
Abstract
The detection of pre-extensively (pre-XDR) and extensively drug-resistant tuberculosis (XDR-TB) is challenging. Drug-susceptibility tests for some anti-TB drugs, especially ethambutol (ETH) and ethionamide (ETO), are problematic due to overlapping thresholds to differentiate between susceptible and resistant phenotypes. We aimed to identify possible metabolomic markers to detect Mycobacterium tuberculosis (Mtb) strains causing pre-XDR and XDR-TB. The metabolic patterns of ETH- and ETO-resistant Mtb isolates were also investigated. Metabolomics of 150 Mtb isolates (54 pre-XDR, 63 XDR-TB and 33 pan-susceptible; pan-S) were investigated. Metabolomics of ETH and ETO phenotypically resistant subgroups were analyzed using UHPLC-ESI-QTOF-MS/MS. Orthogonal partial least-squares discriminant analysis revealed distinct separation in all pairwise comparisons among groups. Two metabolites (meso-hydroxyheme and itaconic anhydride) were able to differentiate the pre-XDR and XDR-TB groups from the pan-S group with 100% sensitivity and 100% specificity. In comparisons of the ETH and ETO phenotypically resistant subsets, sets of increased (ETH = 15, ETO = 7) and decreased (ETH = 1, ETO = 6) metabolites specific for the resistance phenotype of each drug were found. We demonstrated the potential for metabolomics of Mtb to differentiate among types of DR-TB as well as between isolates that were phenotypically resistant to ETO and ETH. Thus, metabolomics might be further applied for DR-TB diagnosis and patient management.
Similar content being viewed by others

Introduction
Tuberculosis (TB) is a major infectious disease caused by Mycobacterium tuberculosis (Mtb)1. Treatment of TB is becoming more difficult and challenging with the emergence of drug resistance. In 2020, 25,681 cases were reported of pre-extensively drug-resistant tuberculosis (pre-XDR-TB; resistant to isoniazid/rifampicin and any fluoroquinolone) or XDR-TB (resistant to rifampicin, plus any fluoroquinolone, plus bedaquiline (BDQ) and/or linezolid (LZD))1. The global treatment success rate for DR-TB remains low, at 59 percent2. In particular, the treatment success rate for XDR-TB was only 39%1. Selection of appropriate anti-TB drugs for treatment of each patient requires evaluation of the drug-resistance properties of their strain of Mtb: this is crucial for DR-TB treatment3.
Improper diagnosis of DR-TB not only raises the chance of drug resistance developing, but it can also result in lower therapeutic efficacy, increased side effects, and decreased patient compliance. Furthermore, according to a prior epidemiological investigation, the recurrence rate of DR-TB is around 61.3%, which is significantly higher than for drug-susceptible TB (27.9%)4. Transmission of primary DR-TB, especially pre-XDR and XDR-TB5,6 is a serious situation requiring novel tools for detection and classification. Therefore, the investigation of specific markers to rapidly identify DR-TB, especially pre-XDR/XDR-TB, is essential for early diagnosis and timely drug-regimen adjustment in patients.
Metabolomics, an emerging science of the “-omics” era, can be used in the identification and quantification of low-molecular-weight metabolites (< 1500 Da). Metabolic fingerprints can potentially be used to discriminate between states of health and disease7. In TB, metabolomics analysis of Mtb isolates is one such form of precision medicine offering personalized management of TB patients. Recently, metabolomics has been applied to generate metabolite patterns that can differentiate among DR-TB types8. Key metabolites associated with specific forms of TB disease have the potential for use as diagnostic biomarkers or indicators9,10. Therefore, this approach could strengthen the performance of drug-susceptibility testing (DST) and can also be used in personalized medicine for TB patient management. However, the number of studies applying this technology is limited.
For DST, discrepancies between genotypic and phenotypic test results can occur11,12,13,14. Determination of resistance status against some anti-TB drugs, especially ethambutol (ETH) and ethionamide (ETO), can be problematic due to overlapping minimum inhibitory concentration (MIC) thresholds to differentiate between susceptible and resistant genotypes5. Only 38% of isolates phenotypically resistant to ETO (and 35% for ETH) were also genotypically resistant5. In particular, there was an 80% disagreement for ETO between genotypic DST and the phenotypic test provided by the Mycobacteria Growth Indicator Tube (MGIT)15. Metabolomic analysis might provide additional markers to help resolve such discrepancies.
Here, we aimed to determine the metabolomic profiles of Mtb using ultra-high performance liquid chromatography coupled with the electrospray ionization-quadrupole-time of flight-mass spectrometry (UHPLC-ESI-QTOF-MS/MS) approach to distinguish pre-XDR and XDR-TB isolates from drug-susceptible isolates. We also determined the specific metabolites present in ETO- and ETH-resistant isolates of Mtb.
Results
Sample characteristics
One hundred and fifty Mtb isolates were included for metabolomic analysis. These comprised 54 pre-XDR and 63 XDR-TB isolates identified as such based on the previous WHO definitions1. Thirty-three pan-S isolates were used as the control group. Phenotypic DST results for all isolates were available for 14 anti-tuberculosis drugs (isoniazid (INH), rifampicin (RIF), streptomycin (STM), ethambutol (EMB), Kanamycin (KAN), para-amino salicylic acid (PAS), levofloxacin (LFX), ethionamide (ETO), gatifloxacin (GAT), moxifloxacin (MOX), linezolid (LND), clarithromycin (CLA), azithromycin (AZM) and amikacin (AMK). Lineage data were available for 54/150 (36%) of the isolates (Supplementary Table 1). There were statistically significant differences (p = 0.00006) in the proportions of lineage 2 (East-Asian) isolates represented among groups; pan-S (3/7 isolates, 42.85%), pre-XDR (27/31 isolates, 87.1%) and XDR-TB (16/16, 100%).
Overall metabolic profiles
The metabolites (n = 4071) of Mtb isolates with different susceptibility profiles are shown in Supplementary Table 2. The raw mass spectral data were processed by peak labeling, baseline filtering, retention-time correction, normalization and other standard procedures. In the positive and negative spectrum modes, 2526 and 1545 characteristic ion peaks were detected, respectively. Metabolites identified in both positive and negative modes were used for the downstream analysis. The standard quality-control (QC) strategy was applied, and their coefficient of variation (CV) is shown in Supplementary Table 3. The mean %CV across the 17 QC repeats showing the variation at 33.12%.
