
Abstract
Accumulating evidence shows that pseudogenes can function as microRNAs (miRNAs) sponges and regulate gene expression. Mining potential interactions between pseudogenes and miRNAs will facilitate the clinical diagnosis and treatment of complex diseases. However, identifying their interactions through biological experiments is time-consuming and labor intensive. In this study, an ensemble learning framework with similarity kernel fusion is proposed to predict pseudogene–miRNA associations, named ELPMA. First, four pseudogene similarity profiles and five miRNA similarity profiles are measured based on the biological and topology properties. Subsequently, similarity kernel fusion method is used to integrate the similarity profiles. Then, the feature representation for pseudogenes and miRNAs is obtained by combining the pseudogene–pseudogene similarities, miRNA–miRNA similarities. Lastly, individual learners are performed on each training subset, and the soft voting is used to yield final decision based on the prediction results of individual learners. The k-fold cross validation is implemented to evaluate the prediction performance of ELPMA method. Besides, case studies are conducted on three investigated pseudogenes to validate the predict performance of ELPMA method for predicting pseudogene–miRNA interactions. Therefore, all experiment results show that ELPMA model is a feasible and effective tool to predict interactions between pseudogenes and miRNAs.
Similar content being viewed by others

Introduction
Non-coding RNAs (ncRNAs) refer to the RNA molecules that could not translate into proteins, which composed up to about 98% of the human genome. These ncRNAs play an essential role in epigenetic regulation of gene expression at transcriptional and post-transcriptional levels. Pseudogenes are defined as incomplete copies of genes that code for proteins, but lack of coding function. However, pseudogenes could be transcribed into ncRNAs and be considered as regulators in organisms. MicroRNAs (miRNAs) are a class of small, single stranded, non-coding RNAs, which are involved gene expression at post-transcriptional level1. By binding to targeting mRNAs, miRNAs cause degradation and translation repression of mRNAs2. The fine-tuning of gene regulation by pseudogenes and miRNAs has attracted attentions in many biological processes.
Pseudogenes and miRNA are essential components of competing endogenous RNAs (ceRNAs) network. ceRNA hypothesis is proposed to describe the interactions among ceRNAs members and miRNAs3. The ceRNAs members include pseudogenes, long noncoding RNAs (lncRNAs), circular RNA (circRNAs), and protein-coding RNAs, etc. The ceRNAs could form a ceRNA network modulate mRNA expression and regulate protein levels. Recent experimental results show that abnormal expression and dysregulations of both pseudogenes and miRNAs are related to complex diseases. For example, pseudogene GBAP1 contributes to the development and progression of gastric cancer by sequestering the miR-212-3p from binding to GBA4. Therefore, pseudogenes and miRNAs can interact with each other, which jointly associated the occurrence of human diseases. However, it is very laborious and time-consuming to verify the associations between pseudogenes and miRNAs through biological experiments. So reasonable and effective computational methods is urgently need to mine the associations between pseudogenes and miRNAs.
Identifying pseudogene–miRNA associations contribute to discover more biological mechanisms in biological process and disease states. Compared with biological methods, the computational approaches are less time consumption. In the area of miRNA research, mining the potential miRNA-disease associations is a high hop topic5,6,7,8. For example, RWRMMDA model is proposed to predict the miRNA-disease associations by integrating multiple similarities, which also used improved extended random walk with restart algorithm based on miRNA similarity-based and disease similarity-based heterogeneous networks9. Zhou et al.10 proposed GBDT-LR method to prioritize miRNA candidates for diseases by combining gradient boosting decision tree with logistic regression. Besides, a large number of computational models are also developed to forecast other ncRNA associations and disease-biomolecule associations, for example, predicting the lncRNA–miRNA11,12, circRNA–miRNA13,14, lncRNA–disease15,16, circRNA–disease17,18,19, drug–disease20 interactions. Motived by these ncRNA interaction prediction, Zhou et al.21 incorporates feature fusion and graph auto-encoder to predict pseudogene–miRNA associations. In the model, various perspective attribute information for pseudogenes and miRNAs is obtained as their similarity features, and graph auto-encoder is used to obtain the low-dimensional representation of nodes. Then, the low-dimensional vector is fed into Extreme Gradient Boosting (XGBoost) to predict the pseudogene–miRNA associations. Compared with these ncRNA-miRNA and ncRNA-disease association prediction, only one computational model is developed to predict pseudogene–miRNA associations. Therefore, it still exists some limitations for further improvement. Especially, there is an urgent need to develop more accurate and efficient computational methods to infer associations between pseudogenes and miRNAs.
