Patients
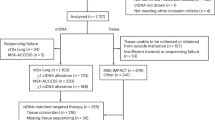
From May 2014 to April 2017, 391 patients were enrolled in the first part of the study, and 320 patients (81.84%) had detectable molecular abnormalities in their tumors. Seven patients had no abnormalities on tumor molecular testing. The remaining patients had inadequate tumor cells for analysis (n = 19), had disease that was non-measurable/biopsy that was not feasible (n = 15), withdrew consent (n = 12), had worsening performance status (PS) (n = 8), had no evidence of cancer (n = 2), were lost to follow-up (n = 2), had two tumor types (n = 1), or were ineligible for logistic reasons (n = 5). Overall, 69 of the 320 patients were randomized (Fig. 1).

Patient enrollment and randomization by time.
Demographics
The baseline clinical characteristics of the 320 patients are shown in Table 1. The median age was 63 years (range, 25–83 years). Fifty-three percent were women. Twelve percent of patients had PS 0 and 88% had PS 1. The median time from diagnosis to enrollment on the study was 25.6 months (95% CI, 21.8–29.8 months). The median number of prior therapies was three (range, 0–14). Overall, 95% of patients had received standard therapy. Forty percent of patients had liver metastases; 24% had elevated lactate dehydrogenase (LDH) levels (≥618 IU/L, the upper limit of normal [ULN]); 15% had abnormal (above or below the normal limits) platelet counts; and 8% had low albumin levels (<lower limit of normal [LLN]). The most common cancer types were head and neck, 19%; gastrointestinal, 16%; lung, 11%; gynecological, 9%; and colorectal, 9%.
Of 320 patients, 276 (86.25%) had molecular testing results using formalin-fixed, paraffin-embedded (FFPE) specimens derived from fresh tumor biopsies. The sites of tumor biopsy are summarized in Supplementary Table 1. For 44 (13.75%) patients, archival tumor tissue, obtained <2 years before enrollment on the study, was used. Of 276 patients, 22 patients had a second biopsy because tumor tissue was inadequate for analysis. The median time from enrollment to tumor biopsy was 8 days (range, 0–87 days); from tumor biopsy/shipment to Foundation Medicine to time of results, 20 days (range, 9–59 days); and from availability of results to initiation of treatment, 11 days (range, 5–52 days). The median time from enrollment to initiation of treatment was 2.3 months (95% CI, 1.9–2.9 months).
Molecular testing
The hotspot mutations are summarized in Supplementary Fig. 1. The median number of variants per tumor sample was three (range, 0–31). The most common variants were missense mutations, n = 741; amplifications, n = 138; nonsense mutations, n = 101; and copy losses, n = 83. The most common variant types were single nucleotide variants (SNVs), 74%; copy gains, 17%; and copy losses, 7%. In total, 245 genes were found to be mutated in at least one patient, and the most frequently mutated genes were TP53, 42%; KRAS,16%; PIK3CA,12%; and CDKN2A, 11%.
Next, we investigated the enriched abnormalities, including hotspot mutations and SNVs, according to the primary tumor types (Fig. 2). The proportion of patients with mutations in certain genes was calculated within specific tumor types and compared to the remaining tumor types. Alterations that were significantly enriched included APC, KRAS, and SMAD4 in patients with colorectal cancer (CRC); TP53 and CCNE1 in patients with ovarian cancer; PIK3CA and ESR1 mutations in patients with breast cancer; and CDK4 mutations in patients with sarcoma.

Enrichment of hotspot mutations per tumor type.
Statistically significant mutation interactions, representing the co-occurrence or exclusiveness of all the detected hotspot mutations, are shown in Fig. 3. For example, APC and KRAS showed strong co-occurrence. Additionally, FGF4 and FGF3 tended to co-occur with CCND1, FGF19, and CDKN2A across all patients. Notably, TP53 showed strong mutual exclusiveness with SDHA.

Interaction of hotspot mutations.
Treatment
Of the 69 patients who met the criteria for randomization, 60 patients received treatment and nine patients were not treated because their insurance would not cover the cost of the assigned treatment. Of the 251 patients who were not randomized, 153 (61%) were treated with investigational (n = 98; 64%) or standard (n = 55; 36%) therapy. Overall, 213 (66.56%) of the 320 patients with detectable molecular abnormalities received anticancer therapy. Fifty-six patients received treatment with immuno-oncology therapy (IO) and 157 received treatment that excluded immuno-oncology therapy (non-IO).
Overall survival
Of the 320 patients, 202 had died at the time of the survival analysis. The median OS duration was 10.9 months (95% C.I., 8.8–12.9) and the mean OS duration was 17.1 months (95% C.I., 15.1–19.1) (Supplementary Fig. 2). Results of univariate and multivariate analyses for OS are shown in Table 2. In the univariate analysis, factors that showed significant association with shorter OS were hepatic metastases, low (<LLN) albumin level, elevated (>ULN) LDH level, older age, KRAS mutations, TP53 mutations, CDKN2A mutations, P53 pathway abnormalities, PDGF signaling pathway abnormalities, apoptosis signaling pathway abnormalities, Ras pathway abnormalities, and T-cell activation pathway abnormalities. The following factors were not statistically significant in the univariate analyses: sex (p = 0.069), performance status (0 vs. 1; p = 0.095), number of prior therapies (continuous variable, p = 0.52; 0–3 vs. ≥4 lines of therapy, p = 0.15; Supplementary Fig. 3), number of metastatic sites (continuous variable, p = 0.15), platelet counts (continuous variable, p = 0.49), FGF/FGFR amplifications (p = 0.591), and tumor mutational burden (TMB) (continuous variable, p = 0.85). Longer time from diagnosis to enrollment on the study was associated with poorer OS (p < 0.0001) (Supplementary Fig. 4).