Metabolomic comparisons among pan-S, pre-XDR and XDR-TB isolates
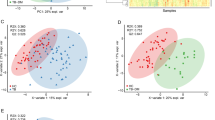
The patterns of metabolomes among pan-S, pre-XDR and XDR-TB isolates were analyzed using 3D-PCA. There was a distinct separation between pan-S (green) and drug-resistant isolates (blue and red) based on 3D-PCA (Fig. 1a). The top twelve metabolic markers for differentiation among pan-S, pre-XDR and XDR-TB groups are shown (Table 1). We then further analyzed the metabolomic pattern with heat-map analysis (Fig. 1b) using the twelve metabolites with the greatest variation between groups. The dendrogram shows the correlation between the relative intensities of metabolites in each sample. The decision tree for classification among pan-S, pre-XDR and XDR-TB groups is shown in Fig. 2. The probability of assigning each sample to the correct group was 100% (Table 2) and only two metabolites (meso-hydroxyheme and itaconic anhydride) were required. These two metabolites were not found in the human metabolite database (HMDB).

3D-PCA of pan-S, pre-XDR and XDR Mtb isolates (a). 3D-PCA was conducted to determine whether the groups could be distinguished based on metabolomics. There is a distinct separation between pan-S (green) and drug-resistant groups (blue and red). The pan-S Mtb isolates in this study can be separated from drug-resistant groups using metabolomic data. The heat map (b) shows the relative expression levels of the twelve metabolites with the greatest differences in levels among pan-S, pre-XDR and XDR-TB groups (n = 150 samples).

Decision tree for classification of drug-resistance status based on levels of two metabolomic markers (meso-hydroxyheme and itaconic anhydride). Sensitivity and specificity were both 100% for assignment of any sample to the correct group.
O-PLS-DA among pan-S, pre-XDR and XDR-TB isolates
Besides 3D-PCA, supervised O-PLS-DA was used for pairwise comparisons (pan-S vs. pre-XDR, pan-S vs. XDR-TB and pre-XDR vs. XDR-TB). The O-PLS-DA score plot showed a clear separation among the three Mtb groups (Fig. 3). When compared with pan-S, 1-carboxyvinylcarboxyphosphonate, C23H45O9P and L-iodopyranuronate are the most significantly increased metabolites in the pre-XDR group (Fig. 4, (upper row)). Glycerol arsenosugar, C17H34O4 and N-acetyl-D-muramate are the most significantly increased metabolites in the XDR-TB compared with the pan-S group (Fig. 4, (middle row)). Lacty (2) diphospho-(5′) adenosine, 1-carboxyvinylcarboxyphosphonate and C23H45O9P are the most significantly increased metabolites in XDR-TB compared with pre-XDR (Fig. 4, (lower row)).

Orthogonal partial least-squares discriminant analysis (O-PLS-DA) cross-validated score plots showing significant separation between all pairs of groups; (a) pan-S vs pre-XDR, (b) pan-S vs XDR-TB and (c) pre-XDR vs XDR-TB. Green circles depict pan-S isolates, orange triangles depict pre-XDR isolates and red squares depict XDR isolates. All three comparisons show p-value < 0.01 and predictive abilities of (a) are (R2X = 0.288, R2Y = 0.737 and Q2 = 0.729) (b) are (R2X = 0.365, R2Y = 0.946 and Q2 = 0.944) and (c) are (R2X = 0.232, R2Y = 0.789 and Q2 = 0.779).

Metabolites differing significantly among pan-S, pre-XDR and XDR-TB groups based on O-PLS-DA. The green color shows the pan-S group, the orange color shows the pre-XDR group, the red color shows the XDR-TB group. Each black dot represents a single M. tuberculosis isolate. Only the top three markers in each pairwise comparison are shown.
Metabolomic analysis of ethionamide- and ethambutol-resistant isolates
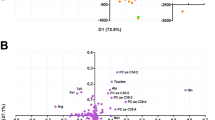
To ensure that identification of the metabolic markers found in ETO- and ETH-resistant isolates were not confounded by lineage-specific factors, the proportion of each lineage was compared between the resistant and susceptible groups. There was no significant difference (p = 0.875) in the proportion of lineage 2 isolates that were ETO resistant (12/12 isolates with available lineage data) versus those that were ETO susceptible (15/17 isolates with available lineage data) (Supplementary Table 4). Seven metabolites had increased levels (N-acetyl-D-muramate, 2,4,6-trinitrobenzene sulfonate, C24H50N1O7P, glycerophospholipids, C33H61O17P, C54H102O13 and C61H115O19P) and six had decreased levels (L-histidinol phosphate, cyclic-AMP, 2-iodophenol, 6-deoxy-5-ketofructose-1-phosphate, glycerol arsenosugar and fatty acyls) relative to ETO-susceptible isolates (Fig. 5 and Supplementary Table 5).

Metabolite markers to identify ethionamide resistance. The comparison is based on the five subsets of matched test and control subgroups (A to E) that contain various drug-resistance patterns. Only matched test strains (n = 12 from among ETO-resistant strains) and control strains (n = 17) were used. Then, a Venn diagram was created using the Venn function in R-programming. The specific controls (any isolates without ETO resistance) were used to create a new comparison. Metabolites present at higher or lower levels were analyzed after a comparison of metabolite expression levels between test and control. Only intersecting results found in all five comparisons were filtered. The numbers in brackets of each group refer to the number of isolates included. For ETO, seven increased and six decreased metabolites were identified.
Similarly, there was no significant difference (p = 0.905) in the proportion of lineage 2 isolates that were ETH resistant (11/12 isolates with available lineage data) versus those that were ETH susceptible (13/13 isolates with available lineage data) (Supplementary Table 4). Metabolomic analysis of ETH-resistant isolates revealed 15 metabolites with increased levels; O-acetyl-L-homoserine, (indol-3-yl)pyruvate, (7,8-dihydropterin-6-yl) methyl diphosphate, (D-alanyl)adenylate, S-(hydroxymethyl) bacillithiol, 2,4-dichlorotoluene, 3-bromopropanesulfonate, metosulam, C19H38O4, C21H42O4, C27H54O2, gycerophospholipids, C21H44N1O7P, C76H146O6 and glycerolipids (Fig. 6 and Supplementary Table 6). The level of one metabolite (bromoacetate) was lower in ETH-resistant isolates.

