
Abstract
One of the leading causes of death (20.1 per 100,000 women per year), breast cancer is the most prevalent cancer in females. Statistically, 95% of breast cancer are categorized as adenocarcinomas, and 55% of all patients may go into invasive phases; however, it can be successfully treated in approximately 70–80% of cases if diagnosed in the nascent stages. The emergence of breast tumor cells which are intensely resistant to conventional therapies, along with the high rate of metastasis occurrence, has highlighted the importance of finding novel strategies and treatments. One of the most advantageous schemes to alleviate this complication is to identify the common differentially expressed genes (DEGs) among primary and metastatic cancerous cells to use resultants for designing new therapeutic agents which are able to target both primary and metastatic breast tumor cells. In this study, the gene expression dataset with accession number GSE55715 was analyzed containing two primary tumor samples, three bone-metastatic samples, and three normal samples to distinguish the up- and down regulated genes in each stage compared to normal cells as control. In the next step, the common upregulated genes between the two experimental groups were detected by Venny online tool. Moreover, gene ontology, functions and pathways, gene-targeting microRNA, and influential metabolites were determined using EnrichR 2021 GO, KEGG pathways miRTarbase 2017, and HMDB 2021, respectively. Furthermore, elicited from STRING protein–protein interaction networks were imported to Cytoscape software to identify the hub genes. Then, identified hub genes were checked to validate the study using oncological databases. The results of the present article disclosed 1263 critical common DEGs (573 upregulated + 690 downregulated), including 35 hub genes that can be broadly used as new targets for cancer treatment and as biomarkers for cancer detection by evaluation of expression level. Besides, this study opens a new horizon to reveal unknown aspects of cancer signaling pathways by providing raw data evoked from in silico experiments. This study’s outcomes can also be widely utilized in further lab research since it contains diverse information on common DEGs of varied stages and metastases of breast cancer, their functions, structures, interactions, and associations.
Similar content being viewed by others

Introduction
As a complicated disease in which uncontrolled, abnormal, and rapid growth of mutated cells cause tissues and organs dysfunctions1, cancer should not be counted as a singular, one-facet disease but as an amalgam of more than 100 various diseases2. Cancer can be examined from different perspectives with a gamut from immunosurveillance3 to epigenetic studies4. The immunological survey has demonstrated that both adaptive and innate immune systems, particularly lymphocytes, have a critical role in cancer incidence. The in vivo genomic studies have also proved that the lack of some important genes such as recombinase activating gene (RAG)-2 can diminish the lymphocytes’ capacity to combat malignant cells5. That’s because this gene plays an influential role in the production of peripheral αβ T cells, B cells, and Natural Killer (NK) cells6.
Mutations and, consequently, cancerous cells’ incidents can occur in different human tissues and organs. Breast cancer is the most common malignancy in females; however, it can be successfully treated in ~ 70–80% of patients if diagnosed timely7. Breast cancer or mammary carcinoma is initially caused by the uncontrolled growth of abnormal cells in the epithelium of ducts (85%) or lobules (15%)8. Figure 1 illustrates and compares the cancerous and normal breast tissue simultaneously.

Comparison of healthy breast biopsy with a cancerous mammary biopsy (samples from a 68-year-old female with ductal carcinoma grade 1, Elston-Ellis score 4): While malignant cells can be explicitly observed in the tumor tissue biopsy, there is no cancerous cell in normal breast. Obtained from: The Human Protein Atlas (https://www.proteinatlas.org/learn/dictionary/pathology/breast+cancer#Breast-cancer-1,-ductal-carcinoma)9,10.
As aforesaid, it has been reported that 95% of breast malignancies are adenocarcinomas11, and 55% of cases of breast cancer are diagnosed with invasive ductal carcinoma (IDC)12. Various symptoms have been counted as mammary carcinoma signs, but admittedly breast lump is the most prevalent symptom in patients diagnosed with breast cancer13. Both short and long diagnostic intervals are recorded in women with breast cancer14, nonetheless, evidence indicates that most patients usually experience short diagnostic intervals15. Long diagnostic intervals are the most concerning cases, whereas it is testified that they are associated with lower than five-year survival rates in patients16. Various treatment strategies are currently adopted for breast cancer, including the removal of tumors and their adjacent lymph nodes by surgical resection17, chemotherapy18, radiotherapy (which is typically associated with numerous complications)19, and immunotherapy. Chemotherapy agents such as Ribociclib, a cyclin-dependent kinase 4 and 6 (CDK4/6) inhibitor, are shown to be effective in breast cancer treatment and improving the survival rate. In a study by Seock-Ah Im et al.20, the Ribociclib treatment group had a higher survival rate than that of the placebo group (75.2% vs. 61.7%, respectively). Such studies denote the importance of detecting the contributing genes to breast cancer and utilizing appropriate therapeutic agents for the disease treatment20. Genetic studies have revealed the most critical anti-oncogenes and oncogenes that are related to breast cancer21. For instance, breast cancer-associated genes 1 and 2 (BRCA 1 and BRCA 2), located on chromosomes 17q21 and 13q12 sequentially, are the coding genes that produce anti-tumor proteins suppressing malignant cells in breast tissue22,23. Human epidermal growth factor receptor 2 (HER2), located on chromosome 17q12, has also been recognized as a primary oncogene that stimulates cancer signaling pathways in cells by binding to family members of epithelial growth factor receptor (EGFR)7. Once again, these findings highlight the significance of identifying the genes which are mainly pertaining to breast cancer incidents.