In this study, an ensemble learning framework with similarity kernel fusion (SKF) method is developed to mine the pseudogene–miRNA associations, named ELPMA. First, GIP kernel similarity, hamming profile similarity, cosine similarity for pseudogenes and miRNAs is calculated based on the known pseudogene–miRNA associations. Then, pseudogene expression similarity and miRNA function similarity are computed based on the pseudogene expression profiles and miRNA–target information, respectively. Besides, the pseudogene similarities and miRNA similarities are fused using SKF method. Then, the feature representation of pseudogene–miRNA interactions is constructed by combing the pseudogene–pseudogene similarity, miRNA–miRNA similarity, and experimentally validated pseudogene–miRNA associations. Next, resampling method is used to build multiple different balanced pseudogene–miRNA association training subsets, which could reduce the bias of small-scale samples. Finally, individual learners are performed on each subset to obtain the primitive outcomes, and the soft voting is used to yield final decision based on the prediction results of individual learners. To assess the effectiveness of ELPMA model, five-fold cross validation is implemented applied to assess the prediction performance of our proposed method. As a result, the mean area under the ROC curve (AUC) and mean area under the precision-recall curve (AUPR) of ELPMA method achieved 0.9896 and 0.9913, respectively. According to comparison with other four methods, assessment results shown that ELPMA method obtain comparable performance. In the case studies, the predicted miRNAs for the three investigated pseudogenes are also used to validate the prediction performance of ELPMA method. All the results shown that our proposed model could serve as a recommendable tool for predicting pseudogene–miRNA associations.
Materials and methods
Gold standard data set
The pseudogene–miRNA associations are obtained from starBase v2.0, in which very high stringency of pseudogene symbol is selected22. After screening and removing redundancy, 1570 experimentally supported pseudogene–miRNA associations is sorted out, covering 318 pseudogenes and 260 miRNAs. In this study, a pseudogene–miRNA adjacency matrix PM(i, j) is constructed based on the validated associations between pseudogenes and miRNAs. If there is an association between pseudogenes p(i) and miRNAs m(j), PM(i, j) is assigned as 1, otherwise 0.
Expression similarity for pseudogenes
The expression level of pseudogenes in various cancers and normal tissues is obtained from dreamBase database23. In dreamBase database, expression information of pseudogenes is selected as the characteristic information of pseudogenes. When two pseudogenes have a higher correlation score tend to be more similarity expressed. The pseudogene expression profiles are measures as follows:
where N is the number of properties of the expression profiles, xk and yk denote the expression values in different cancers and normal tissues.
Function similarity for miRNAs
Given that miRNAs targeting more of the same genes tend to be involved in similar biological function. The interactions between miRNA and target gene information are obtained from miRTarBase24. The miRNA–target interactions are employed to measure the miRNA function similarity for each pair of miRNAs. If two sets of target genes (say Gi and Gj) respectively have relationship with miRNA Mi and miRNA Mj, the miRNA function similarity is calculated as follows:
where Gi and Gj represent the sets of target gene that related with miRNAs.
GIP kernel similarity for pseudogenes and miRNAs
The GIP kernel similarity is applied to calculate the similarity between pseudogenes and miRNAs based on the known pseudogene–miRNA association adjacency matrix25. The GIP kernel similarity for pseudogenes can be calculated as follows:
where p(i) represents the pseudogene interaction profiles, which is a binary vector that encode the interaction between pseudogene i and all miRNAs, i.e., the i-th row of the gold standard pseudogenes-miRNA adjacency matrix PM. The parameter γp controls the kernel bandwidth. np is the number of pseudogenes.
Similar to pseudogenes, the GIP kernel similarity for miRNAs is defined as:
where m(i) represents the miRNA interaction profiles, which is a binary vector that encode the interaction between miRNA i and each pseudogene, i.e., the i-th column of adjacency matrix PM. The parameter γm is also used to control the kernel bandwidth. nm is the number of miRNAs.
