
Abstract
The hydroxyl (OH) radical is the key oxidant in the global atmosphere as it controls the concentrations of toxic gases like carbon monoxide and climate relevant gases like methane. In some regions, oxidation by chlorine (Cl) radical is also important, and in the stratosphere both OH and Cl radicals impact ozone. An empirical method is presented to determine effective OH concentrations in the troposphere and lower stratosphere, based on CH4, CH3Cl, and SF6 data from aircraft measurements (IAGOS-CARIBIC) and a ground-based station (NOAA). Tropospheric OH average values of 10.9 × 105 (σ = 9.6 × 105) molecules cm−3 and stratospheric OH average values of 1.1 × 105 (σ = 0.8 × 105) molecules cm−3 were derived over mean ages derived from SF6. Using CH4 led to higher OH estimates due to the temperature dependence of the CH4 + OH reaction in the troposphere and due to the presence of Cl in the stratosphere. Exploiting the difference in effective OH calculated from CH3Cl and CH4 we determine the main altitude for tropospheric CH4 oxidation to be 4.5 ~ 10.5 km and the average Cl radical concentration in the lower stratosphere to be 1.1 × 104 (σ = 0.6 × 104) molecules cm−3 (with a 35% measurement uncertainty). Furthermore, the data are used to examine the temporal trend in annual average stratospheric OH and Cl radical concentrations between 2010 and 2015. The year 2013 showed highest stratospheric OH and lowest Cl but no clear temporal trend was observed in the data in this period. These data serve as a baseline for future studies of stratospheric circulation changes.
Similar content being viewed by others
Introduction
The hydroxyl radical (OH) is the most important oxidant in the troposphere and lower stratosphere. It initiates removal from the atmosphere of toxic gases such as carbon monoxide (CO), radiatively active gases such as methane (CH4), tropospheric ozone precursors such as volatile organic compounds (VOCs), and NOx (NO + NO2), and stratospheric ozone-depleting compounds such as hydrochlorofluorocarbons (HCFCs).1,2 Therefore, it plays a key role in the atmospheric oxidation capacity, air quality, and climate.3
One source of atmospheric OH is the reaction of O1D, a minor product in the photolysis of ozone, with H2O.4,5 An even larger source in terms of gross OH formation is recycling from its reaction products, which maintains the atmospheric oxidation efficiency.6 Despite its low ambient concentration (<1 pptv), and its short atmospheric lifetime (<1 s), the first valid OH measurements (by fluorescence assay with gas expansion, FAGE) were reported as early as the mid-1980s at ground level.7 Measurements were later extended to the upper troposphere and lower stratosphere with balloons8 and the high-altitude ER2 research aircraft,9 in parallel with alternative measurement methods such as differential optical absorption spectroscopy DOAS,10 mass spectrometry via H234SO4 (ref. 11), and 14CO radioactive counting techniques.12 These in-situ measurements are broadly consistent with regional scale indirect OH determinations using the depletion of hydrocarbons over multi-hour timescales13,14, and with empirical assessments based on the variability-lifetime relationship.15,16
Although OH concentrations at a given point in space and time can now be measured reliably at high frequency to examine local photochemistry, it is difficult to relate such OH concentrations that vary strongly with actinic flux and H2O, to longer lived species transported zonally in the troposphere and eventually into the stratosphere. To address this, advanced models have been applied to make indirect estimates of the global OH concentration based on methyl chloroform (CH3CCl3) and 14CO data.2,3,6,17,18,19 However, such models inevitably contain in-built assumptions, including uncertain emissions inventories as well as transport and deposition parameterizations that may differ from real world conditions.
In this study, we have developed and applied an empirical data-based method to estimate the “effective OH concentration” that has acted in the troposphere and lower stratosphere over longer (yearly) timescales between 2008–2015. Our approach uses long-term measurements of three species (SF6, CH3Cl, and CH4) made at the ground and monthly at 10–12 km altitude during long distance commercial aircraft flights. SF6 measurements are used to derive the mean air age of the airborne samples, i.e., of the trace compounds contained therein, so that the initial surface mixing ratios of CH3Cl and CH4, upon emission, can be determined from the surface network and compared with the OH affected samples taken at altitude. Knowing the age of the sampled air, the net change in concentration and the reaction rate coefficient allows an “effective OH” concentration to be calculated assuming OH is the only sink. Both CH3Cl and CH4 are predominately removed from the atmosphere by OH although reaction rates are relatively slow (atmospheric lifetimes are about 1 year20 and 8–10 years18, respectively). CH3Cl is mostly emitted from tropical vegetation21 and is the most abundant natural source of stratospheric chlorine.22,23 The greenhouse gas CH4 is emitted from wetlands, ruminants, rice fields, landfills and fossil fuel use, and atmospheric concentrations have increased strongly over the past 200 years.24,25 In the context of this study an important difference between CH3Cl and CH4 is that the rates of reaction with OH and Cl are much more dependent on temperature in the case of CH4. Using this data-based approach we may empirically determine a representative annual “effective OH concentration” for the troposphere and the lower stratosphere. Furthermore, we may look for indications of temporal trends in OH and hence changes in global oxidation capacity. Finally, by exploiting the reaction rate differences between CH3Cl and CH4 we can even attempt to determine the height in the troposphere where CH4 oxidation by OH is largest and estimate Cl radical concentration in the lowermost stratosphere.