There are currently various bioinformatics approaches, such as RNA-seq24 and Microarray25, to investigate the transcriptomes and analyze the gene expression level in different tissues and diseases. Such approaches are utilized to compose large quantities of gene expression analysis to simplify the detection of critical DEGs, pathways, and biomarkers correlated with a particular disease26. Microarray analysis can be performed using online databases such as National Center for Biotechnology Information (NCBI), Gene Expression Omnibus (GEO), and the outcomes can be downloaded as text files freely27. The GEO2R tool provides an opportunity to define and compare the results of high-throughput genomic data; hence, this database has been adopted to evaluate the genes’ expression in the current study.
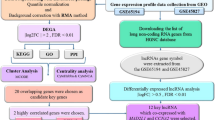
In the present study, the gene expression dataset GSE55715, in which the gene expression of three various states, including healthy mammary epithelial cells, primary tumor cells from the right breast, and P7731 cell line (ER-/PR-/HER2-; triple negative bone metastasis breast cancer cell line) was characterized, is used for detecting the upregulated and downregulated genes in different breast cancer stages in the first step. In the second step, after exposing two cut-offs, the common DEGs in primary and metastatic tumor cells were detected using Venny to investigate these common gene profiles. Other online bioinformatics databases were also utilized to explore the TFs, miRNAs, and metabolites which are associated with such genes. Upstream and downstream regulatory pathways, along with protein–protein interactions, were identified for the protein encoding genes. The importance of this study is due to the detection of common up- and downregulated genes in both primary and metastatic cancerous cells; hence, the results can be adapted for other clinical and experimental conductions like drug designing by docking tools or pharmacological studies.
Materials and methods
Gene profile and microarray data
A comprehensive review was conducted to find a proper study that has suitably analyzed the transcriptome data for normal control cells, primary tumor cells, and metastatic cancerous cells. A study by Dilara Savci-Heijink et al.28, which has the microarray analysis of all desired states, was chosen for further consideration28. This study has the gene expression dataset with accession number GSE55715 analyzing the transcriptome pattern of 8 samples, including three samples of normal mammary epithelial cells, two samples of primary tumor cells, and three samples of the P7731 cell line. GSE55715 was obtained from the NCBI database’s GEO (http://www.ncbi.nlm.nih.gov/geo/). The platform used for array data in this analysis was the GPL6947 (Illumina HumanHT-12 V3.0 expression beadchip), and it contained eight samples, as previously mentioned28.
Identification of differentially expressed genes
After selecting the appropriate study, the GEO online analyzer (http://www.ncbi.nlm.nih.gov/geo/geo2r/) was adopted to analyze the genes’ profiles and to detect DEGs29. GEO2R, as an online bioinformatics analyzer, allows the users to compare two or more defined groups in terms of gene expression. In order to distinguish the up- and downregulated genes in primary and tumor cells in comparison to normal breast cells, three certain groups were defined and two various analyses were implemented. In the first analysis, the gene expression pattern of 2 samples of primary tumor cells (as a first test group) was compared with the normal mammary epithelial cells (as the control group). In the next round, the expression profile of 3 metastatic tumor cells (as the second test group) was analyzed to be compared to the control group. It is also plausible to detect the genes which have a pivotal role in driving breast cancer from primary tumor to metastatic stage by doing a GEO2R transcriptome analysis where the metastatic group is the test group while the primary group is defined as the control. Zhao et al.30 conducted such analysis on three various GSE datasets to identify these genes for early detection of bone metastasis.
The cut-off conditions for detecting upregulated and downregulated genes were defined according to univariate tests. Adjusted p value ≤ 0.05 and |Log fold-change (Fc)|≥ 2 were defined as cut-off criteria, respectively. After applying the cut-off criteria to determine the DEGs, specific symbols of upregulated and downregulated genes were elicited using their gene code. Repetitive/duplicate genes were then deleted, and others were utilized for further analysis.