Hamming profile similarity for pseudogenes and miRNAs
Given the length for a pair of vectors are same, hamming profile is the number of elements of which corresponding values are different. The higher Hamming profile value represents the two vector has lower similarity. Hamming profile similarity for pseudogenes is calculated as follows:
where IP(pi) is the i-th row of the pseudogene–miRNA adjacency matrix PM.
Similarly, the hamming profile similarity for miRNA is defined as follows:
where IP(mi) is the i-th column of the pseudogene–miRNA adjacency matrix PM.
Cosine similarity for pseudogenes and miRNAs
Cosine similarity algorithm has been widely used in the collaborative filtering recommendation algorithm. Here, based on known pseudogene–miRNA associations, the similarity of pseudogenes pi and pj is defined as follows:
where r represents the number of pseudogenes. The binary vector PM(pi) indicates whether exist an association between pseudogene pi and each miRNA (the row i of the PM matrix, if pi is related to miRNA, otherwise 0). Meanwhile, SP_cos(pi, pj) represents the cosine similarity between pseudogene pi and pj. SP_cos is the pseudogene cosine similarity matrix.
Similarly, the cosine similarity of miRNA mi and miRNA mj is computed as follows:
where MP(mi) denotes whether there is an association between miRNA mi and each pseudogene (the column of MP matrix, if mj is related to pseudogene, otherwise 0). SM_cos(mi, mj) is the cosine similarity between miRNA mi and miRNA mj. The SM_cos is the miRNA cosine similarity matrix. n is the number of miRNAs.
Integrated similarity by similarity kernel fusion method
In this study, four kinds of pseudogene similarities and five miRNA similarities are calculated. The integrated pseudogene similarity is measured by combining pseudogene expression similarity, pseudogene GIP kernel similarity, pseudogene hamming profile similarity, pseudogene cosine similarity. The integrated miRNA similarity is calculated by combining miRNA function similarity, miRNA GIP kernel similarity, miRNA hamming profile similarity and cosine similarity. Here, similarity kernel fusion method is used to fuse the four pseudogene similarities and five miRNA similarities26. Let Sp,r (r = 1,2,…,4) represents the four pseudogene similarities and Sm,n (n = 1,2,…,5) represents the five miRNA similarities, respectively.
Firstly, each original kernel for pseudogenes is normalized by Eq. (9).
where when NSp,r satisfies \(\sum\nolimits_{{c_{k} \in C}} {NS_{c,m} (c_{k} ,c_{j} )} = 1\), NSp,r is the normalized pseudogene similarity.
Then, a sparse kernel for each pseudogene similarity is computed by Eq. (10).
where Fc,m is a sparse kernel and it satisfies \(\sum\nolimits_{{c_{j} \in C}} {F_{c,m} (c_{k} ,c_{j} )} = 1\). Ni is a set of pi’s neighbors including ci itself.
Therefore, four pseudogene similarities could be computed as Eq. (11).
where \(SP_{p,r}^{t + 1}\) is the status matrix of r-th pseudogene similarity kernel after t + 1 iterations.\(SP_{p,k}^{0}\) denotes the initial status of Sp,k.
After t + 1 steps, the overall kernel for pseudogenes is calculated as Eq. (12).
Finally, a weight matrix wp is used to remove the noise in the matrix Sp.
The fused pseudogene similarity is computed as Eq. (14).
Similarly, the integrated miRNA similarity as Sm* is computed, in which involved five miRNA similarities to be fused.
Ensemble learning framework with resampling method
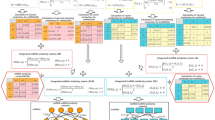
To predict the potential pseudogene–miRNA associations, an ensemble learning framework with similarity kernel fusion method is proposed. Inspired by the previous research27,28, ELPMA model is proposed through the following steps: (1) using the resampling method to obtain multiple different training subsets, and the diversity of individual learners is increased; (2) to integrate the prediction results of individual learners, soft voting is employed to obtain the final prediction. The process of constructing the ensemble learning framework is shown in Fig. 1.

Ensemble learning framework for the pseudogene–miRNA association prediction.