Results
OH concentration as a function of mean age
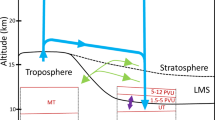
The derived “effective OH concentration” experienced by an air parcel will be hereafter shortened to OHeff, and depending on the air age be referred to as tropospheric OHeff (age < 100 days) and stratospheric OHeff (age > 200 days). The rationale is that an air sample collected in the upper troposphere—lower stratosphere (UTLS) region can be regarded as a mixture of two major large-scale airflows.26,27,28 The first is the fast transport of air from the tropical tropopause layer (TTL) to the extra tropics (Fig. 1 blue lines), which normally takes place within 0.3 years (~ 100 days), while the second pathway (Fig. 1, red lines) is the slower downwelling transport from the “overworld” (potential temperature > 380 K) into the lowermost stratosphere, which is associated with the lower branch of the Brewer-Dobson circulation (BDC). Note that the compounds within the lower stratosphere of the northern hemisphere are predominately influenced by the BDC and not the northern mid-latitude emissions (~ 10%).29 In order to simplify interpretation of the results and to apply our analysis to the region of highest airborne data coverage we use only samples collected between 30°–60°N in the UTLS region (Fig. 1, box).

a A schematic representation of the major atmospheric transport pathways associated with CARIBIC samples between 30°–60°N. The blue arrows indicate fast transport from ground to the tropical tropopause, the red arrows indicate downward transport from the stratosphere into the lowermost stratosphere. The box indicates CARIBIC flight sampling altitude. The NOAA observation station at Mauna Loa (MLO) is marked in blue. b Scatterplot of all samples’ latitude vs. potential temperature, color-coded with mean age
Figure 2 shows the OHeff derived from both CH3Cl (black boxes) and CH4 (red boxes) and OHr (real OH, blue boxes) derived from OHeff and stratospheric Cl (eq. 4, see Methods) as a function of mean air age. The tropospheric (mean age < 100 days) mean OHeff from both species is significantly larger (by a factor of 6 on average) than the lower stratospheric (mean age > 200 days) mean OHeff. Air with a mean age of between 100–200 days appears to be influenced by both troposphere and stratosphere, and accordingly OHeff for both species in this age range lies between the younger (tropospheric) and older (stratospheric) values. Median tropospheric OHeff were 9.93 × 105 and 2.63 × 106 molecules cm−3 for CH3Cl and CH4, respectively, whereas median stratospheric values from 200–1100 days were 1.69 × 105 and 3.35 × 105 molecules cm−3. Tropospheric OHeff values exhibit a much larger variability than stratospheric values, likely due to the strong sources and sinks of both molecules in the tropical troposphere, including weak seasonal variations that impact the term [A]g − [A]C in Eq.1. Stratospheric OHr exhibits smaller variation compared to stratospheric OHeff and is rather constant for samples with mean age between 200 and 700 days with a median value of 1.0 × 105 molecules cm−3, whereas for samples older than 700 days, OHr decreases due to the decrease of ozone, water vapor, and molecule density in the upper stratosphere. Median, average, and standard deviation data are given numerically in Table 1(a).