Metabolite marker to identify ethambutol (ETH) resistance. The comparison is based on each of the six subsets of the matched test and control (A to F) that contain various drug resistance patterns. Only the matched Test and Control; Test (n = 12 from 17 ETH resistant strains) and Control (n = 13) were used. The subset of isolates with (test) and without (control) ETH phenotypic resistance was filtered. Then, a Venn diagram was created using the Venn function in R-programming. The specific controls (any isolate without ETH resistance) were used to create a new comparison. Metabolites present at higher or lower levels were analyzed after a comparison of metabolite expression levels between test and control. Only intersect results found in all six comparisons were filtered. The numbers in the brackets of each group confer to the number of isolates. For ETH, fifteen increased and one decreased metabolite were found.
Discussion
Only one previous study has investigated metabolic markers of Mtb that might distinguish pan-S (n = 18), MDR (n = 17) and XDR (n = 18) isolates16. That study used a relatively small sample size and did not identify any significant metabolites that could differentiate between their MDR and XDR isolates. Another study investigated TB-patient serum metabolomics and discovered four potential biomarkers: N1-methyl-2-pyridone-5-carboxamide (N1M2P5C), 1-myristoyl-sn-glycerol-3-phosphocholine (MG3P), caprylic acid (CA), and D-xylulose (DX) that, in combination, could discriminate between MDR-TB (n = 30) and pan-S (n = 30) with both sensitivity and specificity of 86.7%8. Here, we analyzed metabolomic profiles of Mtb isolates of known drug-resistance status to differentiate between pre-XDR and XDR-TB. We also specifically analyzed the metabolic pattern of isolates resistant or sensitive to ETO and ETH, two drugs for which there is frequently a discrepancy between genotypic and phenotypic DST results5,17,18.
We expected that Mtb isolates with different drug-susceptibility profiles would be metabolically diverse, and that untargeted metabolomics should show metabolic patterns correlated with drug susceptibility or resistance. Based on 3D-PCA, clear differentiation between the pan-S and drug-resistant isolates (pre-XDR and XDR-TB) was found. However, the pre-XDR and XDR-TB isolates were not totally separated. Then, we used a decision tree as an approach to differentiate among the three TB groups. Interestingly, only two metabolites (meso-hydroxyheme and itaconic anhydride) were required to provide 100% sensitivity and 100% specificity for distinguishing among the three groups. Meso-hydroxyheme is a key intermediate of the Mycobacterium heme utilization degrader (MhuD) reaction19. MhuD converts host-derived heme into iron by degrading it19,20,21. Iron is required for numerous essential biological processes and is associated with the aminoglycoside-resistance mechanism of Mtb22. Iron and heme are thought to be potential targets for future drug development due to their uptake into pathogens23. The higher level of meso-hydroxyheme in resistant isolates could explain the difference among drug-resistance types. However, our analysis revealed that XDR-TB isolates had lower meso-hydroxyheme compared to pan-S and pre-XDR. Possibly, XDR-TB isolates require less iron for survival than pan-S and pre-XDR isolates. The limiting of available iron in the human host, which is sequestered in high-affinity binding proteins such as heme, is an important part of the innate immune response to bacterial infections24. Lowering the amount of iron required for survival in the host could be one of the adaptations of XDR-TB isolates. Another metabolite that was differentially expressed among DR-TB types is itaconic anhydride. This metabolite is an inhibitor of isocitrate lyase, a key enzyme that enables the bacilli to persist under oxidative-stress conditions25. Possibly, a drug-resistant isolate is more fit to survive in hostile environments26. Neither of these two metabolites has any matches in the human metabolic database. Therefore, besides being diagnostic markers for DST, these metabolites might potentially find a use in treatment monitoring to detect any change of drug susceptibility of the pathogen in patients. To confirm the clinical application of these markers, further study investigating these metabolites in patient serum during the course of treatment is needed.
We also used O-PLS-DA for pairwise analyses among the three groups (pan-S, pre-XDR and XDR-TB) to identify metabolic markers. All pairwise comparisons yielded clear distinctions, so we focused on the three most-significantly increased metabolites in each case. Interestingly, levels of C23H45O9P differentiated between pre-XDR and pan-S and also between pre-XDR and XDR-TB isolates. This metabolite therefore shows great potential for discrimination among drug-resistance types. The metabolites identified as important using O-PLS-DA differed from those identified using the decision tree because the two methods use different algorithms: one identifies markers that can be used to differentiate among three groups whereas the other makes only pairwise comparisons. Nonetheless, based on the metabolic patterns that are clearly different between groups, the potential applicability remains for the metabolome to distinguish isolates with different drug-resistance properties.
Phenotypic DST of Mtb for certain drugs can be problematic due to the uncertain MIC cut-off values for some drugs and the extent of reproducibility of such tests27. Previously, our research group reported that 65% of isolates that were phenotypically ETH resistant and 62% that were phenotypically ETO resistant did not yield the same results according to genotypic resistance analysis5. Phenotypic drug-resistance profiles are not always associated with distinctive metabolic fingerprints16. Rego et al. (2021) compared a relatively low number of Mtb isolates (n = 53) and did not investigate specific drugs, especially ETH and ETO. In our study, we attempted to identify the metabolomic patterns associated with phenotypic resistance against ETH and ETO using subsets of DR Mtb isolates. Due to the high level of genotypic /phenotypic discrepancy for ETO and ETH (Supplementary Table 4), we relied on the gold standard of drug-susceptibility testing using the agar proportion method28.