Recognition of common up- and downregulated genes
A number of 889 and 1131 genes were detected as upregulated and downregulated genes in primary and metastatic cancerous cells compared to normal cells, respectively. In order to determine the number of common upregulated genes between these two types of cancerous cells, the Venny online tool (https://bioinfogp.cnb.csic.es/tools/venny/) was used31. The same strategy was adopted to recognize common downregulated genes among 946 and 1180 downregulated genes of the primary tumor and bone-metastatic cells sequentially. In the end, 573 and 690 genes were ascertained as common up- and downregulated genes sequentially. The further analysis concentrated on the common DEGs,but not the first DEGs.
Transcription factors and gene regulatory networks analysis
The expression of encoding genes has an apparent correlation with the cis and trans-regulatory elements32. Trans-regulatory elements (TRE) code the particular proteins called transcription factors which can highly influence the genes’ expression33. ChIP enrichment analysis (ChEA) database (https://maayanlab.cloud/chea3/) was used to identify the transcriptional factors that control the expression of common up- and downregulated genes of primary and metastatic tumor cells in comparison to normal cells. The information provided in the ChEA results from analysis of ChIP-based experimental methods such as ChIP-chip, ChIP-seq, ChIP-PET, and DamID which are used to profile the transcription factors that can bind to DNA and affect genes’ expression34. After selecting the most effective TFs overruling common DEGs regarding the adjusted p-value, the number of target genes and false discovery rate (FDR) were also calculated.
The common DEGs were also submitted to eXpression2Kinases (X2K) (https://amp.pharm.mssm.edu/X2K/) to find the gene regulatory network. X2K is an online bioinformatics resource that is generated to predict the relationship amongst upstream kinase pathways, the most influential TFs, and target genes35. It designs a diagram demonstrating the association of TFs, protein complexes, and protein kinases, which are responsible for the changes in the expression level of common up- and downregulated genes and transcriptome35. The inferred network of regulatory factors was downloaded and then visualized using Cytoscape software version 3.9.1 (https://cytoscape.org/).
Detecting upregulated transcription factors
In order to discern common upregulated TFs, 573 common upregulated gene symbols as well as human TFs list including 1640 TFs’ names from The Human Transcription Factors database (http://humantfs.ccbr.utoronto.ca/)36 were uploaded to Venny (https://bioinfogp.cnb.csic.es/tools/venny/)31. The gene symbols common in both groups showed the common upregulated TFs. Since such TFs may play a crucial role in PPIs and other genes expression, they were separately detected for further prospective research. The same process was conducted to discover common downregulated TFs by uploading 690 common downregulated genes and human TFs to the Venny tool.
Gene ontology, pathways, and functional analysis of DEGs
Enrichment analysis was conducted to probe a group of genes’ functions and their related pathways. This analysis also helped to understand the biological characteristics of the candidate genes, such as gene ontology (GO)37. Gene ontology analysis was applied to investigate the associated biological process (BP), cellular component (CC), and molecular function (MF) of common DEGs between primary and metastatic cancerous cells. This analysis was carried out using EnrichR (https://amp.pharm.mssm.edu/Enrichr/), a free online and web-based bioinformatics tool that comprehensively investigates the BP, CC, and MF related to submitted genes38.
The signaling pathways, in which upregulated and downregulated genes are contributed, are the other factors that can make a basis for understanding the tumorigenesis and metastasis process39. The Kyoto Encyclopedia of Genes and Genomes (KEGG, www.genome.jp/kegg/)40,41,42 website was used to analyze the particular pathways where the detected DEGs may play a crucial role. The KEGG database is an online resource for analyzing the pathways and functions of the inserted genes40. An adjusted p value < 0.05 was applied as a statistical index to detect the most meaningful GOs and pathways.
Detection of DEG-targeting MicroRNAs and DEGs-related metabolites
Evidently, microRNAs (miRNA) are responsible for controlling the expression of various genes by targeting them in eukaryotic cells; therefore, they play one of the most crucial roles in transcriptome changes and can influence the level of gene expression in any disease43. miRTarbase (http://amp.pharm.mssm.edu) is a comprehensive free database which its 2020 update contained more than 13,404 validated miRNA-target interactions (MTIs)44. It provides diverse information on miRNAs which probably influence the submitted genes. It is widely used to detect transcriptome-affecting miRNAs, and its data are validated by experimental methods such as molecular assay, Northern blot, Microarray, and next-generation sequencing (NGS)45. The common DEGs between primary tumor and metastatic cancerous cells were submitted to this database and the outcomes were imported to Microsoft Excel (https://www.microsoft.com/en-us/download/details.aspx?id=56547) for further analysis. The top 10 miRNAs targeting up- and downregulated genes were selected based on their adjusted p-value.