OH abundance (OHeff, OHr) derived from CH3Cl and CH4 for different mean age groups over all years (2008–2015 for OHeff, 2010–2015 for OHr), the inserted figure excludes the troposphere to expand the concentration scale. Box plots with the median (horizontal solid line in the box) and the mean (square in the box) give the 25 and 75% percentiles and whisker in dashed line indicates one standard deviation. Number of samples > 12 for each group
Interestingly, OHeff calculated using the CH4 data was consistently higher than using CH3Cl data. The tropospheric median OHeff derived from CH3Cl for years 2008–2015 is 9.9 × 105 molecules cm−3, which matches the tropospheric mean OH concentration of 1.1 × 10 6 molecules cm−3 reported from a recent global modeling study.6 However, the median tropospheric OHeff derived from CH4, 2.6 × 106 molecules cm−3, is between two and three times larger. Intuitively this seems unreasonable since both measurements stem from the same air sample and, therefore, must render the same “effective OH concentration” assuming there is no additional reagent inputs. One possible explanation for this apparent discrepancy is that loss processes besides OH remove CH4 from the atmosphere but are then calculated as OH in this approach. For example, the reaction with chlorine radicals (Cl) with CH4 has been shown to be significant in Asian pollution outflow.30 The aforementioned paper reported the presence of radicals in the ratio [Cl]:[OH] of 9 ~ 16 Cl:103 OH, and since the reaction rate of CH4 with Cl is ~ 32 times faster than that with OH (for CH3Cl it is only ten times faster),31 an apparent overestimation of OHeff by about 30–50% can ensue. However, such high Cl radical concentrations have only been seen under very specific conditions (lofted coastal pollution outflow) and so this cannot explain the OHeff difference in the global tropospheric data. An alternative explanation for the discrepancy is the strong temperature dependence of reaction 2, as CH4 oxidation by OH occurs much more rapidly in the warmer lower atmosphere. Assuming that the OHeff from CH3Cl is correct (as it is lower and in agreement with previous global estimates), we may determine at which temperature (and therefore which altitude) CH4 oxidation mainly occurs. By this method, we calculate that most tropospheric oxidation of CH4 occurs at circa 7 km (4.5 ~ 10.5 km, 220 ~ 260 K). OH radical abundance reaches an optimum just above the boundary layer (2–4 km) where the water concentration and photon fluxes are high and the flux of reactive sink species from the surface is decreasing. In addition, a second OH optimum occurs in the outflow region of clouds (10 km)32 where uplifted and lightning generated NOx enhances OH levels through the reaction of HO2 with NO. Therefore the 4.5 ~ 10.5 km height derived in this study likely represents an average of the effects of both OH maxima regions. This empirical estimate concurs with the model derived report that OH concentrations are at maximum in the free troposphere owing to recycling by NOx.6 A recent global modeling study using comprehensive chemistry6 suggests that OH is relatively high in the 4.5–10.5 km altitude range, which was attributed to OH recycling rather than primary formation. The model derived distribution is supported by this work.
Stratospheric Cl radical
Figure 3 shows the annual Cl radical concentration derived from air samples with age larger than 200 days in the stratosphere over the time period 2010–2015. The average Cl concentration over all periods is 1.1 (±0.6) × 104 molecules cm−3. Even though this result is five times higher compared to a 24 h average Cl concentration of 2.40 × 103 molecules cm−3 for 14–18.5 km height,33 it is more comparable to lower stratospheric Cl concentration of 5 × 103–3 × 104 molecules cm−3 estimated by CO/C2H6 ratio.34 Our result is also in close agreement with a recent model estimation35 based on isotopic ratios of methane and CO that yielded 1.6 × 104 molecules cm−3 for the lowermost stratosphere. No significant variation during this timeframe is observed in our dataset, although in 2013 Cl concentrations were lower than in the other years. Note that due to other minor loss processes (e.g., reaction with O1D, photodissociation36) in the stratosphere and mesosphere for both molecules, there is a slight overestimation of stratospheric Cl concentration. Mean, median, and one standard deviation data are given in Table 1(b).

Annual trend (2010–2015) of stratospheric chlorine radical concentration derived from samples with mean age larger than 200 days. Box plots with the median (horizontal solid line in the box) and the mean (square in the box) give the 25 and 75% percentiles and whisker in dashed line indicates one standard deviation. Number of samples > 12 for each group
Stratospheric OH radical
Figure 4 presents the annual average stratospheric OHr as a function of sampling year rather than the mean air age shown in Fig. 2. Data are from samples with age larger than 200 days to be classed as stratospheric. The overall stratospheric OHr of 1.1 (±0.8) × 105 molecules cm−3 for the years 2010–2015 is derived. Median average OH values generally vary within 0.8 – 1.3 × 105 molecules cm−3 with an exceptionally low median value of 5.4 × 104 in the year 2014, statistical details see Table 1(c). No clear trend is apparent in the dataset, and the highest value was found for 2013.

Annual trend (2010–2015) of stratospheric OHr concentration derived from samples with mean age larger than 200 days. Box plots with the median (horizontal solid line in the box) and the mean (square in the box) give the 25 and 75% percentiles and whisker in dashed line indicates one standard deviation. Number of samples > 12 for each group
Discussion
This study presents a new empirical method of monitoring the effective OH concentration in the global troposphere and lower stratosphere over long periods. The method also delivers the mean altitude range for tropospheric CH4 oxidation and the effective Cl radical concentration in the lower stratosphere. These parameters are all useful measures of the overall atmospheric oxidizing capacity and markers for future circulation changes. Provided that high quality, long-term monitoring of these three gases continues, the impact of future global events can be assessed in these terms. For example, a volcanic eruption, a change in global CFC emission rates, or a change in stratospheric circulation patterns can lead to changes in ultraviolet radiation, stratospheric chlorine loading, and water vapor, all of which can significantly impact the derived metrics.