In the multivariate analysis, independent factors associated with shorter OS were age ≥60 years, liver metastases, low (<LLN) albumin levels, high (>ULN) LDH levels, and KRAS and TP53 mutations (Fig. 4 and Supplementary Table 2).

Independent risk factors predicting overall survival (multivariate analysis).
The association between patients’ OS and TMB is shown in Supplementary Fig. 5. The median OS durations for patients in the high, intermediate, and low TMB groups were 7.69, 10.68, and 15.25, respectively. The low TMB group had longer OS compared to the others (p = 0.008, hazard ratio = 0.59). There was no significant difference in OS between patients in the high TMB and intermediate TMB groups (p = 0.30; hazard ratio = 1.21). We also performed OS analysis in patients with head and neck cancer by TMB. The median OS by TMB group was as follows: high, 7.23 months (95% CI, 1.18–NA); intermediate, 11.90 months (95% CI, 8.22–NA); low, not reached (95% CI, 9.07–NA) (Supplementary Fig. 6). In the remaining patients, the median OS by TMB group was as follows: high, 8.58 months (95% CI, 5.88–14.50); intermediate, 9.67 months (95% CI, 7.79–13.22); low, 13.78 months (95% CI, 6.38–29.29) (Supplementary Fig. 7).
Since “head and neck” cancers and “gastrointestinal, other” cancers were the most common tumor types, we evaluated the distribution of TMB across all tumor types (Supplementary Fig. 8). Although “head and neck” and “gastrointestinal, other” represent the tumors with the highest numbers of patients, there was no statistical difference in the distribution of the TMB groups between tumor types (“head and neck” vs. remaining tumors, “gastrointestinal, other” vs. remaining tumors, or “head and neck” + “gastrointestinal, other” vs. remaining tumors).
OS by tumor type is shown in Supplementary Table 3 and Supplementary Fig. 9. The longest OS was observed in head and neck cancer (median, 21.1 months; 95% C.I., 8.5–33.6), followed by breast cancer (median, 18.8 months; 95% C.I., 5.7–31.8), sarcoma (median, 13.8 months; 95% C.I., 6.7–21.0), and lung cancer (median, 13.5 months; 95% C.I., 7.0–20.1). Patients with cancer of unknown primary had the shortest OS (median, 1.5 months; 95% C.I., 0.0–3.7).
There was a trend towards longer OS in patients treated with IO-containing therapy compared to those treated with non-IO containing therapy (p = 0.069; HR = 0.68). The median OS of patients who received IO-containing therapy was 21.9 months (95% CI, 13.5–33.7 months), and the median OS of patients who received non-IO treatment was 13.3 months (95% CI, 10.8–15.8 months) (Supplementary Fig. 10).
Discussion
IMPACT2 was initiated after the first IMPACT study demonstrated superior response, PFS, and OS for matched targeted therapy compared with unmatched therapy in consecutive patients who were referred for phase I clinical trials and had tumor molecular profiling. The aim of IMPACT2 is to assess the personalized medicine approach in a randomized study across tumor types using adaptive design to overcome the limitations of IMPACT. The execution of the trial is arduous and includes tumor biopsies for molecular testing, annotation of the genomic results, and availability of multiple clinical trials with matched and unmatched treatments. Other essential elements of IMPACT2 are randomization based on patient status on the date of clinic visit; real-time patient monitoring; efficient communication between patients, sponsors, and investigators; and accurate data collection and assessment of patient outcomes.
In this interim analysis, next-generation sequencing (NGS) testing using FFPE specimens from fresh tumor biopsies and treatment of patients with advanced metastatic cancer prospectively was feasible. Overall, 81.84% of the enrolled patients had detectable molecular abnormalities, and 66.56% of 320 patients received anticancer therapy. The median OS duration was 10.9 months (95% C.I., 8.8–12.9) and the mean OS duration was 17.1 months (95% C.I., 15.1–19.1). These survival data are compatible with previous reports on patients who were treated in our Phase I Program1,2,3,4,5.
The relatively small proportion (21.6%) of patients who met criteria for randomization was attributed to lack of clinical trials with targeted therapies against genetic alterations in individual patients; worsening performance status; ineligibility for clinical trials with targeted therapy, particularly due to comorbidities; and logistics issues (drugs were not provided at no cost to patients, as in the NCI MATCH or American Society of Clinical Oncology TAPUR [Targeted Agent and Profiling Utilization Registry] studies).
The median number of variants per tumor type (3, range, 1–31) and the distribution of the alterations (741 missense mutations, 138 amplifications, 101 nonsense mutations, and 83 copy losses) reflect the advanced stage and complexity of the patients’ tumors. Most alterations were SNVs (74%) followed by copy gains (17%) and copy losses (7%), with C > T being the most frequent base substitution. The large number of genes (n = 245) that were mutated in ≥1 patient emphasizes the need to develop effective targeted agents against each gene that may drive carcinogenesis in humans. The distribution of the most frequently mutated genes (TP53, 42%; KRAS, 16%; PIK3CA, 12%; and CDKN2A, 11%) is consistent with that in our published data in patients with advanced cancer referred for investigational therapy.