We analyzed the relative amounts of metabolites in ETO-resistant isolates (that were also resistant to other drugs) and subtracted from these the expression levels in isolates that were not resistant to ETO (but some may have been resistant to other drugs). Only the common metabolites found in ETO-resistant isolates and not in any ETO-susceptible isolates were counted as ETO resistance-specific metabolites. We found seven increased and six decreased metabolites specific to ETO-resistant isolates. Changes in these metabolites might be associated with the ETO-resistance mechanism. The most increased metabolites included glycerophospholipids (GPL) and C61H115O19P. The literature suggests that the bacterial stress sensor may respond directly to GPL concentration29. Previously, comparative lipidomic analysis revealed an increased amount of fatty acyls and GPL in DR-TB: both metabolites are important for Mtb virulence and pathogenicity30. The most important metabolite showing decreased levels in ETO-resistant isolates was cyclic-AMP (cAMP). A variety of cyclic nucleotides are utilized by Mtb, including cAMP, cyclic-di-AMP (c-di-AMP) and cyclic-di-GMP (c-di-GMP) that regulate bacterial cell physiology and disrupt signaling in host cells31. The cAMP is important for gene regulation in mycobacteria, and the ability to secrete cAMP into host macrophages during infection contributes to Mtb pathogenesis32. C-di-GMP has been reported to promote the resistance of Mtb to ETH, possibly because increased Ethr activity suppresses ethA expression, lowering the amount of active ETH in the bacterial cytoplasm33. However, the role of cAMP in the drug-resistance mechanism in Mtb is unknown and needs to be further investigated. Most of the ETH- and ETO-resistant isolates used in our study belonged to lineage 2. This lineage is strongly associated with MDR-TB phenotypes34,35 and acquisition of resistance34. Due to the lineage 2 proportion between ETH/ETO resistant and susceptible isolates are relatively comparable, the metabolomic markers we found is therefore not confounded by the lineages of Mtb.
We used a similar approach to identify the metabolites associated with ETH resistance. In ETH-resistant isolates, fifteen metabolites exhibited increased expression levels and only one had decreased levels. The most increased metabolites included (indol-3-yl)pyruvate and 3-bromopropanesulfonate. The latter is a specific inhibitor of methyl-CoM reductase and completely inhibits dechlorination of 1,2-DCA but has not previously been considered in the context of drug resistance36. The most decreased metabolite was bromoacetate, used by researchers as a toxic small molecule to model the selective pressures imposed by antibiotics and anthropogenic toxins in Escherichia coli. Further study of these changed metabolites is needed to explain the ETH-resistance mechanism of Mtb. A previous study used untargeted urine metabolomics with gas chromatography-time of flight mass spectrometry (GC-TOF–MS) to investigate the drug metabolism of a TB patient cohort (n = 20)37. They identified 2-aminobutyric acid (AABA) as the specific metabolite associated with ethambutol resistance. However, AABA was not on our list of ETH-resistance metabolites. This might be due to differences in study design, especially given that our model investigated pathogen metabolites compared to human metabolites.
Like ETO resistance-associated metabolites, no information is available on whether the metabolites specifically found in ETH-resistant isolates are actually associated with mechanisms of resistance. Although statistically significant, changes in metabolite levels in each group were usually less than two-fold, which might or might not be biologically meaningful and/or reproducible. Additional studies are needed to fill this knowledge gap.
In laboratory diagnosis, pathogen detection using microscopic and/or molecular techniques including drug-susceptibility tests are used to identify DR-TB38. However, these techniques are laborious, and the DST results are sometimes discordant between methods. In the advanced “omics” era, our findings might support the future development of metabolomics-based TB diagnosis. In a clinical setting, the metabolite patterns of Mtb could also be useful. Acquired resistance might occur, defined as resistance to one or more anti-TB drugs in isolates recovered from patients who had received previous anti-TB treatment39. In acquired resistance, metabolomics of Mtb can change according to the resistance phenotype40. Here, we focused on untargeted LC–MS-based metabolomics. The major advantage of this approach is the discovery of novel metabolites in relation to the study context. We showed that metabolomics of Mtb could be used to distinguish between various DR-TB strains as well as between isolates that were phenotypically resistant to ETO and ETH. However, the reproducibility of the metabolomic analysis from the machine is still one of the concerns as reflected with %CV over 30% in the QC sample set. This technical limitation could be managed by repeated sampling, an approach which would increase the cost per test for an already expensive technique. Therefore, the application of metabolomics as a diagnostic aid for personalized treatment and monitoring of TB patients is still limited due to the high cost, low reproducibility, and requirement for sophisticated equipment. As with other advanced methods, such as high-throughput DNA sequencing, we can expect the cost of mass spectrometry to decrease in the future, making it suitable for affordable and routine use. Much more research and development are needed to make this technology cost effective, easy to use and practical in real-world settings. The work reported here has laid the foundation for further study and validation.
Limitations of our study should be discussed. We analyzed only pre-XDR and XDR-TB strains compared to pan-S. As MDR-TB Mtb isolates are resistant to INH and RIF, a state that is shared by pre-XDR and XDR-TB, we included only highly resistant isolates in the study. We assumed that metabolomics changed more as resistance increased. No clinical data such as the treatment regimens of TB patient were available. The proportions of isolates of different lineages among groups were significantly different. Therefore, the unequal representation of lineages might have influenced the metabolite markers found. Additional study controlling for lineage before testing drug-resistance pattern should be done. For some Mtb isolates, there were no available phenotypic DST data. Therefore, these isolates could not be included for metabolomic analysis relating to ETO and ETH resistance. Although the sample size for ETO and ETH metabolomic analysis was limited, lineage representation did not significantly differ among the datasets used and the analysis approach was very stringent, giving us confidence in the findings. Because of the limited number of samples available, a testing/validation set analysis approach could not be used. Therefore, the random sampling approach indicating 100% accuracy might not truly reflect the actual performance of metabolomic analysis for DR-TB identification. The metabolomic data used in this study was from normal, cultured cells that were not subject to stresses such as presence of drugs. To increase the safety of the analysis protocol, we used only dead bacterial cells for sample preparation. We avoided experiments that included activation of the bacteria with anti-TB drugs to stimulate the expression of bacterial metabolites.