This method is built on three key assumptions. The first is that both CH3Cl and CH4 are predominantly oxidized by OH. While this is true for the troposphere and the lower stratosphere, over longer timescales (>500 days) air masses can be expected to also enter the upper stratosphere as part of the Brewer-Dobson circulation. At these higher altitudes a minor photolysis sink for CH3Cl and the reaction of O1D radicals with CH4 could lead to a small overestimation of the calculated effective OH. Indeed, in Fig. 2 a tendency to higher OHeff in older air can be seen in the CH4 derived OH results. By only considering data within certain age ranges (0–100 days troposphere and 200–400 days lower stratosphere) the impact of additional loss mechanisms is limited. The second assumption is that this time segregation does delineate troposphere (0–100 days), mixed troposphere–stratosphere (100–200 days) and stratosphere (>200 days). This assumption is supported by the trend in OH variability measured in the three categories. The third assumption made here is that the tropospheric effective OH using CH3Cl is correct and CH4 derived OH is high because of either Cl radicals (in the stratosphere) or the temperature dependence of the reaction (in the troposphere). Support for this assumption comes from the fact that the CH3Cl estimate is lower than that of CH4 (additional chemistry leads to overestimation), no other significant loss rates are known, and the CH3Cl estimate more closely matches the most recent global model studies.
Uncertainties and sensitivities of this method are important to consider. Different to variations which show statistical distributions of the results, uncertainties are the differences between the measured values and the true values, whereas sensitivities represent how much output values will be affected by the input values. Firstly, there is an instrumental uncertainty. By considering the measurement errors of SF6, CH4, CH3Cl measurements at ground level and by aircraft and reaction rates kOH and kCl, measurement uncertainties are calculated by error propagation for mean age, OHeff derived from CH4 and CH3Cl, stratospheric Cl and stratospheric OHr of 8%, 10%, 22%, 35%, and 37%, respectively. Secondly the choice of the source region could influence the calculation. An ideal tracer for the mean age calculation should be well mixed in the source region so that its mixing ratio time series can be used to derive initial concentrations. Since SF6 is not perfectly well mixed globally (i.e., there is a weak NH/SH gradient and a slight latitude dependence in each hemisphere), an average mixing ratio should be applied to represent the source region. Therefore, we chose the average SF6 mixing ratio in the northern hemisphere (NH) as determined by the NOAA network since our samples were collected in northern mid-latitudes. The mean age results were also derived from individual source regions, namely Mauna Loa (MLO, 19.5°N), Cape Matatula (SMO, 14.3°S), Niwot Ridge (NWR, 40.052°N), Pt. Barrow (BRW, 71.3°N), Mace Head (MHD, 53°N) and 20°S to 20°N region, but the overall average NH SF6 observation gave the most positive mean age values for our aircraft measurements. A ± 0.05 ppt difference of SF6 mixing ratios in different tropospheric source regions corresponds to 0.17 years difference for the derived mean age. Thirdly, we consider the sensitivity of calculation to the temperature-dependent reaction rates. We use two temperatures (216 and 250 K) to represent the average temperatures in the troposphere and stratosphere. By varying these temperatures by ±5 K, the reaction rate of the reaction CH3Cl + OH is less sensitive (variation of −14 ~ 11%) than that of the reaction CH4 + OH (variation of −17 ~ 33%) (see Table S1 in the Supplementary Material). Fourthly, sensitivity of seasonal cycles of CH4, CH3Cl in source region. Both CH4, CH3Cl have seasonal cycles in the troposphere with amplitudes of ca. 1% and 5%, respectively. When examining their temporal concentration in the lowermost stratosphere (Fig.S2 in the Supplementary Material) with the N2O concentration,37 seasonal variations of CH4 have been damped out (linear correlation in each N2O group) while those of CH3Cl are attenuated but still visible. By varying ±5% of CH3Cl in the source region, the OHeff of samples with age ~ 70 days and larger than 700 days change ~ 80% and ~ 25%, respectively. Finally, in this study OH is calculated as a second-order reaction (see Eqs. 2 and 3 in Method). Alternatively one can use the pseudo first order approach in which OH is assumed to be constant Ac = Ag*exp( – Γ*k*OH) (6) and this produces fractionally higher values (<1%).
It should be noted that the now well established methyl chloroform (MCF)-derived global mean OH requires several important assumptions, for example, concerning MCF emissions (at least before 2000), ocean uptake and stratospheric loss. Furthermore, MCF is declining rapidly as emissions have stopped, and it does not provide height information, nor indications about Cl abundance. Other relatively long-lived halocarbons have been considered to complement the MCF method, but these OH estimates are also dependent on the source estimates. Therefore, our new, independent method should be acknowledged as an important complementary source of information on OH and Cl.