Findings of the enrichment analysis of hotspot mutations and CNVs per tumor type are in line with published data (APC, KRAS, and SMAD4 mutations in CRC; TP53 and CCNE1 in ovarian cancer; PIK3CA and ESR1 in breast cancer; and CDK4 in sarcoma [Fig. 2]). The co-occurrence of mutations may be associated with their presence in specific tumor types, i.e., the APC and KRAS alterations (p < 0.01) in CRC. A trend towards co-occurrence of FGF4 and FGF3 with CCND1, FGF19, and CDKN2A was also noted across all patients. The strong mutual exclusiveness (p < 0.01) of TP53 hotspot mutations (42% of 320 patients) and SDHA hotspot mutations (7% of 320 patients) may provide insights regarding the role of these genes in carcinogenesis (Fig. 3).
The duration of OS decreased as TMB increased (median: 15.25 months with low TMB; 10.68 months with intermediate; and 7.69 months with high; Supplementary Fig. 5). The longer OS of patients with low TMB compared to those with intermediate or high TMB is likely attributable to their relatively favorable tumor biology owing to fewer molecular abnormalities. Biomarkers for selection of immunotherapy and immuno-oncology clinical trials were limited at the time of initiation of IMPACT2, and therefore, very few patients received immunotherapy.
Whereas we used the top and bottom 20% of TMB values to define high and low TMB, respectively, other investigators have used different cut-off values6,7,8,9,10,11. High TMB has been defined as >23.1 mutations/Mb in patients with melanoma9; >100 mutations per tumor in patients with melanoma treated with antibodies against cytotoxic T-lymphocyte antigen 4 (CTLA-4)11; >10 mutations/Mb in patients with non-small cell lung cancer7; and ≥ 20 mutations/Mb in patients with various tumor types treated with immuno-oncology therapy10. In the latter study, high TMB was associated with better clinical outcomes compared to patients with lower TMB. High TMB has been associated with response to immune checkpoint inhibitors. Although the relatively small number of patients precluded robust analyses, and patients were treated on numerous clinical trials, the OS analysis by TMB demonstrated a better separation of the survival curves in the head and neck patient group compared to others (Supplementary Figs. 6 and 7). Caution is warranted in the interpretation of these results that indicate that the individual tumor types should be taken into consideration in assessing the role of TMB in OS. Taking into consideration our findings and published results, it is plausible that the clinical significance of TMB is at least partially associated with driver molecular alterations (that may impact clinical outcomes more than TMB), tumor type, test performance, cut-off point, and/or type of therapy, including immuno-oncology therapy. Ongoing clinical trials are testing the importance of this biomarker. Standardization and consensus about the use of TMB would be useful.
Independent factors associated with shorter OS in the multivariate analysis were elevated LDH levels (p < 0.0001), low albumin levels (p = 0.002), liver metastases (p = 0.02), age ≥60 years (p = 0.009), KRAS mutations (p < 0.0001), and P53 mutations (p = 0.025) (Fig. 4 and Supplementary Table 2). The first three factors indicate advanced disease and the first four are established markers associated with poorer outcomes. The association of KRAS and P53 mutations with shorter OS may be explained at least in part by the essential role of these biomarkers in carcinogenesis and the lack of effective targeted therapies against these alterations.
Several other trials have investigated the role of precision medicine in treating cancer. In SHIVA, a multicenter French trial, 293 of 741 enrolled patients had ≥1 molecular alteration and were treated with one of 11 targeted therapies. No difference in PFS was noted between the two arms, although the study design was suboptimal12,13. The TAPUR study is evaluating U.S. Food and Drug Administration-approved treatments in patients with advanced cancer and potentially actionable molecular alterations, providing real-world data. As of April 2020, nine arms of that study were expanded, and seven arms were closed14. NCI-MATCH, a phase II non-randomized trial, evaluates the clinical benefit of targeted treatments matched to tumor molecular alterations in patients with refractory malignancies15. Although few of the enrolled patients (16 of 645) were treated in the initial analysis, the study was amended to allow patients with clinical laboratory improvement amendments (CLIA)-certified NGS testing available at study entry. Some subprotocols demonstrated encouraging results, i.e., in patients with deficient mismatch repair and advanced non-colorectal tumors treated with nivolumab, the ORR was 36% (all partial responses) and the median OS was 7.3 months16. In patients with tumor AKT1 E17K mutation (0.77% frequency) treated with the pan-AKT inhibitor capivasertib, the ORR was 23% (all partial responses) and the 6-month PFS was 52%17. In contrast to IMPACT2, where the regulatory institutional committees required a change in the eligibility criteria from 0 to 2 prior therapies to enroll patients who have exhausted all standard options, NCI-MATCH enrolls patients with ≥1 standard systemic therapy and no other treatments available that are known to prolong OS18.