Resampling strategy
There are 1570 experimentally confirmed pseudogene–miRNA associations as positive samples, and 81,110 unconfirmed pseudogene–miRNA pairs as unlabeled samples. So only a small part of experimentally confirmed pseudogene–miRNA associations. To settle the problem caused by the imbalanced dataset, the resample strategy is employed to build multiple different balanced training subsets. The negative samples are guaranteed to have the same number with positive samples. When construct a subset, all positive samples are sort out, and same unlabeled samples are randomly selected as negative samples. Then, the negative samples and positive training sample are combined to balance the positive and negative samples. The training set of positive sample P and the unlabeled sample set U are defined as follows:
where P represents the positive samples, and U denotes the unknown pseudogene–miRNA association samples.
In each training subset, the number of unlabeled pseudogene–miRNA associations is the same as the number of positive samples. The set N (N ∈ U) represents the negative samples selected from U, and the number of N is same as the number of P. The set of T = P ⋃ N is the training set in base learning.
Sample representation
To learn the pseudogenes and miRNAs potential feature representation, multiple data source is incorporated to obtain the integrated similarities for pseudogenes and miRNAs. Here, a pseudogene–miRNA pair was taken as a sample. The feature vector of i-th pseudogene, FP(p(i)), is defined as follows:
where Np represents the number of pseudogenes. Similarly, the feature vector of jth miRNA, FM(m(j)), is defined as follows:
where Nm represents the number of miRNAs. Then, the feature vector of each pseudogene–miRNA pair (p(i),m(j)) is defined by combining the FP(p(i)) and FM(m(j)) as follows:
Soft voting for pseudogene–miRNA association prediction
Ensemble learning combines multiple individual learners to increase the prediction performance compared to individual models. Owing to the training subsets are different and the feature spaces of the subsets are heterogenous, the trained individual learners are also different from each other. In this study, an ensemble learning framework is developed by using the XGBoost as individual learner on the multiple sample subsets. XGBoost is a machine learning algorithm in which regression trees is used as functions in gradient boosting to optimize trees29.
Set the output of a tree as shown below:
where xi is the input vector, q represents the structure of each tree and wq represents the score of the leaf node q. The output of the set of K trees is:
where K is the number of regression functions, the objective function for learning the set of fk is shown as follows:
where l represents the loss function between the observed value yi and predict value \(\hat{y}_{i}\). Ω(fk) is the regularization term to avoid overfitting. γ is the pseudo-regularization hyperparameter. λ is the L2 norm for leaf weights. T is the total number of leaf nodes.
The optimal objective function value could be written as:
where I is the set of leaf nodes, gi is the first derivative of l and hi is the second derivative of l.
Here, the outputs of XGBoost are taken as primitive results. Then, the soft voting is used to make the final decision. The prediction scores of individual learners are averaged, and confirmed whether the pseudogene is associated with each other. Take an unknown pseudogene–miRNA association as sample input, n individual learners could produce n prediction results, and then the n prediction results are integrated by using the soft voting strategy30. Specifically, the output of the i-th sample by soft voting is defined as follows:
where O(i,j) is the prediction scores of the j-th individual learners for the i-th sample. n represents the number of training subsets. O(i) > 0.5 represents the pseudogene–miRNA pair is associated; otherwise, it is considered to be not associated with each other.
Results
Performance evaluation
In this work, k-fold cross validation is employed to evaluate the performance of the ELPMA model. The validated pseudogene–miRNA associations are regarded as the positive set, and equal number of samples are randomly selected from the negative sample set as negative samples. For each cross validation, (k-1) positive subsets and the same number of negative subsets took from k subsets to train the models; the remaining one positive subset and one negative subset are used for testing to evaluate the prediction performance. Specifically, fivefold and tenfold cross validation are used to evaluate the prediction performance of ELPMA model. Moreover, several metrics are used to measure the prediction performance of ELPMA method, including precision (Pre), sensitivity (Sen), accuracy (Acc), F1-score, AUC (Area under the receiver operating characteristic curve), AUPR (Area under the precision-recall curve), and MCC (Matthews’s correlation coefficient). The calculation formulas of these metrics are shown as follows:
where TP and TN represent the number of true positives and true negatives, respectively. FP and FN represent the number of positives and negatives, respectively, that are wrongly predicted.