The radical abundances derived here represent multiday average effective concentrations that are derived directly from long lived atmospheric gas measurements. Therefore, this work provides an important ground truth dataset for comparison with modeling approaches, and oxidant levels that can be used in conjunction with rate coefficients to derive lifetime estimates for other atmospheric species. This valuable information is obtained from relatively inaccessible regions using measurements of only three molecules. Provided the aircraft and ground-based measurements continue, then future OH and Cl oxidant changes induced by major volcanos or stratospheric circulation changes should be captured by applying this method. This work provides the empirical methodological approach and the baseline for future studies.
Methods
Dataset
Whole air samples of IAGOS-CARIBIC (In-service Aircraft for a Global Observing System-Civil Aircraft for the Regular Investigation of the atmosphere Based on an Instrument Container) were collected at 10–12 km in canisters during four flights monthly since 2008 and non-methane hydrocarbons and greenhouse gases were analyzed in laboratory at the Max Planck Institute for Chemistry, Mainz, Germany.38 Flight paths are shown in Fig. 1(b). SF6, CH4, and CH3Cl data of CARIBIC project were analyzed23,38,39,40 using GC-ECD (for SF6) and GC-FID (for CH4 and CH3Cl) with measurement precisions of 1.5%, 0.17%, and 1%, respectively. Ground station SF6 data was taken from the monthly northern hemispheric NOAA/ESRL halocarbons flask program. CH4 data is from the hourly NOAA ESRL Carbon Cycle Cooperative Global Air Sampling Network at Mauna Loa41 and CH3Cl data from the daily NOAA/ESRL halocarbons in situ program. NOAA used GC-FID for CH4 and GC-ECD for SF6 with precisions of 0.5% (SF6),42 0.2% (CH4),43 respectively.
Mean age calculation
SF6 is a long-lived industrial tracer with estimated global emissions of 7.4 ± 0.6 Gg/year44 and has negligible sinks in the troposphere and stratosphere. Consequently, the atmospheric mixing ratio of SF6 has been observed to increase steadily for the past three decades45 with a growth rate of 0.27 ppt/year. In several previous studies, this species has been used to determine the “mean age” of air samples collected at altitude.27,46,47,48 This “mean age” is the term given to the time since the SF6 mixing ratio measured from the aircraft is equivalent to the SF6 measured in the surface source region, or in other words it indicates the average transit time between air leaving the surface until it is measured. We used the monthly northern hemispheric SF6 observations from NOAA/ESRL halocarbons flask program as the initial SF6 concentrations and a second-order polynomial fitting was applied for the time series (years 2005–2016) of SF6 mixing ratio. Then we applied SF6 mixing ratios by aircraft measurements to the fitting and found out the initial emission time. The difference of sampling time and initial emission time is the mean age. We excluded samples with calculated age less than 0 day (2.3% of total samples) that arise due to the higher mixing ratios of SF6 in extratropical troposphere than that in the tropics.26 The spatial distribution of mean age can be found in Fig. 1(b). Absolute counts for each mean age group are shown in Fig. S1 in the Supplementary Material.
Effective OH calculation
The effective OH concentration [OH]eff was calculated according to equation (1).
Where Γ is the mean age, [A]c is the mixing ratio of a compound A (in this case either CH4 or CH3Cl) sampled aloft by the CARIBIC aircraft at time t, and [A]g is the mixing ratio of A observed from the ground station at time t – Γ. Thus, [A]g−[A]C equates to the average loss of A during a time period of Γ, and kOH+A is the second-order reaction rate of compound A with the OH radical, which is calculated for two temperatures (216 K for Γ < 100 days and 250 K for Γ > 100 days) corresponding to 11 km and 40 km height.49 In this calculation we assume that air with Γ < 100 days has experienced predominately tropospheric conditions (average temperature of 216 K), whereas air with Γ > 100 days will have been transported to the stratosphere (average temperature of 250 K).
The reactions and associated rates31,50,51 are as follows:
(\(k_{{\mathrm{OH}} + {\mathrm{CH}}_4,\,216{\mathrm{K}}} = 6.9 \times 10^{ - 16}\,{\mathrm{cm}^{3}}{\mathrm{s}^{{ - 1}}};\,k_{{\mathrm{OH}} + {\mathrm{CH}_{4}},\,250{\mathrm{K}}}\) = \(2.06 \times 10^{ - 15}\,{\mathrm{cm}^{3}}{\mathrm{s}^{{ - 1}}}\))
(\(k_{{\mathrm{OH}} + {\mathrm{CH}_{3}{\mathrm{Cl}}},\,216{\mathrm{K}}} = 6.13 \times 10^{ - 15}\,{\mathrm{cm}^{3}}{\mathrm{s}^{{ - 1}}};\,k_{{\mathrm{OH}} + {\mathrm{CH}_{3}{\mathrm{Cl}}},\,250{\mathrm{K}}}\) = \(1.48 \times 10^{ - 14}\,{\mathrm{cm}^{3}}{\mathrm{s}^{{ - 1}}}\))
Stratospheric Cl and OH radical calculation
Effective OH concentrations for the lower stratosphere, see Fig. 1, were consistently higher when calculated from CH4, similar to the tropospheric values described above. However, in the stratosphere, the differential reaction rate of Cl with CH4 and CH3Cl is a viable explanation for the relatively constant offset in calculated OHeff as chlorine radical production at high altitudes (>10 km) occurs over wide areas due to photolysis of chlorine containing compounds. For this calculation, we assume loss of CH4 (or CH3Cl):
where [OH]r is the real OH concentration in the stratosphere, [Cl] is the chlorine radical concentration, [OH]eff-A is the effective OH derived from compound A in this study. Applying Eq. 4 for CH4 and CH3Cl, then [Cl] can be expressed as:
The ratios of k(Cl)/k(OH) are taken as 24 and 10 for CH4 and CH3Cl at 250 K, respectively.