It is testified that human biological metabolites can potentially affect the expression of different genes contributing to disorders like cancer46. This issue highlights the importance of surveying the most influential metabolites when investigating the transcriptome changes in diverse diseases47. The EnrichR database that is linked to Human Metabolome Database (HMDB)48 provides a broad domain of information on various metabolites affecting gene expression, along with their biological roles and disease associations. Therefore, this database was used to identify the top 10 important metabolites. A table was designed for the top 10 metabolites relevant to common up- and downregulated genes in primary and metastatic tumor cells by considering their p- value/adjusted p-value.
Construction of PPI network, detection of hub genes, and modular analysis
The protein–protein interactions (PPI) among coding genes may have an essential role in cancer incidents and can also be utilized as targets for cancer treatment49. The common DEGs were inserted in the Search Tool for Retrieval of Interacting Genes database (STRING) (version 11.0; https://string-db.org), which gives remarkable information on both known and predicted PPI among coding DEGs50, to identify and anticipate functional interactions among expressed proteins by transcriptome (the medium confidence ≥ 0.400 was set as a cut-off to form the PPI network). The PPI networks for up- and downregulated genes were imported to Cytoscape software (version 3.9.1; www.cytoscape.org) for further visualization, modification, and analysis, such as module construction and hub genes detection.
Modules are described as a group of closely-associated genes that cooperate in arranging a particular function in PPI networks51. A Cytoscape plug-in named Molecular Complex Detection (MCODE) (version 2.0.0) was applied on PPI networks to illustrate the most critical gene modules by considering Degree Cutoff = 2, Node Score Cut-off = 0.2, K-Core = 2, Max. Depth = 100 and haircut cluster finding setting as visualization criteria52. The Cyohubba (version; 0.1), another Cytoscape software plug-in, was utilized for ranking and discerning the critical nodes (which are equivalent to hub genes) in the PPI networks53. The top 15 and 18 nodes (i.e., coding genes) of PPIs ranked by Maximal Clique Centrality (MCC) were ascertained as hub genes for common up- and downregulated genes between primary tumor and metastatic cancerous cells, respectively.
Survival analysis, hub genes characterization, and study validation
In order to determine the prognostic importance of the detected hub genes and validate the study, the five top hub genes having the most meaningful relations with both primary and metastatic cancer cell lines (according to MCC analysis by Cytohubba) was submitted to the Kaplan–Meier plotter (https://kmplot.com/analysis/). Kaplan–Meier plotter is an online and free gadget primarily used for survival analysis in various cancers54. The GEPIA (http://gepia.cancer-pku.cn/), a bioinformatics web resource based on TCGA and GTEx data which provides comprehensive information on the level of different gene expression55, was also applied to investigate the expression of common hub genes in normal and tumor tissues and to authorize the results. The Human Protein Atlas database (https://www.proteinatlas.org/), a Swedish biological program which started in 2003 aiming at visualizing human histology by considering the expression of varied proteins in cells and tissues and integrating diverse omics data56, was used to illustrate the effect of the expression of proteins encoded by 5 top upregulated hub genes on normal and cancerous tissues.
Cancer gene dependency and gene-disease association analysis
One crucial analysis that reveals a utile comprehensive outcome on cell lines’ survival at the gene level is cancer gene dependency. To evaluate the dependency rate of various breast cancer cell lines to identified upregulated hub genes on one hand, and validate the upshots on the other hand, the online platform DepMap, which can be accessed from https://depmap.org/portal/download was adopted57. To dive into details, the CRISPRGeneDependency.csv file was downloaded from DepMap Public 22Q4 Primary Files58. Then, the list of breast cancer cell lines was searched in the mentioned file, and the dependency score of 15 upregulated hub genes was extracted from it. In the next step, a text (.txt) file was composed of found data59. To draw the hub-gene dependency heat map, after using advanced options, the text file was uploaded to CIMMiner (one matrix CIM) (https://discover.nci.nih.gov/cimminer/oneMatrix.do)60. Eventually, the gene dependency heat map for 15 upregulated hub genes in primary and metastatic breast cancer cell lines was interpreted after downloading the final heat map61.
In the final step, to find out more about the potential role of detected upregulated hub genes, such genes’ characteristics were analyzed in a breast carcinoma file (UMLS CUI: C0678222; MeSH: D001943) downloaded from the DisGeNET database (https://www.disgenet.org/dbinfo)62.