The strengths of IMPACT2 include the prospective nature of the study; the implementation of FFPE specimens derived from fresh tumor biopsies for NGS, which was not the standard of care when IMPACT2 was initiated; the inclusion of state-of-the-art NGS testing (315-gene panel, 27-gene amplification testing); and discussion of the clinical significance of NGS results at the study’s tumor molecular board and at a multidisciplinary conference to optimize treatment selection. The most important strength is access to a broad portfolio of cutting-edge early phase clinical trials against multiple targets offered by health care providers with expertise in drug development. The weaknesses of our trial include the variety of tumor types, the multiplicity and complexity of molecular alterations, the inherent limitations of treating advanced, metastatic cancer, and the variety of investigational agents that change over time. Although the availability and efficiency of molecularly driven studies increases over time, our study—like other clinical trials across tumor types—cannot systematically account for all differences in tumor biology and characteristics of individual patients for optimal treatment selection.
In conclusion, IMPACT2 establishes the feasibility of tumor biopsies for genomic profiling in patients with solid tumors—that was not the standard practice—and prospective treatment of patients. Tumor board and multidisciplinary review of molecular alterations and available clinical trials optimizes personalized treatment selection. In the study patient population, age <60 years, no liver metastases, normal albumin and LDH levels, and absence of KRAS or TP53 mutations were independent factors predicting longer OS. Outcomes for randomized patients will be reported after completion of the study, which continues to accrue patients. Our data contribute to evolving clinical research that offers comprehensive molecular testing to help select efficacious targeted therapy to accelerate drug approval. Optimization of biomarker testing using tumor and cell-free DNA and integration of innovative targeted therapeutic approaches will advance the landscape of precision oncology, enabling delivery of personalized care to more patients with cancer.
Methods
Eligibility criteria
Patients were eligible if they were ≥18 years of age and had advanced or metastatic cancer that was refractory to standard-of-care therapy, had declined to receive standard-of-care therapy, or had no standard-of-care therapy available for their tumor type. The study was registered in www.clinicaltrials.gov (NCT02152254). Patients could have received unlimited lines of prior therapy. Other eligibility criteria included a European Cooperative Oncology Group PS of 0-1 and adequate bone marrow (absolute neutrophil count ≥1000/µL; platelets ≥100,000/µL), hepatic (total bilirubin level ≤1.5 times the ULN, unless the patient has known Gilbert’s disease, and alanine aminotransferase/serum glutamic pyruvic transaminase levels ≤2.5 times the ULN without liver metastases), and renal (serum creatinine clearance ≥50 mL/min by the Cockcroft-Gault formula) function. Patients with brain metastases were eligible to participate in the study if the metastases were stable (treated and asymptomatic) and the patient was off steroids for at least 2 weeks. Patients with a previous malignancy (other than the patients’ known cancer) who were treated successfully and were disease-free for at least 3 years and patients with a history of basal cell carcinoma of the skin or pre-invasive carcinoma of the cervix were not excluded from the study. Women of childbearing potential were required to use adequate contraception (hormonal or barrier method of birth control; abstinence) prior to study entry and for the duration of study participation.
Exclusion criteria included anticancer treatment within 3 weeks of initiating study treatment, ≥grade 2 adverse events associated with prior therapy, uncontrolled hypertension, angina, ventricular arrhythmias, congestive heart failure (New York Heart Association Class ≥II), prior or current cardiomyopathy, atrial fibrillation with heart rate >100 bpm, unstable ischemic heart disease, peripheral neuropathy ≥grade 2, pregnancy, concurrent severe and/or uncontrolled medical disease that could compromise participation in the study (i.e., uncontrolled diabetes, severe infection requiring active treatment, severe malnutrition, chronic severe liver or renal disease), and any other condition that would, in the investigators’ judgment, contraindicate the patient’s participation in the clinical study due to concerns about safety or compliance with clinical study procedures. For oral therapy only, patients were excluded from the study if they had gastrointestinal diseases that would preclude adequate absorption.
Although the study was optimally designed to enroll patients with 0–2 prior therapies, the initial accrual rate was too low, prompting an amendment to allow patients with unlimited lines of therapy. Therefore, the original criterion allowing patients with 0–2 prior therapies was updated according to evolving institutional guidelines to require that the patient’s cancer was refractory to standard-of-care therapy, the patient declined to receive standard-of-care therapy, or there was no standard-of-care therapy available for the patient’s tumor type. All patients signed informed consent forms stating that they were aware of the investigational nature of the study. The study adhered to the guidelines of the Institutional Review Board, which approved the study. The study was activated in May 2014.
Tumor molecular profiling
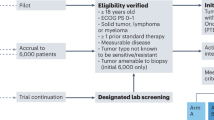
The study schema is shown in Fig. 5. Patients who were eligible for the study underwent tumor biopsy. Tumor samples were obtained by core biopsy performed by an interventional radiologist or bronchoscopy. FFPE specimens derived from fresh tumor biopsies were sent to Foundation Medicine for molecular profiling. It should be noted that at the sponsor’s request, the study was placed on hold in April 2017. Subsequently, patients enrolled from trial initiation until April 2017 constituted the “first part” of the study.

Study schema.
Next-generation sequencing
Prior to shipment to Foundation Medicine, tumor specimens were reviewed by an MD Anderson pathologist to ensure adequate tumor cellularity (≥20%) for analysis. All procedures were performed in a CLIA-compliant environment.