Data availability
The CARIBIC dataset that support the findings of this study are available from the corresponding author upon reasonable request. Ground station data can be found under URL: ftp://ftp.cmdl.noaa.gov/hats/sf6/combined/HATS_global_SF6.txt (for SF6), ftp://aftp.cmdl.noaa.gov/data/trace_gases/ch4/in-situ/surface/mlo/ch4_mlo_surface-insitu_1_ccgg_HourlyData.txt (for CH4), and ftp://aftp.cmdl.noaa.gov/data/hats/methylhalides/ch3cl/insituGCs/CATS/hourly/mlo_CH3Cl_All.dat (for CH3Cl).
References
Lelieveld, J. et al. Atmospheric oxidation capacity sustained by a tropical forest. Nature 452, 737–740 (2008).
Montzka, S. A. et al. Small interannual variability of global atmospheric hydroxyl. Science 331, 67–69 (2011).
Patra, P. K. et al. Observational evidence for interhemispheric hydroxyl-radical parity. Nature 513, 219–223 (2014).
Levy, H. Normal atmosphere: Large radical and formaldehyde concentrations predicted. Science 173, 141–143 (1971).
Heard, D. E. & Pilling, M. J. Measurement of OH and HO2 in the Troposphere. Chem. Rev. 103, 5163–5198 (2003).
Lelieveld, J., Gromov, S., Pozzer, A. & Taraborrelli, D. Global tropospheric hydroxyl distribution, budget and reactivity. Atmos. Chem. Phys. 16, 12477–12493 (2016).
Hard, T. M., Chan, C. Y., Mehrabzadeh, A. A., Pan, W. H. & O’Brien, R. J. Diurnal cycle of tropospheric OH. Nature 322, 617 (1986).
Stimpfle, R. M. & Anderson, J. G. In-situ detection of OH in the lower stratosphere with a balloon borne high repetition rate laser system. J. Geophys. Res. 15, 1503–1506 (1988).
Wennberg, P. O. et al. Aircraft-borne, laser-induced fluorescence instrument for the in situ detection of hydroxyl and hydroperoxyl radicals. Rev. Sci. Instrum. 65, 1858–1876 (1994).
Perner, D. E. et al. OH-Radicals in the lower troposphere. Geophys. Res. Lett. 3, 466–468 (1976).
Eisele, F. L. & Tanner, D. J. J. Ion-assisted tropospheric OH measurements. J. Geophys. Res. 96, 9295–9308 (1991).
Felton, C. C., Sheppard, J. C. & Campbell, M. J. Measurements of the diurnal OH cycle by a 14C-tracer method. Nature 335, 53–55 (1988).
Mckeen, S. A., Trainer, M., Hsie, E. Y., Tallamraju, R. K. & Liu, S. C. On the indirect determination of atmospheric OH radical concentrations from reactive hydrocarbon measurements. J. Geophys. Res. 95, 7493–7500 (1990).
Blake, N. J. et al. Estimates of atmospheric hydroxyl radical concentrations from the observed decay of many reactive hydrocarbons in well-defined urban plumes. J. Geophys. Res.: Atmos. 98, 2851–2864 (1993).
Williams, J. et al. Variability-lifetime relationship for organic trace gases: A novel aid to compound identification and estimation of HO concentrations. J. Geophys. Res.: Atmos. 105, 20473–20486 (2000).
Williams, J., Gros, V., Bonsang, B. & Kazan, V. HO cycle in 1997 and 1998 over the southern Indian Ocean derived from CO, radon, and hydrocarbon measurements made at Amsterdam Island. J. Geophys. Res.: Atmos. 106, 12719–12725 (2001).
Rigby, M. et al. Role of atmospheric oxidation in recent methane growth. Proc. Natl Acad. Sci. USA 114, 5373–5377 (2017).
Prinn, R. G. et al. Evidence for variability of atmospheric hydroxyl radicals over the past quarter century. Geophys. Res. Lett. 32, https://doi.org/10.1029/2004gl022228 (2005).