Performance analysis of ELPMA method with different individual learners
To assess the ability of the ELPMA method to predict the associations between pseudogenes and miRNAs, fivefold cross validation is implemented on the gold standard data set. In the ensemble framework, different individual learners could affect the prediction performance. Here, AdaBoost, Random Forest (RF), Extreme Gradient Boosting (XGB) and Extremely Randomized Trees (ERT) are used as the individual learners, respectively. The individual learners are represented as ELPMA-AB, ELPMA-RF, ELPMA-XGB and ELPMA-ERT, respectively. In the ELPMA model, parameter selection are important factors, and the hyper-parameters of each model are tuned. For example, the number of individual learners of ELPMA is range from 2 to 20 with steps of 1. Furthermore, the range of hyper-parameter turning of ELPMA-XGB is as that n_estimators are selected from [50, 100, 200, 300, 400, 500], the learning rate is set from 0.1 to 0.9 with an interval of 0.1. The range of hyper-parameter turning of ELPMA-ERT is as that the value of max_depth is selected from [10, 20, 30, 40, 50] and the n_estimators are selected from [50, 100, 200, 300, 400, 500]. In addition, different hyper-parameters of ELPMA-AB and ELPMA-RF model are selected to obtain optimal performance. Finally, the prediction performance of the ELPMA model that using different individual learners is listed in Table 1. When the number of individual learners, n_estimators, learning rate are respectively set as 10, 400, 0.2, ELPMA-XGB yields the Precision of 0.9716, the Recall of 0.9369, the F1-score of 0.9540, the Acc of 0.9548, the AUC of 0.9897, the AUPR of 0.9914. As shown in Table 1, ELPMA-XGB is higher than other models in these seven metrics.
In addition, the ROC curves of the k-fold cross validation are plotted by the proposed ELPMA-XGB method, respectively. The experimental results show that ELPMA-XGB achieves mean AUC values of 0.9897 and 0.9906 for the fivefold and tenfold cross validation (Fig. 2). Therefore, ELPMA-XGB model is appropriate as the individual learners of ELPMA method for the prediction of pseudogene–miRNA associations.

ROC curves under k-fold cross validation performed by the ELPMA-XGB framework. (a) ROC curves under fivefold cross validation; (b) ROC curves under tenfold cross validation.
Influence of training data on model performance
In the task, experimentally validated pseudogene-miRNA associations are selected as the only information source for model construction. The number of known pseudogene-miRNA associations may influence the prediction of our method ELPMA. To evaluate the impact of the number of training data on the performance, we used different proportions of training data to implement the ELPMA model. The fivefold and tenfold cross-validation results obtained by ELPMA is shown in Table S1. The results shown that the performance of ELPMA model getting better with the training data increasing. Therefore, the size of the training data has a great influence on the prediction performance of ELPMA model. With the number of training data increasing, the prediction performance of is also increased.
Effectiveness of soft voting for the ensemble learning framework
To demonstrate the effectiveness of the soft voting for the ensemble learning method, the soft voting performance is compared with individual learners on ELPMA model. Detailed results of the comparison are shown in Fig. 3. In the figures, the horizontal axis represents the index number of individual learners, and the vertical axis are the AUC values and AUPR values. From the Fig. 3, we also seen that the AUC of individual learners is between 0.9823 and 0.9849, and the AUPR of individual learners is between 0.9849 and 0.9873 under fivefold cross validation. The results indicate that soft voting in the proposed method could improve the prediction performance of ELPMA model. It also indicates that ELPMA is an effective framework to predict the pseudogene–miRNA interactions.

Performance comparison of ELPMA method and individual learners.