The WHO Laboratory Biosafety Manual, has categorized drug-susceptible, drug-resistant, and multidrug-resistant Mtb strains into biohazard risk category 3 whereas XDR Mtb strains were assigned to risk group 4, the highest risk category for human and community health41. We used heat inactivation of the bacteria to ensure biosafety while working with XDR Mtb strains and applied the same method to all other strains for consistency. However, this approach could potentially alter the metabolome of bacteria subjected to heat and also cause the degradation of some metabolites. Therefore, the metabolomic analysis of heat-inactivated Mtb might not accurately reflect the normal metabolism and might give misleading information on the biology of Mtb. This is the key limitation of our study that should be considered when interpreting the results. Metabolomic profiles obtained following extraction methods that do not use heat inactivation42 should be investigated in the future.
The high number of bacterial cells obtained in culture yields a high concentration of metabolites, thus increasing the sensitivity for UHPLC-ESI-QTOF-MS/MS detection and analysis. However, it is uncertain that large amounts of these same metabolites would be released in clinical samples. The significant metabolomic markers we found were less than two-fold different between test and control groups. It is unclear whether this difference between the Mtb groups is biological meaningful and/or clinically significant. This needs to be further investigated. We attempted to analyze the metabolites associated with para-aminosalicylic acid resistance, but too few resistant and control isolates were available to reveal any significant metabolite marker.
In conclusion, we reported that Mtb metabolomics could distinguish among pan-S, pre-XDR and XDR according to levels of two metabolites (meso-hydroxyheme and itaconic anhydride). We also demonstrated the potential for metabolomics of Mtb to differentiate between isolates that were phenotypically resistant to ETO and ETH.
Methods
Mtb isolates and setting
Mtb isolates from our previous project43, for which phenotypic/genotypic drug-susceptibility results were available (total n = 150; 33 pan-S, 54 pre-XDR and 63 XDR-TB), were used in this study. The previous definition of XDR-TB, (multidrug resistant-TB (MDR-TB)) was that such isolates were resistant to any fluoroquinolone and at least one of three second-line injectable drugs (capreomycin, kanamycin and amikacin)44. We used this definition to classify TB groups in our previous drug-susceptibility test (DST) system. These isolates were collected from 1998 to 2013 from various provinces in Thailand. From 150 isolates, whole-genome sequence datasets were available for 54 isolates43,45 and lineage classification of these was done using RD-Analyzer46. Notably, some isolates exhibit resistance to more than one drug. This study used bacteria cultured from stored stock. Each Mtb isolate was cultured on Lowenstein-Jensen medium at 37 °C for 4 weeks, multiple colonies were scraped from the tubes, resuspended in sterile distilled water, stored at − 70 °C and material for the metabolomic analysis was extracted on the next day. Each sample was associated with information including the UHPLC-MS/MS-based metabolomics data (.d files) but without information that could lead to identification of any participant: no informed consent was required.
Drug-susceptibility testing
Phenotypic DSTs for anti-TB drugs were performed using the standard agar proportional method47. Drug critical concentrations (CCs) used were 0.2 mg/L for isoniazid, 1.0 mg/L for rifampicin, 5.0 mg/L for ethambutol and ethionamide, 6.0 mg/L for amikacin and kanamycin, and 2.0 mg/L for streptomycin, p-amino salicylic acid, ofloxacin, levofloxacin, moxifloxacin and gatifloxacin. A critical proportion value of 1% was used. A culture that had 1% or more growth on the medium containing the critical concentration of the anti-TB drug is considered as resistant. Genotypic drug susceptibility test results of ETO and ETH was based on our previous study5 that analyzed using TB-profiler48.
Sample preparation
Stock cultures of Mtb inactivated by heat at 95 °C for 30 min were used. Colonies were suspended in HPLC grade water, and optical density was adjusted at 600 nm to ODs equal to 5, and 1500 μL of the resulting bacterial suspension were used. The metabolomics extraction was performed following the protocol of P A. Vorkas et al.49. Ten microliters aliquots from each sample were pooled and mixed in a 1.5 ml tube to make the quality-control (QC) sample and 120 µl of this was transferred to an HPLC glass insert.
UHPLC-ESI-QTOF-MS/MS analysis
The aqueous-phase extracts of each sample were analyzed on a reverse-phase platform. The separation part was performed using the UHPLC system (Bruker, Germany) Bruker intensity solo HPLC C18 2.1 × 100 mm, 2 μm column (Bruker, Germany). The column temperature was set at 40 °C and the autosampler temperature was set at 4 °C. Mobile phase A was water 100% with 0.1% formic acid (FA) and mobile phase B was acetonitrile 100% with 0.1% FA. The flow rate was set at 0.35 ml/min and the elution gradient was set as follows: 99% A (0.0–2.0 min, 0.25 ml/min), 99–1% A (2.0–17.0 min, 0.25 ml/min), 1% A (17.0–20.0 min, 0.25 ml/min), 1–99% A (20.0–20.1 min, 0.25–0.35 ml/min), 99% A (20.1–28.3 min, 0.35 ml/min), 99% A (28.3–28.5 min, 0.35–0.25 ml/min), 99% A (28.5–30.0 min, 0.25 ml/min). Injection volume of sample (7 μl) was applied for both positive and negative ionization polarity modes. The mass spectroscopy part was performed using the compact ESI-Q-TOF system (Bruker, Germany). Sodium formate (2 mM sodium hydroxide, 0.1% FA, 50% isopropyl alcohol) was directly injected as an external calibrant with flow rate 0.5 μl/min. The condition in positive ionization polarity mode: mass range 50–1300 m/z, cone voltage 35 V, capillary voltage 4500 V, source temperature 220 °C, desolvation temperature 220 °C, desolvation gas flow 8 L/min. The conditions in negative ionization polarity mode: m/z range: 50–1300 m/z, cone voltage 31 V, capillary voltage 4500 V, source temperature 220 °C, desolvation temperature 220 °C, desolvation gas 8 L/min. The standard QC strategy was applied for the UHPLC-MS analysis. A pool of all samples was prepared as for QC. This QC sample was injected at the beginning, following every 10 sample injections, and at the end to estimate the instrument stability and determine reproducibility. Following sample analysis, QC sample dilutions, 1:2, 1:4, 1:8 and 1:16 in the reconstitution buffer, were run in the MS/MS mode, followed by extraction of a blank sample and reconstitution blank in MS mode to estimate the complements and impurity of the extraction and reconstitution solvent. The UHPLC-MS/MS-based metabolomics dataset is shown in Supplementary Table 2.