Patient samples were sequenced by Foundation Medicine, Inc. (Cambridge, MA), using FoundationOne CDx™, a comprehensive NGS-based in vitro diagnostic device designed to capture cancer genes. CDx™ detects substitutions, insertion and deletion alterations (indels), and copy number alterations in 324 genes and selected gene rearrangements in DNA isolated from FFPE tumor tissue samples. Genomic DNA was extracted from 40 µm of tissue using the Promega Maxwell 16 FFPE Plus LEV DNA Purification kit (Madison, WI) according to the manufacturer’s instructions and quantified using a standardized PicoGreen fluorescence assay (Invitrogen, Carlsbad, CA). At least 50 ng and up to 200 ng of extracted DNA was sheared to ~100–400 bp by sonication before end-repair, dA addition, and ligation of indexed, Illumina-sequencing adaptors (San Diego, CA). Sequencing libraries were hybridization-captured using a pool of >24,000 custom-designed and individually synthesized 5′-biotinylated DNA oligonucleotides (Integrated DNA Technologies, Coralville, IA). These baits were designed to target ~1.5 Mb of the human genome, including 4,604 exons of 324 genes related to cancer therapy, 47 introns of 19 genes frequently re-arranged in cancer, and 3549 SNPs throughout the genome. DNA sequencing was performed using the HiSeq-2000 instrument (Illumina), with 49 × 49 paired-end reads.
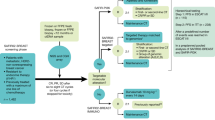
Treatment
Results of molecular testing as provided by Foundation Medicine were presented at weekly or bimonthly meetings of the study’s tumor board, which consisted of the study oncologists, statistician, radiologist, and molecular biologists. Patients were also presented at a weekly multidisciplinary conference to optimize treatment selection. Subsequently, they were seen in clinic and were randomized to receive matched targeted therapy selected on the basis of genomic alteration analysis or treatment not selected on the basis of alteration analysis. Treatment was also determined on the basis of whether the patient had ≥1 targetable alteration, clinical trials were available, the patient met eligibility criteria for the clinical trials under consideration, and insurance approved coverage of the associated cost. Stratification factors for randomization (determined in 2013) included alterations in the following genes: KRAS, BRAF, ERBB2 (Her2), EGFR, PIK3CA, PTEN, MET, or “Other” (i.e., remaining tumor alterations). Patients who did not meet the criteria for study randomization were treated with therapies chosen by their treating physician.
Exploratory analyses
Results, provided by Foundation Medicine at the time of completion of molecular testing, were used to guide therapy. We also performed the following exploratory analyses.
Variant annotation and pathway analysis
A customized workflow pipeline was applied to analyze the sequencing results; the pipeline was adapted from tools that are applied to cancer genome sequencing projects such as TCGA but implemented with further optimization for deep clinical sequencing. Briefly, we aligned the reads to human reference assembly hg19 using BWA and Picard. MuTect was used to identify somatic point mutations, and Pindel was used to identify somatic insertions and deletions. To eliminate artifact calls and germline contamination, a series of post-call filtering algorithms for somatic mutations were applied: (a) total read count in tumor sample ≥20, (b) log of odds score ≥20, (c) variant allele frequency ≥0.02 in tumor sample, and (d) population frequency threshold of 0.15% for filtering out common variants in the databases dbSNP, 1000 Genome Project, Exome Aggregation Consortium, and ESP6500. Mutations were annotated using ANNOVAR and hotspot mutations were annotated using the COSMIC database. The variants that passed the filtering but were not reported in COSMIC were annotated as variants of unknown significance. An unbiased pathway enrichment analysis was performed using the Panther pathway database (http://pantherdb.org/) (Supplementary Table 4). In this analysis, we evaluated the association between patient’s OS and patient tumor alterations on both the gene level and pathway level (Table 2). As the alterations on the pathway level are highly dependent on the genes included in the panel, our goal was to select the pathways without introducing any bias. Therefore, we performed the pathway enrichment analysis on the gene panel to identify the pathways that are reflected, and then we performed the univariate analysis on the pathways. The pathways reflected by this gene panel that were included in the univariate analysis are listed in Supplementary Table 4. Table 2 shows the gene/pathway alterations that were significantly associated with patient OS.
Tumor mutational burden analysis
Patients were divided into three groups according to the TMB identified in DNA sequencing. The TMB cut-off points were determined taking into consideration the following two criteria: (1) the burden of the high TMB group should be at least two-fold higher than that of the low TMB group; (2) the cutoff should reflect the curve of TMB distribution. Using these criteria, we set the cutoff of the top 20% (mutation load ≥18) as the high TMB group, the bottom 20% (mutation load <9) as the low TMB group, and the remaining as the intermediate TMB group. The majority of patients were in the intermediate TMB group.
Statistical methods
Response Evaluation Criteria In Solid Tumors (RECIST) guidelines were used to evaluate tumor response and disease progression every two cycles19,20. OS was calculated from the date of consent to the date of death from any cause or last follow-up. Cox regression analysis was used to determine the association between OS and patient pretreatment characteristics. The risk factors that were statistically significant in the univariate analysis were further selected for the multivariate analysis, using the backward stepwise selection elimination method (likelihood ratio). According to this method, removal testing is based on the probability of the likelihood-ratio statistic, which is based on the maximum partial likelihood estimates. The cut-off time for this analysis was June 2019.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The next-generation sequencing data generated during the study, are available in the European Genome-phenome Archive (EGA) (data are subject to controlled-access): https://identifiers.org/ega.dataset:EGAD00001006887 (dataset ID) and https://identifiers.org/ega.study:EGAS00001004964 (study ID)21. The dataset IMPACT2_supporting_data.xlsx, supporting Fig. 4, Tables 1 and 2, Supplementary Tables 1–3 and Supplementary Figs. 2–10, is part of the supplementary files that accompany the article.