Krol, M. & Lelieveld, J. Can the variability in tropospheric OH be deduced from measurements of 1,1,1-trichloroethane (methyl chloroform)? J Geophys. Res.: Atmos. 108, https://doi.org/10.1029/2002jd002423 (2003).
Montzka, S. & Reimann, S. (Coordinating Lead Authors) et al. Ozone-Depleting Substances (ODSs)and Related Chemicals, in Scientific Assessment of Ozone Depletion: 2010, Ch. 1 (World Meteorological Organization: Geneva, Switzerland, 2011).
Yokouchi, Y., Ikeda, M., Inuzuka, Y. & Yukawa, T. Strong emission of methyl chloride from tropical plants. Nature 416, 163–165 (2002).
Santee, M. L., Livesey, N. J., Manney, G. L., Lambert, A. & Read, W. G. Methyl chloride from the Aura Microwave Limb Sounder: First global climatology and assessment of variability in the upper troposphere and stratosphere. J. Geophys. Res.: Atmos. 118, 13,532–513,560 (2013).
Umezawa, T. et al. Methyl chloride in the upper troposphere observed by the CARIBIC passenger aircraft observatory: Large-scale distributions and Asian summer monsoon outflow. J. Geophys. Res.: Atmos. 119, 5542–5558 (2014).
Baker, A. K. et al. Estimating the contribution of monsoon-related biogenic production to methane emissions from South Asia using CARIBIC observations. Geophys. Res. Lett. 39, https://doi.org/10.1029/2012gl051756 (2012).
Turner, A. J., Frankenberg, C., Wennberg, P. O. & Jacob, D. J. Ambiguity in the causes for decadal trends in atmospheric methane and hydroxyl. Proc. Natl Acad. Sci. USA 114, 5367–5372 (2017).
Bönisch, H., Engel, A., Curtius, J., Birner, T. & Hoor, P. Quantifying transport into the lowermost stratosphere using simultaneous in-situ measurements of SF6 and CO2. Atmos. Chem. Phys. 9, 5905–5919 (2009).
Ray, E. A. et al. Improving stratospheric transport trend analysis based on SF6 and CO2 measurements. J. Geophys. Res.: Atmos. 119, 14110–114128 (2014).
Garny, H., Birner, T., Bönisch, H. & Bunzel, F. The effects of mixing on age of air. J. Geophys. Res.: Atmos. 119, 7015–7034 (2014).
Orbe, C. et al. Airmass origin in the Arctic. Part I: Seasonality. J. Clim. 28, 4997–5014 (2015).
Baker, A. K. et al. Evidence for strong, widespread chlorine radical chemistry associated with pollution outflow from continental Asia. Sci. Rep. 6, 36821 (2016).
DeMore, W. B. S. et al. Chemical kinetics and photochemical data for use in stratospheric modeling. Eval. Number 12, 1–266 (1997).
Lange, L. et al. Detection of lightning-produced NO in the midlatitude upper troposphere during STREAM 1998. J. Geophys. Res.: Atmos. 106, 27777–27785 (2001).
Park, S. et al. Vertical transport rates and concentrations of OH and Cl radicals in the Tropical Tropopause Layer from observations of CO2 and halocarbons: implications for distributions of long- and short-lived chemical species. Atmos. Chem. Phys. 10, 6669–6684 (2010).
Lelieveld, J. et al. Chlorine activation and ozone destruction in the northern lowermost stratosphere. J. Geophys. Res.: Atmos. 104, 8201–8213 (1999).
Gromov, S., Brenninkmeijer, C. A. M. & Jöckel, P. A very limited role of tropospheric chlorine as a sink of the greenhouse gas methane. Atmos. Chem. Phys. 18, 9831–9843 (2018).
Minschwaner, K. & Manney, G. L. Derived methane in the stratosphere and lower mesosphere from Aura Microwave Limb Sounder measurements of nitrous oxide, water vapor, and carbon monoxide. J. Atmos. Chem. 71, 253–267 (2015).
Boering, K. A. et al. Stratospheric mean ages and transport rates from observations of carbon dioxide and nitrous oxide. Science 274, 1340 (1996).
Brenninkmeijer, C. A. M. et al. Civil aircraft for the regular investigation of the atmosphere based on an instrumented container: The new CARIBIC system. Atmos. Chem. Phys. 7, 4953–4976 (2007).
Schuck, T. J., Brenninkmeijer, C. A. M., Slemr, F., Xueref-Remy, I. & Zahn, A. Greenhouse gas analysis of air samples collected onboard the CARIBIC passenger aircraft. Atmos. Meas. Tech. 2, 449–464 (2009).
Baker, A. K., Slemr, F. & Brenninkmeijer, C. A. M. Analysis of non-methane hydrocarbons in air samples collected aboard the CARIBIC passenger aircraft. Atmos. Meas. Tech. 3, 311–321 (2010).