Comparison with other existing methods
To comparatively illustrate the superiority of ELPMA method, GBDT-LR10, ABMDA31, CD_LNLP17, and LAGCN20 are compared with ELPMA method to predict the pseudogene–miRNA interactions. These five methods are individual evaluated based on gold standard data set with k-fold cross validation and recommended hyperparameters. As show in Fig. 4, ELPMA shows the best performance in term of the average AUC values under fivefold and tenfold cross validation. It shows that the ROC curves of ELPMA model is above those of GBDT-LR, ABMDA, CD_LNLP and LAGCN method in most cases. The average AUC scores of ELPMA method are up to 0.9897 and 0.9906 for the fivefold and tenfold cross validation, respectively, which is superior to the other four methods (Fig. 4). In addition, the results of performance evaluation indicators such as F1-score, Acc, MCC are shown in Table 2 for fivefold and tenfold cross validation. Although the Precision of ELPMA is inferior to ABMDA and Acc of ELPMA is inferior to CD_LNLP and LAGCN, the evaluation metrics of ELPMA are higher than others (Table 2). Furthermore, we used the paired t-test based on 10 runs of fivefold and tenfold cross-validation to test the performance of the ELPMA method and the comparison methods. Table 3 shows that ELPMA is significantly preferred to other computational methods in terms of Sensitivity, F1-score, AUC, AUPR and MCC (Table 3). Therefore, all the above results show that ELPMA method provides a great improvement in predict the pseudogene–miRNA interactions.

ROC curves of different methods under k-fold cross validation. (a) ROC curves under fivefold cross validation; (b) ROC curves under tenfold cross validation.
Case studies
To illustration the prediction performance of ELPMA method in screening pseudogene–miRNA interactions, case studies of three pseudogene related miRNA are conduct for further validation. Given the investigated pseudogene–miRNA interaction to be unknown in all known associations. In this section, the pseudogene MSTO2P, MTND4P12 related miRNAs are removed in the known associations, and then use other associations to train the model and predict the probability of all miRNAs associated with the investigated pseudogenes. Through the calculation of ELPMA method, the candidate associations between pseudogene and miRNAs are sorted in descending order. Then, the top 10 rank results are selected with high probability scores for the three investigated pseudogenes, and the predicted associations are verified with the starBase database.
Pseudogene MSTO2P is found to be implicated in several diseases including lung cancer32, colorectal cancer33, etc. MSTO2P could function as a miR-128-3p sponge in non-small cell lung cancer cells (NSCLC), and MSTO2P/miR-128-3p to regulate coptisine sensitivity of NSCLC cells via TGF-β pathway. In addition, MSTO2P related top 10 miRNAs, in which 9 of the top10 is proved by starBase (Table 4).
MTND4P12 is considered as an oncogenic pseudogene upregulated in skin cutaneous melanoma, and it can upregulate the expression of oncogene AURKB by serving as ceRNA34. Hsa-let-7e-5p is also identified as candidate miRNA that regulated by MTND4P12, hsa-let-7e-5p and MTND4P12 is co-expression in skin cutaneous melanoma. As shown in Table 4, the MTND4P12 related top 10 miRNAs is supported by starBase.
Conclusion
Increasing evidences show that both pseudogenes and miRNAs play oncogenic or tumor-suppressive roles in disease progression. Predicting pseudogene–miRNA associations will contribute to understanding the pathological mechanisms, diagnosis, and treatment of diseases. In this work, a computational method is proposed to infer the associations between pseudogenes and miRNAs, which employed an ensemble learning framework with similarity kernel fusion, named ELPMA. By comparing with other four models, the prediction performance of our proposed method is powerful to predict the pseudogene–miRNA interactions. The case study of investigated MSTO2P and MTND4P12 related miRNAs also proved the ELPMA method is reliable and effective.
The good performance of ELPMA method is attributed to three main factors: (1) ELPMA integrates the biological information including pseudogene expression profiles and miRNA–targets interactions. (2) ELPMA introduces the resampling method to settle the problem caused by the imbalanced pseudogene–miRNA dataset. (3) The application of XGBoost as individual learner of the ensemble learning framework guarantees the effectiveness of learning the meaning of combinations of features from feature representation.
There are also some limitations in the ELPMA method. First, the gold standard pseudogene-miRNA associations may have nosy, and the negative samples are randomly selected from the unconfirmed associations, limiting the prediction performance. In addition, the ELPMA method relies on the known pseudogene–miRNA interaction network, and it could not predict novel pseudogene-miRNA interactions without any known associations. Therefore, developing more effective framework is essential to infer the associations between pseudogenes and miRNAs.
Data availability
The data will be made available on request from the corresponding author.
References
Bartel, D. P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 116, 281–297. https://doi.org/10.1016/s0092-8674(04)00045-5 (2004).