For data inquiries, please contact the corresponding author Dr. Apostolia-Matia Tsimberidou, email address: atsimber@mdanderson.org. The data generated and analyzed during this study are described in the following metadata record: https://doi.org/10.6084/m9.figshare.1364342022.
References
Tsimberidou, A. M. et al. Personalized medicine in a phase I clinical trials program: the MD Anderson Cancer Center initiative. Clin. Cancer Res. 18, 6373–6383 (2012).
Tsimberidou, A. M. et al. Personalized medicine for patients with advanced cancer in the phase I program at MD Anderson: validation and landmark analyses. Clin. Cancer Res. 20, 4827–4836 (2014).
Tsimberidou, A. M. et al. Initiative for Molecular Profiling and Advanced Cancer Therapy (IMPACT): an MD Anderson Precision Medicine Study. JCO Precis. Oncol. Epub 2017 (2017).
Tsimberidou, A. M. et al. Long-term overall survival and prognostic score predicting survival: the IMPACT study in precision medicine. J. Hematol. Oncol. 12, 145 (2019).
Wheler, J. J. et al. Cancer therapy directed by comprehensive genomic profiling: a single center study. Cancer Res. 76, 3690–3701 (2016).
Alexandrov, L. B. et al. Signatures of mutational processes in human cancer. Nature 500, 415–421 (2013).
Ready, N. et al. First-line nivolumab plus ipilimumab in advanced non-small-cell lung cancer (CheckMate 568): outcomes by programmed death ligand 1 and tumor mutational burden as biomarkers. J. Clin. Oncol. 37, 992–1000 (2019).
Rizvi, N. A. et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348, 124–128 (2015).
Johnson, D. B. et al. Targeted next generation sequencing identifies markers of response to PD-1 blockade. Cancer Immunol. Res. 4, 959–967 (2016).
Goodman, A. M. et al. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol. Cancer Ther. 16, 2598–2608 (2017).
Snyder, A. et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 371, 2189–2199 (2014).
Le Tourneau, C. et al. Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): a multicentre, open-label, proof-of-concept, randomised, controlled phase 2 trial. Lancet Oncol. 16, 1324–1334 (2015).
Tsimberidou, A. M. & Kurzrock, R. Precision medicine: lessons learned from the SHIVA trial. Lancet Oncol. 16, e579–e580 (2015).
Mangat, P. K. et al. Rationale and design of the Targeted Agent and Profiling Utilization Registry (TAPUR) study. JCO Precis. Oncol. Epub 2018 (2018).
Flaherty, K. T. et al. The Molecular Analysis for Therapy Choice (NCI-MATCH) trial: lessons for genomic trial design [published online ahead of print Janurary 10, 2020]. J. Natl. Cancer Inst. 112, djz245 (2020).
Azad, N. S. et al. Nivolumab is effective in mismatch repair-deficient noncolorectal cancers: results from arm Z1D-A subprotocol of the NCI-MATCH (EAY131) study. J. Clin. Oncol. 38, 214–222 (2020).
Kalinsky, K. et al. AZD5363 in patients (Pts) with tumors with AKT mutations: NCI-MATCH subprotocol EAY131-Y, a trial of the ECOG-ACRIN cancer research group (EAY131-Y). Eur. J. Cancer 103, e15 (2018).
Clinicaltrials.gov. Targeted Therapy Directed by Genetic Testing in Treating Patients With Advanced Refractory Solid Tumors, Lymphomas, or Multiple Myeloma (The MATCH Screening Trial). Accessed August 2020; https://www.clinicaltrials.gov/ct2/show/NCT02465060.
Eisenhauer, E. A. et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur. J. Cancer 45, 228–247 (2009).
Therasse, P. et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J. Natl. Cancer Inst. 92, 205–216 (2000).
Tsimberidou, A. M. et al. Preliminary results from the Initiative for Molecular Profiling and Advanced Cancer Therapy 2 (IMPACT 2) study. Eur. Genome-phenome Arch. https://identifiers.org/ega.study:EGAS00001004964 (2021).
Tsimberidou, A. M. et al. Metadata supporting the article: precision medicine: preliminary results from the Initiative for Molecular Profiling and Advanced Cancer Therapy 2 (IMPACT 2) study. figshare https://doi.org/10.6084/m9.figshare.13643420 (2021).
Acknowledgements
The authors wish to thank Henry (Hiep) Vo, PhD, for editorial assistance with the manuscript. This work was primarily supported by a research grant from Foundation Medicine to Dr. Tsimberidou (Institution) and from donor funds from Jamie’s Hope, Mr. and Mrs. Zane W. Arrott, and Mr. and Mrs. Steven McKenzie for Dr. Tsimberidou’s Personalized Medicine Program. This work was also supported in part by the National Institutes of Health/National Cancer Institute award number P30 CA016672 (University of Texas MD Anderson Cancer Center), the Clinical Translational Science Award 1UL1 TR003167; the Cancer Prevention Research Institute of Texas (CPRIT) Precision Oncology Decision Support Core (RP150535), and the Sheikh Khalifa Bin Zayed Al Nahyan Institute for Personalized Cancer Therapy.