More investigations to find biological patterns in various transcriptomic data of breast cancer
In order to investigate the similar or different patterns that may be present among various types of biological samples, an identical analysis was conducted on a GSE65216 (GPL570) dataset. This dataset, publicly available since Jan 23, 201563, contains expression profiling of diverse kinds of breast cancer from Institut Curie (Maire cohort). To find any similar or different patterns between this dataset (GSE65216) and the primary dataset of the study (GSE55715), the same analysis approach was carried out using the GEO2R tool by defining the triple-negative breast cancer (TNBC) samples as test group and healthy samples of this dataset as the control group. The results were then imported to Excel to find the upregulated genes in TNBC samples compared to healthy samples considering the same cut-offs (Log (adjusted p-value) < 0.05 and LogFc > 2). Afterward, the PPI network of the upregulated genes was elicited from the STRING database to find the hub genes for final comparison by exporting the data to the Cytoscape application and analyzing it by Cytohubba plug-in. The 20 top upregulated hub genes of GSE65216 were then aligned with 15 top upregulated hub genes of GSE55715 (as the primary dataset of this study) to discover any consistent results and similar patterns between the two datasets.
Furthermore, to make a comprehensive analysis of different types of breast cancer considering the varied PAM50 subtypes and to enhance the reliability of the found outcomes, one more analysis was performed on the GSE45827 dataset64. To dive into detail, PAM50 is one prominent categorization in which a 50-gene signature is utilized to classify the breast cancer in five different types in oncological studies65,66. Firstly, different breast cancer subtypes were divided into three separate test groups: Basal-like, Luminal B, and HER2-enriched. Shortly, Luminal B is a type of breast tumor with estrogen receptors, which can be progesterone negative or positive. The only difference between these two types is that Luminal B has HER2 receptors while Luminal B lacks them. HER2-enriched and basal-like tumors are both ER and PR negative, but HER2-enriched tumors are HER2 positive, while basal-like (also known as triple-negative breast cancer) is HER2 negative67. Subsequently, these three test groups were compared to the healthy samples as the control group. Then, the identical approach and cut-offs were followed to identify the upregulated genes, draw the PPI network, and indicate the top hub genes.
In both analyses, the volcano plot, as well as the UMAP diagram, were reviewed to identify the separation index of the test and control groups. Briefly, the more test and control samples are separated, the more reliable the analysis is due to the statistical principles.
Breast cancer metastasis to various organs: Is there any rational relevance?
To investigate any rational relation between breast cancer metastasis to various organs, a comprehensive comparison ran among the common upregulated hub genes designated to have a critical role in primary and bone-metastatic breast cancer and the hub genes which were responsible for breast cancer metastasis to skin. The GSE56493 dataset appropriate samples were divided into two groups: 1. Skin-metastasis breast cancer as the test group, and 2. normal breast cells as the control group. Then, the same cut-offs (Log (adjusted p-value) < 0.05 and LogFc > 2), database (STRING), and plug-in (Cytohubba) were used to draw and detect the PPI network of upregulated hub genes, respectively. Afterward, the top 20 upregulated hub genes playing an essential role in breast cancer metastasis were compared to the top 15 common upregulated hub genes of GSE55715.
The tables, figures, and other data generated by applying mentioned methods are entirely presented in the “Results” section.
Results
Data analysis and common DEGs Identification
As mentioned before, this study was carried out to detect the most crucial genes that play a vital role in both primary tumor and bone-metastatic cancerous cells compared to normal mammary cells in breast cancer. The recognition of the most critical upregulated coding genes in both cancerous tissues can widely be used in Ducking and pharmacological studies to design an efficient therapeutic agents which are able to target both tumor cells simultaneously.
The gene expression (transcriptomes) changes in two test groups (primary (as Group 1) and bone-metastatic (as Group 2)) were compared with normal cells as a control group through the GEO2R tool in NCBI. The boxplot, volcano plot, and expression density plot (based on normal distribution) are available in Supplementary 1. The common DEGs of experimental groups, determined through cut-off condition adjusted p-value ≤ 0.05 and │(Log Fc)│ ≥ 2, were distinguished using the Venny online tool. Table 1 demonstrates the exact number of upregulated and downregulated genes in primary and metastatic tumor cells after applying the cut-off criteria. Venn diagram of the common DEGs among groups 1 and 2 can be seen in Supplementary 2.
10 TFs associated with common up- and down regulated genes and gene regulatory networks
Since TFs can widely impact gene expression, the common up- and downregulated genes between groups 1 and 2 were separately submitted to ChEA 2016 database to find the potential TFs. All TFs that had adjusted p-value ≤ 0.05 were considered effective TFs having meaningful relationships with the submitted genes. This indicates that 59 human TFs are capable of targeting both primary and metastatic up regulated genes. In contrast, there were only 15 potential human TFs that could target common down regulated genes between the defined groups. ETS1 20019798, GABP 19822575, and AR 21909140 were the top 3 most influential TFs for common upregulated genes, while VDR 23849224, SOX2 20726797, and CHD1 26751641 were detected as the top 3 important TFs for common downregulated genes influencing at least 106 genes. The top 10 transcription factors associated with common DEGs between groups 1 and 2 are displayed in Table 2.