Data analysis
The metabolomics data from 150 Mtb samples were analyzed. CompassXport.exe v3.0.9.2 was used to convert data to .mzXML format. R-program version 4.1.2 (https://cran.r-project.org) was used in this study. The R-program library “faahKO” was used to convert mzXML to Computable Document Format (CDF)50. The sample-processing function of the MAIT package was applied to take a set of files containing LC/MS sample data and perform peak detection, retention-time correction and peak grouping51. Following this, the peakAnnotation function was used as spectra constructor and peak annotator. Accurate m/z (< 5 ppm) measurements of detected chromatographic peaks were first matched to metabolites from online MS databases (Mycobacterium_lipid3, Mycobacterium metabolite_Mycomass database3, Mtb database52 and NTM metabolite_BIOCYC database52). Study design and flow of the metabolomic analysis is shown (Fig. 7).

Study design and flow of metabolomic analysis. The study included two parts; analysis among pan-S, pre-XDR and XDR-TB isolates (a) and analysis of metabolite markers for identifying ETO/ETH resistance (b). Red letters depict the relevant figure and table results obtained in each step. Pan-S; pan-susceptible, pre-extensively resistance; pre-XDR, extensively resistance; XDR, Ultra-high performance liquid chromatography tandem mass spectrometry; UHPLC MS/MS, Orthogonal partial least-squares discriminant analysis; O-PLS-DA.
To distinguish among pan-S, pre-XDR and XDR-TB isolates based on metabolomics, after cleaning the data and identification of the metabolites, all statistical analysis including multidimensional statistical analysis, (a) intensity heat map (b) 3D principal-component analysis (3D-PCA) (c) relative-intensity box plot (d) and (e) decision-tree analysis, were performed using R-program. Jackknife sampling technique was used for the performance analysis, we sampled one isolate from each group then trained the remaining samples, pan-S (n = 32), pre-XDR (n = 53) and XDR-TB (n = 62) groups, and repeated this step for all samples in each group. We then compared metabolic markers with the human metabolomics database (HMDB). The decision trees for classification among pan-S, pre-XDR and XDR-TB were generated using rpart (R-program)53.
Orthogonal partial least-squares discriminant analysis (O-PLS-DA) was also used for classification among groups. The dataset of samples arranged in columns (pan-S, pre-XDR and XDR-TB) and variables in rows (metabolite intensities) was prepared as a .CSV file and the O-PLS-DA score was calculated and visualized using Metaboanalyst 5.0 (http://www.metaboanalyst.ca/faces/home.xhtml)54. Chi-square or Fisher's exact test was used for comparisons of lineage proportions between groups.
For ETH (or ETO) metabolic markers, the subset of isolates with (test) and without (control) ETH (or ETO) phenotypic resistance was filtered. Then, a Venn diagram for subset analysis was created using the Venn function in R-programming. The specific controls (any isolates without ETH (or ETO) resistance) were used to create a new comparison. Metabolites present at higher or lower levels were analyzed after a comparison of metabolite expression levels between test and control.
Ethical approval
The study protocol was approved by the Center for Ethics in Human Research, Khon Kaen University (HE601249).
Data availability
The datasets generated and analyzed during the current study are available in the MassIVE repository with accession number MSV000091354, [https://massive.ucsd.edu/ProteoSAFe/dataset.jsp?accession=MSV000091354].
References
World Health Organization, Global Tuberculosis Report 2021, Geneva, Switzerland (2021).
Organization, W. H. Factsheet Global TB report 2021.
Layre, E. et al. A comparative lipidomics platform for chemotaxonomic analysis of Mycobacterium tuberculosis. Chem. Biol. 18, 1537–1549. https://doi.org/10.1016/j.chembiol.2011.10.013 (2011).
Sun, Y., Harley, D., Vally, H. & Sleigh, A. Impact of multidrug resistance on tuberculosis recurrence and long-term outcome in China. PLoS ONE 12, e0168865. https://doi.org/10.1371/journal.pone.0168865 (2017).
Nonghanphithak, D. et al. Whole-genome sequence analysis and comparisons between drug-resistance mutations and minimum inhibitory concentrations of Mycobacterium tuberculosis isolates causing M/XDR-TB. PLoS ONE 15, e0244829. https://doi.org/10.1371/journal.pone.0244829 (2020).
Trisakul, K. et al. High clustering rate and genotypic drug-susceptibility screening for the newly recommended anti-tuberculosis drugs among global extensively drug-resistant Mycobacterium tuberculosisisolates.Emerg. Microbes Infect. 11(1), 1857–1866. https://doi.org/10.1080/22221751.2022.2099304 (2022).
Enche-Ady, C. N. A. et al. Metabolomic-guided discovery of Alzheimer’s disease biomarkers from body fluid. J. Neurosci. Res. 95, 2005–2024. https://doi.org/10.1002/jnr.24048 (2017).
Huang, H. et al. The novel potential biomarkers for multidrug-resistance tuberculosis using UPLC-Q-TOF-MS. Exp. Biol. Med. 245, 501–511. https://doi.org/10.1177/1535370220903464 (2020).
Preez, I. D., Luies, L. & Loots, D. T. Metabolomics biomarkers for tuberculosis diagnostics: Current status and future objectives. Biomark. Med. 11, 179–194. https://doi.org/10.2217/bmm-2016-0287 (2017).
Zhang, P. et al. Mass spectrometry-based metabolomics for tuberculosis meningitis. Clin. Chim. Acta Int. J. Clin. Chem. 483, 57–63. https://doi.org/10.1016/j.cca.2018.04.022 (2018).
Aung, W. W. et al. Phenotypic and genotypic analysis of anti-tuberculosis drug resistance in Mycobacterium tuberculosis isolates in Myanmar. Ann. Lab. Med. 35, 494–499. https://doi.org/10.3343/alm.2015.35.5.494 (2015).
Brandao, A. P. et al. Transmission of Mycobacterium tuberculosis presenting unusually high discordance between genotypic and phenotypic resistance to rifampicin in an endemic tuberculosis setting. Tuberculosis 125, 102004. https://doi.org/10.1016/j.tube.2020.102004 (2020).