Author information
Authors and Affiliations
Contributions
A.M.T. (principal investigator) conceptualized the paper, obtained funding and provided oversight, contributed to patient enrollment and treatment, patient assessment, data analysis, data interpretation, and writing the manuscript. D.S.H., S.F., D.D.K., S.P.-P., M.S.K., V.R., V.S., S.M.P., S.-M. T, F.J., J.H., and F.M.-B. contributed to patient assessment and treatment. A.J. contributed to data interpretation, C.C contributed to patient enrollment and data acquisition. L.Z., J.Z., D.J.V., and A.F. contributed to data analysis, and V.A.M provided molecular profiling data. D.A.B. contributed to the statistical design of the paper. All authors read, reviewed the manuscript, and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
Apostolia M. Tsimberidou: Clinical Trial Research Funding (received through the institution) IMMATICS, Parker Institute for Cancer Immunotherapy, Tempus, OBI Pharma, EMD Serono, Baxalta, ONYX, Bayer, Boston Biomedical, Placon Therapeutics, Karus Therapeutics, Tvardi, CPRIT; Travel, Accommodations, Expenses: ASCO, Genentech, Covance, Tempus; Consulting or Advisory Role: Covance, Genentech, Tempus David S Hong Disclosures: (last 36 months) Research/Grant Funding (received through the institution): AbbVie, Adaptimmune, Aldi-Norte, Amgen, Astra-Zeneca, Bayer, Bristol-Myers Squibb, Daiichi-Sankyo, Eisai, Fate Therapeutics, Genentech, Genmab, GlaxoSmithKline, Ignyta, Infinity, Kite, Kyowa, Lilly, LOXO, Merck, MedImmune, Mirati, miRNA, Molecular Templates, Mologen, NCI-CTEP, Novartis, Pfizer, Seattle Genetics, Takeda, Turning Point Therapeutics; Travel, Accommodations, Expenses: Bayer, Genmab, AACR, ASCO, P.O.E.T, CCLO, SITC; Consulting or Advisory Role: Alpha Insights, Amgen, Axiom, Adaptimmune, Baxter, Bayer, eCancer, Genentech, GLG, Group H, Guidepoint, Infinity,Liberium, Medscape, Numab, Oncology Education Project Association, Pfizer, Prime Oncology, Takeda, Trieza Therapeutics, WebMD. Other ownership interests: Molecular Match (Advisor), OncoResponse (Founder), Presagia Inc (Advisor) Siqing Fu: Clinical Trial Research/Grant Funding (Received through the Institution): AstraZeneca; Abbisko, Anaeropharma Science; Arrien Pharmaceuticals; BeiGene; BioAtla, LLC; Boehringer Ingelheim; Eli Lilly & Co.; Hookipa Biotech; Huya Bioscience International; IMV, Inc.; Innovent Biologics, Co., Ltd.; MacroGenics; Medivir AB; Millennium Pharmaceuticals, Inc.; Nerviano Medical Sciences; NeuPharma, Inc.; Novartis; OncoMed Pharmaceuticals; Parexel International, LLC; Sellas Life Sciences Group; Sorcimed Biopharma, Inc.; Tolero Pharmaceuticals; National Institutes of Health/National Cancer Institute; National Cancer Institute/National Institutes of Health P30CA016672 – Core Grant (CCSG Shared Resources) Daniel Dr. Karp: Research/Grant Funding (received through the institution): National Institutes of Health/National Cancer Institute; Myriad Drug Companies. Consulting or Advisory Role: BBLS; Affigen Advisory Board; Black Beret Life Scientist; Phosplatin Therapeutics Advisory Board Sarina A. Piha-Paul: Research/Grant Funding (Received through the Institution): AbbVie, Inc.; ABM therapeutics, Inc; Acepodia, Inc; Alkermes, Inc.; Aminex Therapeutics; Amphivena Therapeutics, Inc.; BioMarin Pharmaceutical, Inc; Boehringer Ingelheim; Bristol Myers Squib; Chugai Pharmaceutical Co., Ltd; Daichi Sankyo, Inc.; Eli Lilly; Five Prime Therapeutics; Genmab A/S; GlaxoSmithKline; Helix BioPharma Corp.; Incyte Corp.; Jacobio Pharmaceuticals Co., Ltd.; Medimmune, LLC.; Medivation, Inc.; Merck Sharp and Dohme Corp.; Novartis Pharmaceuticals; Pieris Pharmaceuticals, Inc.; Pfizer; Principia Biopharma, Inc.; Puma Biotechnology, Inc.; Rapt Therapeutics, Inc.; Seattle Genetics; Taiho Oncology; Tesaro, Inc.; TransThera Bio; National Cancer Institute/National Institutes of Health P30CA016672 – Core Grant (CCSG Shared Resources) Vinod Ravi: Consulting or Advisory Role: Daiichi Sankyo, Inc.; Marvin Health, Inc. Other Ownership Interests: UpToDate Vivek Subbiah: Research funding/Grant support for clinical trials (received through the institution): Roche/ Genentech, Novartis, Bayer, GlaxoSmithKline, Nanocarrier, Vegenics, Celgene, Northwest Biotherapeutics, Berghealth, Incyte, Fujifilm, Pharmamar, D3, Pfizer, Multivir, Amgen, Abbvie, Alfa-sigma, Agensys, Boston Biomedical, Idera Pharma, Inhibrx, Exelixis, Blueprint