X2K web-based bioinformatics resource was used to design the gene regulatory networks and identify the role of the detected TFs by the ChEA 2016 database more accurately. The results of investigating 573 common upregulated genes revealed that the enzymes encoded by Mitogen-Activated Protein Kinase-14 (MAPK14), cell division control-2 (CDC2), and casein kinase two alpha-1 (CK2ALPHA) were the most substantial kinases in the regulatory network of the common upregulated genes. These findings give credence to the results elicited from ChEA 2016 by approving the role of MYC and E2F1 TF families. The gene regulatory network of common upregulated DEGs of groups 1 and 2 is illustrated in Fig. 2.

Gene regulatory network common upregulated genes in primary tumor and metastatic cancerous cells involving TFs and kinases.
The same method was developed for investigating the gene regulatory networks of common downregulated genes, eventually elaborating the importance of the DNA-dependent protein kinase (DNA-PK) and CDC2 kinases in the network. The roles of TFs were also explicitly shown in the X2K database diagram. The modified gene regulatory network for common downregulated genes is depicted in Fig. 3.

Regulatory network of common downregulated genes in both tumor groups containing TFs and kinases.
Upregulated transcription factors
As previous research has indicated, transcription factors–as a group of proteins highly involved in transcribing DNA and RNA synthesis—can broadly influence the expression level of their downstream genes by initiating or regulating such genes’ transcription68. As a result, detecting the upregulated TFs amongst all common upregulated genes can be used to illuminate unknown networks and signaling pathways. As previously mentioned, highlighting the transcription factors among DEGs and other analyses are crucial since such TFs have the regulatory effect (like co-expression, suppression or activation) on the other genes36. Moreover, the mode of regulation of such TFs also reveal the PPI network more accurately69,70. Figure 4 demonstrates the number and percentage of common upregulated (A) and downregulated (B) transcription factors.

Venn diagram of applied method to detect common (A) up- and (B) downregulated TFs. Exact gene symbols of common differentially expressed transcription factors are listed in Supplementary 3.
The results of gene ontology and KEGG pathway analysis
After analyzing the GSE55715 dataset and separating the common DEGs between groups 1 and 2, the GO of 573 common upregulated genes and 690 common downregulated genes were investigated by EnrichR GO 2021 online resource. It was found that common upregulated genes primarily contributed to DNA-related biological processes (BP) such as the double-strand break repair and DNA replication. GO molecular function analysis also revealed that binding to DNA replication origin, cadherin, RNA, and single-strand DNA was the most associated molecular functions to which a considerable number of these 573 genes were contributed. In addition, common upregulated genes were enriched in the different cellular components (CC). Nucleus, focal adhesion, and cytoskeleton are some instances of these CCs. The top 10 most critical GO in which common upregulated genes were contributed, along with the proportional percentage of involved genes, are demonstrated in Fig. 5.

GO enrichment analysis of common upregulated genes between group 1 (primary breast cancer vs. normal) and group 2 (bone-metastatic breast cancer).
The GO investigation of common downregulated genes by the EnrichR 2021 GO database unveiled that gene expression, ncRNA processing, and nuclear-transcribed mRNA catabolic process were the most associated BP with some of the 690 down regulated genes. It was also displayed that the most influential MF and CC in which common downregulated genes were involved. The GO diagram of common downregulated genes can be observed in Fig. 6.

GO enrichment analysis of common downregulated genes between defined groups.
In terms of function and pathways, the analysis of common DEGs by the KEGG database was used, and the most prominent pathways were determined by considering adjusted p-value ≤ 0.05. The findings indicate that common upregulated genes were primarily involved in the cell cycle and bladder cancer-related pathways (Fig. 7A). In contrast, common downregulated genes were remarkably enriched in ribosome or infection-related pathways (Fig. 7B).

Top 10 KEGG pathways in which common upregulated genes (A) and common downregulated genes (B) play an essential role.
Finding miRNA-targeting genes and common DEGs-related metabolites
Due to the importance of miRNA-targeting genes and metabolites at the level of various genes, miRTar base 2017 and EnrichR HMDB online databases were utilized to identify the most effective miRNAs and metabolites, respectively. The complete list of miRNA-targeting genes was downloaded, and the influential ones were detected by considering the adjusted p-value ≤ 0.05. It is shown that hsa-miR-615-3, hsa-miR-124-3p, and hsa-miR-92a-3p have the most meaningful relationships with common upregulated genes by targeting 67, 87, and 83 genes, respectively. In Table 3, the top 10 most critical miRNA-targeting genes, along with the number of their target genes (among the submitted genes) and overlap percentage, are exhibited.