Bartel, D. P. MicroRNAs: Target recognition and regulatory functions. Cell 136, 215–233. https://doi.org/10.1016/j.cell.2009.01.002 (2009).
Salmena, L., Poliseno, L., Tay, Y., Kats, L. & Pandolfi, P. P. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language?. Cell 146, 353–358. https://doi.org/10.1016/j.cell.2011.07.014 (2011).
Ma, G. et al. A genetic variation in the CpG island of pseudogene GBAP1 promoter is associated with gastric cancer susceptibility. Cancer 125, 2465–2473. https://doi.org/10.1002/cncr.32081 (2019).
Huang, L., Zhang, L. & Chen, X. Updated review of advances in microRNAs and complex diseases: Taxonomy, trends and challenges of computational models. Brief. Bioinform. https://doi.org/10.1093/bib/bbac358 (2022).
Huang, L., Zhang, L. & Chen, X. Updated review of advances in microRNAs and complex diseases: Towards systematic evaluation of computational models. Brief. Bioinform. https://doi.org/10.1093/bib/bbac407 (2022).
Huang, L., Zhang, L. & Chen, X. Updated review of advances in microRNAs and complex diseases: Experimental results, databases, webservers and data fusion. Brief. Bioinform. https://doi.org/10.1093/bib/bbac397 (2022).
Chen, X., Xie, D., Zhao, Q. & You, Z. H. MicroRNAs and complex diseases: From experimental results to computational models. Brief. Bioinform. 20, 515–539. https://doi.org/10.1093/bib/bbx130 (2019).
Nguyen, V. T., Le, T. T. K., Than, K. & Tran, D. H. Predicting miRNA–disease associations using improved random walk with restart and integrating multiple similarities. Sci. Rep. 11, 21071. https://doi.org/10.1038/s41598-021-00677-w (2021).
Zhou, S., Wang, S., Wu, Q., Azim, R. & Li, W. Predicting potential miRNA-disease associations by combining gradient boosting decision tree with logistic regression. Comput. Biol. Chem. 85, 107200. https://doi.org/10.1016/j.compbiolchem.2020.107200 (2020).
Xu, M. et al. SPMLMI: Predicting lncRNA-miRNA interactions in humans using a structural perturbation method. PeerJ 9, e11426. https://doi.org/10.7717/peerj.11426 (2021).
Wang, M. N., Lei, L. L., He, W. & Ding, D. W. SPCMLMI: A structural perturbation-based matrix completion method to predict lncRNA-miRNA interactions. Front. Genet. 13, 1032428. https://doi.org/10.3389/fgene.2022.1032428 (2022).
Guo, L. X. et al. A novel circRNA-miRNA association prediction model based on structural deep neural network embedding. Brief. Bioinform. https://doi.org/10.1093/bib/bbac391 (2022).
Wang, X. F. et al. KGDCMI: A new approach for predicting circRNA-miRNA interactions from multi-source information extraction and deep learning. Front. Genet. 13, 958096. https://doi.org/10.3389/fgene.2022.958096 (2022).
Xie, G. B. et al. Predicting lncRNA-disease associations based on combining selective similarity matrix fusion and bidirectional linear neighborhood label propagation. Brief. Bioinform. https://doi.org/10.1093/bib/bbac595 (2023).
Du, X.-X., Liu, Y., Wang, B. & Zhang, J.-F. lncRNA–disease association prediction method based on the nearest neighbor matrix completion model. Sci. Rep. 12, 21653. https://doi.org/10.1038/s41598-022-25730-0 (2022).
Zhang, W., Yu, C., Wang, X. & Liu, F. Predicting CircRNA-disease associations through linear neighborhood label propagation method. IEEE Access https://doi.org/10.1109/ACCESS.2019.2920942 (2019).
Lei, X. & Bian, C. Integrating random walk with restart and k-nearest Neighbor to identify novel circRNA-disease association. Sci. Rep. 10, 1943. https://doi.org/10.1038/s41598-020-59040-0 (2020).
Deng, L., Zhang, W., Shi, Y. & Tang, Y. Fusion of multiple heterogeneous networks for predicting circRNA-disease associations. Sci. Rep. 9, 9605. https://doi.org/10.1038/s41598-019-45954-x (2019).