medicines, Loxo oncology, Medimmune, Altum, Dragonfly Therapeutics, Takeda and, National Comprehensive Cancer Network, NCI-CTEP and UT MD Anderson Cancer Center, Turning point therapeutics, Boston Pharmaceuticals; Travel: Novartis, Pharmamar, ASCO, ESMO, Helsinn, Incyte; Consultancy/ Advisory board: Helsinn, LOXO Oncology/Eli Lilly, R-Pharma US, INCYTE, QED pharma, Medimmune, Novartis. Other: MedscapeShi-Ming Tu: Consulting or Advisory Role: Janssen Biotech (Advisor) Filip Janku: Research/Grant Funding (received through the institution): support from Novartis, Genentech, BioMed Valley Discoveries, Astellas, Astex, Agios, Bicara, Bioxcel Therapeutics, Plexxikon, Deciphera, Piqur, Symphogen, Bristol-Myers Squibb, Asana, Merck, Ideaya Biosciences, JS Innophram, Synthorx, Sanofi, SpringBank Pharmaceuticals, SQZ Biotechnologies, Synlogic, FujiFilm Pharmaceuticals, Sotio, Novellus and Proximagen; is or has been on the Scientific Advisory Boards for Bicara, Guardant Health, Illumina, Ideaya Biosciences, IFM Therapeutics, Synlogic, Sotio, Puretech Health, Petra Pharma, Novellus and Deciphera; is a paid consultant for Cardiff Oncology and Immunomet; and has ownership interests in Cardiff Oncology. John Heymach: Advisory Committee Participation: ARIAD, AstraZeneca, Abbvie, Boehringer Ingelheim, Bristol-Myers Squibb, Calithera Biosciences, Catalyst, EMD Serono, Foundation Medicine, Hengrui Therapeutics, Genentech, Gritstone, GSK, Guardant Health, Eli Lilly, Medivation, Merck, Novartis, Oncomed, Pfizer, Roche, Sanofi, Seattle Genetics, Spectrum, Synta, Takeda. Clinical Research/Grant Funding: AstraZeneca, GlaxoSmithKline, Spectrum. Royalties and Licensing fees: Spectrum. Donald Berry: Other ownership interests: co-owner of Berry Consultants, LLC, a company that designs adaptive Bayesian clinical trials for pharmaceutical and medical device companies, National Institutes of Health cooperative groups, international consortia, and patient advocacy groups. David J. Vining: Other ownership interests: VisionSR, Majority owner and CEO, multimedia structured reporting for use in advancing medical research; Bracco Diagnostics, Inventor, virtual colonoscopy-related products for colorectal cancer imaging Vincent A. Miller: Patent Royalties: Memorial Sloan Kettering Cancer Center, USPO 850141. Stock Holder: Foundation Medicine/ROCHE; EQRX; Mirati Therapeutics. Other Compensation: Board of Directors, Revolution Medicines; EQRX Employee Funda Meric-Bernstam: Consulting: Aduro BioTech Inc., Alkermes, DebioPharm, eFFECTOR Therapeutics, F. Hoffman-La Roche Ltd., Genentech Inc., IBM Watson, Jackson Laboratory, Kolon Life Science, OrigiMed, PACT Pharma, Parexel International, Pfizer Inc., Samsung Bioepis, Seattle Genetics Inc., Tyra Biosciences, Xencor, Zymeworks; Advisory Committee: Immunomedics, Inflection Biosciences, Mersana Therapeutics, Puma Biotechnology Inc., Seattle Genetics, Silverback Therapeutics, Spectrum Pharmaceuticals, Zentalis; Sponsored Research (received through the institution): Aileron Therapeutics, Inc. AstraZeneca, Bayer Healthcare Pharmaceutical, Calithera Biosciences Inc., Curis Inc., CytomX Therapeutics Inc., Daiichi Sankyo Co. Ltd., Debiopharm International, eFFECTOR Therapeutics, Genentech Inc., Guardant Health Inc., Millennium Pharmaceuticals Inc., Novartis, Puma Biotechnology Inc., Taiho Pharmaceutical Co; Honoraria: Chugai Biopharmaceuticals, Mayo Clinic, Rutgers Cancer Institute of New Jersey; Other (Travel Related): Beth Israel Deaconess Medical Center. All remaining authors have declared no conflicts of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tsimberidou, A.M., Hong, D.S., Fu, S. et al. Precision medicine: preliminary results from the Initiative for Molecular Profiling and Advanced Cancer Therapy 2 (IMPACT2) study. npj Precis. Onc. 5, 21 (2021). https://doi.org/10.1038/s41698-021-00159-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41698-021-00159-2
This article is cited by
-
Omics-based molecular classifications empowering in precision oncology
Cellular Oncology (2024)
-
Molecular tumour boards — current and future considerations for precision oncology
Nature Reviews Clinical Oncology (2023)
-
Clinical trial design in the era of precision medicine
Genome Medicine (2022)
-
Challenges and opportunities associated with the MD Anderson IMPACT2 randomized study in precision oncology
npj Precision Oncology (2022)