After the analysis of common DEGs amongst the sorted groups by EnrichR HMDB, the most crucial metabolites influencing the expression of genes were identified according to their p-value (p value ≤ 0.05). Overall, 1080 metabolites were detected to have a reliable association with various common upregulated genes, whereas only three metabolites (i.e., 1H-Indole-3-acetaldehyde, 5-hydroxy-Sulfate, and TYD) unveiled a cogent relationship with common downregulated genes. Guanosine triphosphate, uridine Diphosphate-N-acetyl glucosamine, phosphate, and other phospholipids moiety were detected as the top 10 metabolites that affect gene expression. Table 4 presents the significant metabolites having strongly meaningful association with common DEGs.
Protein–protein interactions, hub genes networks, and modules
The gene symbol of common DEGs was uploaded to STRING online bioinformatics resource to identify the PPI networks. As a result, STRING detected 560 out of 573 common upregulated genes as protein-coding genes, and their PPI enrichment p-value computed to be < 1.0e−16. The PPI network was then imported to Cytoscape for final visualization and analysis (which showed that the PPI network has 493 nodes and 3052 edges). The Cytoscape analysis revealed that 67 nodes (coding genes) were isolated and did not have any edges (interactions) with others, whereas 15 nodes had more than 46 degrees and were surprisingly dense. The PPI network of common upregulated genes is presented in Supplementary 4. The Cytohubba analysis demonstrated that most of these highly-connected genes, such as AURKA, AURKB, MELK, TTK, KIF20A, CDK1, KIF2C, CDCA8, KIAA0101 (also known as PCLAF), MCM4, CDCA5, CDC20, CDC45, PTTG1, and MCM6 are from hub genes. Overall, these 15 genes were detected as the hub genes by Cytohubba MCC analysis (approximately 2.6% of all common upregulated genes). Furthermore, the MCODE plug-in was also applied to identify the most significant modules, which reveals the three important modules, including Module 1 (31 nodes and 418 edges), Module 2 (34 nodes and 208 edges), and Module 3 (17 nodes and 62 edges). These three modules, as well as 15 hub genes, are displayed in Fig. 8.

Common upregulated hub genes network of breast cancer-related genes (A), graphic illustration of Module 1 (B), 2 (C), and 3 (D). The stronger the color is, the more important that gene is.
The all of 690 common downregulated genes among the primary and bone-metastatic tumors were submitted to STRING to find their PPI network. STRING detected 679 out of 690 genes and presented their PPI by reporting their PPI enrichment p-value to be 1.23e−12. The imported PPI network to Cytoscape had 2309 edges and 606 nodes, which indicated there are 73 isolated nodes in PPI, while 17 nodes had 31–86°. The entire PPI network of common downregulated genes is depicted in Supplementary 5. Alike common upregulated genes, 18 genes (about 2.6% of all genes) were identified as hub genes by Cytohubba, and their network was designed. In addition, we could find the most relevant gene groups by analyzing the whole PPI network by MCODE, which provided three critical modules. The common downregulated hub genes and Module 1 (37 nodes and 312 edges), Module 2 (20 nodes and 70 edges), and Module 3 (24 nodes and 75 edges) are shown in Fig. 9.

Common downregulated hub genes network (A), graphic presentation of Module 1 (B), 2 (C), and 3 (D). The bolder the color is, the more critical that gene is.
Results of the survival analysis and study validation
The validation of the results is the most quintessential part of the study, which can endorse the reliability of the data and assure researchers to conduct lab practices based on the results of this study. As explained in the methods section, the top five most pivotal upregulated hub genes were submitted into three various databases, including GEPIA, human protein atlas, and Kaplan–Meier plotter, to investigate the proportional amount of these common hub genes upregulation, their effect on tissue construction and survival rate, respectively. AURKA, AURKB, KIF20A, MELK, and TTK were identified as the most crucial hub genes based on their score after PPI analysis by the MCC method of Cytohubba plug-in. The human atlas protein analysis showed the appearance of abnormalities in tissue after the upregulation of these genes in tumor tissues. Moreover, GEPIA analysis indicates that these five genes were strongly upregulated, and their expression in patients was at least three times more than that of the healthy people. The Kaplan–Meier analysis also validated the reached results and demonstrated that these 5 top common hub genes were identified correctly. It also revealed that the survival rate of patients detected to have a higher level of these genes’ expression was dramatically lower than the control group [Hazard ratio (HR) index and Log (rank p-value) were entirely meaningful]. Overall, evaluating 5 candidate hub genes by these three databases thoroughly validated achieved results. Figure 10 illustrates the results of the validation process by human atlas protein (A), GEPIA (B), and Kaplan–Meier plotter (C).