Dlugokencky, E. J., Crotwell, A. M., Lang, P. M. & Mund, J. W. Atmospheric methane dry air mole fractions from quasi-continuous measurements at Mauna Loa, Hawaii, 1986–2016, Version: 2017-01-20, Path: ftp://aftp.cmdl.noaa.gov/data/trace_gases/ch4/in-situ/surface/. (2017).
Hall, B. D. et al. Improving measurements of SF6 for the study of atmospheric transport and emissions. Atmos. Meas. Tech. 4, 2441–2451 (2011).
Dlugokencky, E. J., Steele, L. P., Lang, P. M. & Masarie, K. A. The growth rate and distribution of atmospheric methane. J. Geophys. Res.: Atmos. 99, 17021–17043 (1994).
Rigby, M. et al. History of atmospheric SF6 from 1973 to 2008. Atmos. Chem. Phys. 10, 10305–10320 (2010).
IPCC. Climate Change 2007: Mitigation. Contribution of Working Group III to the Fourth Assessment Report of the Intergovernmental Panel on Climate Change (eds Metz, B., Davidson, O. R., Bosch P. R., Dave R., Meyer L. A.). (Cambridge, United Kingdom and New York, NY, USA, 2007).
Waugh, D. Age of stratospheric air: Theory, observations, and models. Reviews of Geophysics 40, https://doi.org/10.1029/2000rg000101 (2002).
Engel, A. et al. Age of stratospheric air unchanged within uncertainties over the past 30 years. Nat. Geosci. 2, 28–31 (2008).
Hall, T. M. & Plumb, R. A. Age as a diagnostic of stratospheric transport. J. Geophys. Res.: Atmos. 99, 1059–1070 (1994).
National Oceanic Atmospheric Administration. U.S. Standard Atmosphere, 1976. (U.S. Government Printing Office: Washington, D.C., United States of America, 1976).
Srinivasan, N. K., Su, M. C., Sutherland, J. W. & Michael, J. V. Reflected shock tube studies of high-temperature rate constants for OH+CH4 ->CH3+H2O and CH3+NO2 ->CH3O+NO. J. Phys. Chem. A 109, 1857–1863 (2005).
Atkinson, R. Kinetics of the gas-phase reactions of OH radicals with alkanes and cycloalkanes. Atmos. Chem. Phys. 3, 2233–2307 (2003).
Acknowledgements
We would like to thank Claus Koeppel for the routine maintenance and construction and installation of the whole air sampling system, Angela Baker, Tanja Schuck, Ute Thorenz, and Carina Sauvage for conducting the measurements, Hao Fang (INRIA, France) for the programming support. CARIBIC is part of the European Research Infrastructure IAGOS, which is financed partly by the German Ministry for Education and Research (BMBF 01LK1223). Operation of the CARIBIC observatory is possible through the support and cooperation of Lufthansa and Lufthansa Technik and Frankfurt and Munich airports. We would also like to thank Stephen A. Montzka (for thoughtful comments), Ed Dlugokencky, Geoffrey S. Dutton, and James W. Elkins from NOAA for providing the following data: the NOAA/ESRL GMD network for ground station SF6 data, the NOAA ESRL Carbon Cycle Cooperative Global Air Sampling Network for ground station CH4 data and the NOAA/ESRL halocarbons in situ program for ground station CH3Cl data. This work was funded by the Max Planck Society internal funds.
Author information
Authors and Affiliations
Contributions
M.L. and J.W. developed the idea. M.L., J.W., E.K., H.F., J.L., and C.B. wrote the manuscript. C.B. is the CARIBIC lead scientist and oversaw planning and execution of measurement flights and the provision of aircraft data. All authors discussed the results and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, M., Karu, E., Brenninkmeijer, C. et al. Tropospheric OH and stratospheric OH and Cl concentrations determined from CH4, CH3Cl, and SF6 measurements. npj Clim Atmos Sci 1, 29 (2018). https://doi.org/10.1038/s41612-018-0041-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41612-018-0041-9
This article is cited by
-
Correction to: Long-term spatio-temporal trends in atmospheric aerosols and trace gases over Pakistan using remote sensing
Acta Geophysica (2023)
-
Long-term spatio-temporal trends in atmospheric aerosols and trace gases over Pakistan using remote sensing
Acta Geophysica (2023)
-
Positive feedback mechanism between biogenic volatile organic compounds and the methane lifetime in future climates
npj Climate and Atmospheric Science (2022)
-
Kinetics of IO radicals with C1, C2 aliphatic alcohols in tropospherically relevant conditions
Environmental Science and Pollution Research (2022)
-
Air pollution trends measured from MODIS and TROPOMI: AOD and CO over Pakistan
Journal of Atmospheric Chemistry (2022)