Rafael, L. L. et al. Discordant results between genotypic and phenotypic assays (Xpert MTB/RIF vs. BACTEC MGIT 960 system) for detection of RIF-resistant Mycobacterium tuberculosis isolates in a high burden region. Infect. Genet. Evolut. J. Mol. Epidemiol. Evolut. Genet. Infect. Dis. 96, 105142. https://doi.org/10.1016/j.meegid.2021.105142 (2021).
Kardan-Yamchi, J. et al. Assessment of the GenoType MTBDRsl VER 2.0 compared to the phenotypic drug susceptibility testing and whole genome sequencing for the rapid detection of resistance to fluoroquinolone and second-line injectable drugs among rifampicin-resistant Mycobacterium tuberculosis isolates. Arch. Microbiol. 203, 3989–3996. https://doi.org/10.1007/s00203-021-02387-3 (2021).
Jajou, R. et al. WGS more accurately predicts susceptibility of Mycobacterium tuberculosis to first-line drugs than phenotypic testing. J. Antimicrob. Chemother. 74, 2605–2616. https://doi.org/10.1093/jac/dkz215 (2019).
Rego, A. M. et al. Metabolic profiles of multidrug resistant and extensively drug resistant Mycobacterium tuberculosis unveiled by metabolomics. Tuberculosis 126, 102043. https://doi.org/10.1016/j.tube.2020.102043 (2021).
Faksri, K. et al. Comparisons of whole genome sequencing and phenotypic drug susceptibility testing for Mycobacterium tuberculosis causing MDR-TB and XDR-TB in Thailand. Int. J. Antimicrob. Agents. https://doi.org/10.1016/j.ijantimicag.2019.04.004 (2019).
Pillay, S. et al. Xpert MTB/XDR for detection of pulmonary tuberculosis and resistance to isoniazid, fluoroquinolones, ethionamide, and amikacin. Cochrane Database Syst. Rev. 5, CD014841. https://doi.org/10.1002/14651858.CD014841.pub2 (2022).
Matsui, T. et al. Unique coupling of mono- and dioxygenase chemistries in a single active site promotes heme degradation. Proc. Natl Acad. Sci. U.S.A. 113, 3779–3784. https://doi.org/10.1073/pnas.1523333113 (2016).
Graves, A. B. et al. Crystallographic and spectroscopic insights into heme degradation by Mycobacterium tuberculosis MhuD. Inorg. Chem. 53, 5931–5940. https://doi.org/10.1021/ic500033b (2014).
Thakuri, B., O’Rourke, B. D., Graves, A. B. & Liptak, M. D. A dynamic substrate is required for MhuD-catalyzed degradation of heme to mycobilin. Biochemistry 60, 918–928. https://doi.org/10.1021/acs.biochem.0c00892 (2021).
Sharma, D. & Bisht, D. Role of bacterioferritin & ferritin in M. tuberculosis pathogenesis and drug resistance: A future perspective by interactomic approach. Front. Cellular Infect. Microbiol. 7, 240. https://doi.org/10.3389/fcimb.2017.00240 (2017).
Chao, A., Sieminski, P. J., Owens, C. P. & Goulding, C. W. Iron acquisition in Mycobacterium tuberculosis. Chem. Rev. 119, 1193–1220. https://doi.org/10.1021/acs.chemrev.8b00285 (2019).
Skaar, E. P. The battle for iron between bacterial pathogens and their vertebrate hosts. PLoS Pathog. 6, e1000949. https://doi.org/10.1371/journal.ppat.1000949 (2010).
May, E. E., Leitao, A., Tropsha, A. & Oprea, T. I. A systems chemical biology study of malate synthase and isocitrate lyase inhibition in Mycobacterium tuberculosis during active and NRP growth. Comput. Biol. Chem. 47, 167–180. https://doi.org/10.1016/j.compbiolchem.2013.07.002 (2013).
Lee, J. H. et al. Isoniazid resistance without a loss of fitness in Mycobacterium tuberculosis. Nature Commun. 3, 753. https://doi.org/10.1038/ncomms1724 (2012).
Angeby, K., Jureen, P., Kahlmeter, G., Hoffner, S. E. & Schon, T. Challenging a dogma: Antimicrobial susceptibility testing breakpoints for Mycobacterium tuberculosis. Bull. World Health Organ. 90, 693–698. https://doi.org/10.2471/BLT.11.096644 (2012).
Kent, P. T. Public Health Mycobacteriology: A Guide for the Level III Laboratory (Public Health Service, Centers, 1985).
Grabowicz, M. & Silhavy, T. J. Envelope stress responses: An interconnected safety net. Trends Biochem. Sci. 42, 232–242. https://doi.org/10.1016/j.tibs.2016.10.002 (2017).
Pal, R., Hameed, S., Kumar, P., Singh, S. & Fatima, Z. Comparative lipidomics of drug sensitive and resistant Mycobacterium tuberculosis reveals altered lipid imprints. 3 Biotech 7, 325. https://doi.org/10.1007/s13205-017-0972-6 (2017).
Johnson, R. M. & McDonough, K. A. Cyclic nucleotide signaling in Mycobacterium tuberculosis: An expanding repertoire. Pathog. Dis. https://doi.org/10.1093/femspd/fty048 (2018).
Knapp, G. S. & McDonough, K. A. Cyclic AMP signaling in mycobacteria. Microbiol. Spectr. https://doi.org/10.1128/microbiolspec.MGM2-0011-2013 (2014).
Zhang, H. N. et al. Cyclic di-GMP regulates Mycobacterium tuberculosis resistance to ethionamide. Sci. Rep. 7, 5860. https://doi.org/10.1038/s41598-017-06289-7 (2017).
Klotoe, B. J. et al. Genomic characterization of MDR/XDR-TB in Kazakhstan by a combination of high-throughput methods predominantly shows the ongoing transmission of L2/Beijing 94–32 central Asian/Russian clusters. BMC Infect. Dis. 19, 553. https://doi.org/10.1186/s12879-019-4201-2 (2019).
Thawornwattana, Y. et al. Revised nomenclature and SNP barcode for Mycobacterium tuberculosis lineage 2. Microb. Genomics https://doi.org/10.1099/mgen.0.000697 (2021).