Yu, Z., Huang, F., Zhao, X., Xiao, W. & Zhang, W. Predicting drug-disease associations through layer attention graph convolutional network. Brief. Bioinform. https://doi.org/10.1093/bib/bbaa243 (2021).
Zhou, S., Sun, W., Zhang, P. & Li, L. Predicting pseudogene-miRNA associations based on feature fusion and graph auto-encoder. Front. Genet. 12, 781277. https://doi.org/10.3389/fgene.2021.781277 (2021).
Li, J. H., Liu, S., Zhou, H., Qu, L. H. & Yang, J. H. starBase v2.0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic acids Res. 42, D92–D97. https://doi.org/10.1093/nar/gkt1248 (2014).
Zheng, L. L. et al. dreamBase: DNA modification, RNA regulation and protein binding of expressed pseudogenes in human health and disease. Nucleic Acids Res. 46, D85-d91. https://doi.org/10.1093/nar/gkx972 (2018).
Huang, H. Y. et al. miRTarBase update 2022: An informative resource for experimentally validated miRNA-target interactions. Nucleic Acids Res. 50, D222-d230. https://doi.org/10.1093/nar/gkab1079 (2022).
van Laarhoven, T., Nabuurs, S. B. & Marchiori, E. Gaussian interaction profile kernels for predicting drug-target interaction. Bioinformatics (Oxford, England) 27, 3036–3043. https://doi.org/10.1093/bioinformatics/btr500 (2011).
Jiang, L., Ding, Y., Tang, J. & Guo, F. MDA-SKF: Similarity kernel fusion for accurately discovering miRNA-disease association. Front. Genet. 9, 618. https://doi.org/10.3389/fgene.2018.00618 (2018).
Chen, X., Zhu, C. C. & Yin, J. Ensemble of decision tree reveals potential miRNA-disease associations. PLoS Comput. Biol. 15, e1007209. https://doi.org/10.1371/journal.pcbi.1007209 (2019).
Wei, Z., Yao, D., Zhan, X. & Zhang, S. A clustering-based sampling method for miRNA-disease association prediction. Front. Genet. 13, 995535. https://doi.org/10.3389/fgene.2022.995535 (2022).
Chen, T. & Guestrin, C. XGBoost: A scalable tree boosting system. In Proceedings of the 22nd ACM SIGKDD international conference on knowledge discovery and data mining 785–794. https://doi.org/10.1145/2939672.2939785 (2016).
Dai, Q. et al. Predicting miRNA-disease associations using an ensemble learning framework with resampling method. Brief. Bioinform. https://doi.org/10.1093/bib/bbab543 (2022).
Zhao, Y., Chen, X. & Yin, J. Adaptive boosting-based computational model for predicting potential miRNA-disease associations. Bioinformatics (Oxford, England) 35, 4730–4738. https://doi.org/10.1093/bioinformatics/btz297 (2019).
Gu, M. & Wang, X. Pseudogene MSTO2P interacts with miR-128-3p to regulate coptisine sensitivity of non-small-cell lung cancer (NSCLC) through TGF-β signaling and VEGFC. J. Oncol. 2022, 9864411. https://doi.org/10.1155/2022/9864411 (2022).
Guo, M. & Zhang, X. LncRNA MSTO2P promotes colorectal cancer progression through epigenetically silencing CDKN1A mediated by EZH2. World J. Surg. Oncol. 20, 95. https://doi.org/10.1186/s12957-022-02567-5 (2022).
Guo, Y. et al. Inhibition of AURKB, regulated by pseudogene MTND4P12, confers synthetic lethality to PARP inhibition in skin cutaneous melanoma. Am. J. Cancer Res. 10, 3458–3474 (2020).
Acknowledgements
This research was funded by the Scientific research plan projects of Shaanxi Education Department (Grant No. 21J K0674).
Author information
Authors and Affiliations
Contributions
C.F. conceptualized the study, C.F. and M.D. performed the data collection, designed the method, C.F. drafted the manuscript. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fan, C., Ding, M. Inferring pseudogene–MiRNA associations based on an ensemble learning framework with similarity kernel fusion. Sci Rep 13, 8833 (2023). https://doi.org/10.1038/s41598-023-36054-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-36054-y
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