Holliger, C., Kengen, S. W., Schraa, G., Stams, A. J. & Zehnder, A. J. Methyl-coenzyme M reductase of Methanobacterium thermoautotrophicum delta H catalyzes the reductive dechlorination of 1,2-dichloroethane to ethylene and chloroethane. J. Bacteriol. 174, 4435–4443. https://doi.org/10.1128/jb.174.13.4435-4443.1992 (1992).
Das, M. K. et al. Global urine metabolomics in patients treated with first-line tuberculosis drugs and identification of a novel metabolite of ethambutol. Antimicrob. Agents Chemother. 60, 2257–2264. https://doi.org/10.1128/AAC.02586-15 (2016).
Singh, P., Saket, V. K. & Kachhi, R. Diagnosis of TB: From conventional to modern molecular protocols. Front. Biosci. 11, 38–60. https://doi.org/10.2741/E845 (2019).
The WHO/IUATLD Global project on anti-tuberculosis drug resistance surveillance 1994–1997. Geneva, World Health Organization, 1997 (WHO/TB/97.229).
Idle, J. R. & Gonzalez, F. J. Metabolomics. Cell Metab. 6, 348–351 (2007).
Xu, Y., Wang, G. A. & Xu, M. Biohazard levels and biosafety protection for Mycobacterium tuberculosis strains with different virulence. Biosaf. Health 2, 135–141. https://doi.org/10.1016/j.bsheal.2020.04.001 (2020).
de Carvalho, L. P. et al. Metabolomics of Mycobacterium tuberculosis reveals compartmentalized co-catabolism of carbon substrates. Chem. Biol. 17, 1122–1131. https://doi.org/10.1016/j.chembiol.2010.08.009 (2010).
Faksri, K. et al. Comparative whole-genome sequence analysis of Mycobacterium tuberculosis isolated from tuberculous meningitis and pulmonary tuberculosis patients. Sci. Rep. 8, 4910. https://doi.org/10.1038/s41598-018-23337-y (2018).
World Health Organization. (2013). Definitions and reporting framework for tuberculosis—2013 revision: updated December 2014 and January 2020. World Health Organization. https://apps.who.int/iris/handle/10665/79199.
Nonghanphithak, D. et al. Clusters of drug-resistant Mycobacterium tuberculosis detected by whole-genome sequence analysis of nationwide sample, Thailand, 2014–2017. Emerg. Infect. Dis. 27, 813–822. https://doi.org/10.3201/eid2703.204364 (2021).
Faksri, K., Xia, E., Tan, J. H., Teo, Y. Y. & Ong, R. T. In silico region of difference (RD) analysis of Mycobacterium tuberculosis complex from sequence reads using RD-Analyzer. BMC Genomics 17, 847. https://doi.org/10.1186/s12864-016-3213-1 (2016).
Guidelines for surveillance of drug resistance in tuberculosis. WHO Geneva/IUATLD Paris. International union against tuberculosis and lung disease. Int J Tuberc Lung Dis 2, 72–89 (1998).
Mahe, P., El Azami, M., Barlas, P. & Tournoud, M. A large scale evaluation of TBProfiler and Mykrobe for antibiotic resistance prediction in Mycobacterium tuberculosis. PeerJ 7, e6857. https://doi.org/10.7717/peerj.6857 (2019).
Vorkas, P. A. et al. Untargeted UPLC-MS profiling pipeline to expand tissue metabolome coverage: Application to cardiovascular disease. Anal. Chem. 87, 4184–4193. https://doi.org/10.1021/ac503775m (2015).
Smith, C.A. faahKO: Saghatelian et al. (2004) FAAH knockout LC/MS data. R package version 1.38.0 (2022). https://doi.org/10.1021/bi0480335.
Fernandez-Albert, F., Llorach, R., Andres-Lacueva, C. & Perera, A. An R package to analyse LC/MS metabolomic data: MAIT (Metabolite Automatic Identification Toolkit). Bioinformatics 30, 1937–1939. https://doi.org/10.1093/bioinformatics/btu136 (2014).
Caspi, R. et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 44, D471-480. https://doi.org/10.1093/nar/gkv1164 (2016).
Therneau, T., Atkinson, B., & Ripley, B. Rpart: Recursive Partitioning. R Package Version 4.1-3 (2013). http://CRAN.R-project.org/package=rpart.
Pang, Z. et al. MetaboAnalyst 5.0: Narrowing the gap between raw spectra and functional insights. Nucleic Acids Res. 49, W388–W396. https://doi.org/10.1093/nar/gkab382 (2021).
Acknowledgements
We thank for RCEID and the Faculty of Medicine, KKU for laboratory support during the metabolomics extraction. We would like to thank the Khon Kaen University International Phenome Laboratory for laboratory support during the LC-MS/MS analysis. We would like to acknowledge Prof. David Blair for editing the manuscript via Publication Clinic KKU.
Funding
This work was supported by National Research council of Thailand and Research and Diagnostic Center for Emerging Infectious Diseases (RCEID), Khon Kaen University, Khon Kaen, Thailand. This work was also supported by the Royal Golden Jubilee (RGJ)-Ph.D. program Grant (PHD/0044/2560) of the Thailand Research Fund (TRF). These funding sources had no role in design of the study and collection, analysis, interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
K.F. designed the project. P.C. and D.N. performed Mtb culture and drug susceptibility testing. P.C. performed metabolomics extraction, P.C. and J.P. performed LC-MS instrumentation. K.F. acquired the research fund and supervised research assistants, P.C. B.K. and K.F. performed data analysis and prepared the figures. P.C. and K.F. wrote the manuscript. K.F., A.C., P.K, D.N., J.P., A.S. and R.O.T. provided the resources. K.F., R.O.T., A.S., P.K, J.P., and A.C. discussed and edited the manuscript. All authors reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chaiyachat, P., Kaewseekhao, B., Chaiprasert, A. et al. Metabolomic analysis of Mycobacterium tuberculosis reveals metabolic profiles for identification of drug-resistant tuberculosis. Sci Rep 13, 8655 (2023). https://doi.org/10.1038/s41598-023-35882-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-35882-2
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