The results of validating study: Immunohistochemistry of the top five densest hub genes in breast carcinoma and normal tissue based on (A), the expression level of these five genes in healthy people and patients elicited from GEPIA (B), and survival analysis diagrams for commonly up regulated hub genes designed by Kaplan–Meier (C).
Cancer-gene dependency analysis of hub genes
A cancer genetics dependency for a particular gene denotes how essential that gene is for the survival/proliferation of that cell line58. This is primarily computed by knocking out that gene in a cell line or inhibiting the protein encoded by a gene to measure its effect on blocking that cell line’s survival or inducing its death71. As expounded, the dependency score of 15 upregulated hub genes for available breast carcinoma cell lines amongst all cancer cell lines (CCL) was extracted from CRISPRGeneDependency.csv downloaded from the DepMap portal (https://depmap.org/portal/download/all/). Since cancer cell lines are broadly used as in vitro models for cancer-biology-related topics such as genes’ expressions, drug efficacy etc.72, measuring the genes expression and dependency in such cell lines can be used to validate or reject the findings. In the gene heat map, the redder a common point between a particular gene and a cell line, the more that cell line’s survival depends on that gene. The text of elicited data, including 15 upregulated hub genes, breast cancer primary and metastatic cell lines, and their dependency score (Supplementary 6), was used to draw the heat map in Fig. 11. As can be seen in Fig. 11, the dependency score of many breast carcinoma cell lines for most of the hub genes is high, indicating that those genes are vastly required for cells’ proliferation/survival. This heat map not only validates the integrity of found hub genes but also highlights the significance of discovered PPI networks, gene expression, and cellular signaling.

Heat map of gene dependency for primary and metastatic breast cancer and the 15 upregulated hub genes: while most of the hub genes are highly demanded for cell lines’ survival, some of them, including MELK and KIAA0101 (PCLAF), are reportedly less vital for almost all of the cell lines (Right column: cell lines’ names, Row: hub genes’ symbols). The warmer (redder) a color is, the more vital that gene is for the corresponding cell line.
Gene-disease relations for hub genes
In the final analysis of study validation, the DisGeNet web-based tool (https://www.disgenet.org/search) got used for the assessment of the gene-disease association for the top 15 upregulated hub genes to investigate the Disease-Specificity and Disease Pleiotropy indexes (DSI and DPI, respectively) as well as former articles which have reported that particular gene and breast carcinoma62. The DSI is a value between 0 and 1, indicating the number of diseases the gene is associated with in an inverse proportion, while DPI shows the variety of disease types in which that gene plays a role. In other words, the more DPI is the more types of disorders that gene is associated with, whereas the less DSI is, the more number of diseases are known to be related to that gene73. Table 5 provides a broad range of such information for common upregulated hub genes.
Similar biological patterns in diverse transcriptomic data of breast cancer
As aforesaid, to make the study more comprehensive and endorse its reliability, two more analyses were done on other datasets, including GSE65216 and GSE45827. The results from the first analysis indicated that five upregulated hub genes were shared between GSE65216 and GSE55715, of which 4 of them were previously introduced as the most critical genes having a crucial role in both primary and bone-metastatic breast cancer. This significant overlap emphasizes the primacy of drug discovery or designing for proposed genes in this study. The full results of this analysis are attached in Table 6.
To investigate the possible role of the hub-upregulated genes that were identified as essential drivers in both primary and bone-metastatic breast cancer in various subtypes of breast cancer, different PAM50 subtypes of breast cancer were compared to the healthy samples (Volcano plot and UMAP diagram of samples’ separation are available in Supplementary 7). After eliciting the PPI network for each group, the top 20 hub upregulated genes were highlighted using the Cytohubba plug-in. Then, the common hub upregulated genes among Luminal B, Basal-like, and HER2-enriched breast cancer were discerned by the Venny tool, as shown in Fig. 12. In the final step, these genes were compared to the common hub upregulated genes between primary and bone-metastatic breast cancer to discover any consistent pattern. Three (CDK1, TTK, and MELK) of 12 common upregulated hub genes among Luminal B, Basal-like, and HER2-enriched were also observed in common upregulated hub genes between primary and bone-metastatic breast cancer. Moreover, it was found that some of the other genes were also shared between one particular type of breast cancer and this study's hub gene. For instance, while AURKB was identified as one of the hub genes in Basal-like breast cancer, AURKA was recognized as a critical gene in HER2-enriched breast cancer.

The overlap of upregulated hub genes among Luminal B, HER2-enriched, and Basal-like breast cancer.
These findings show the significance of previously-identified kinases, including AURKB, MELK, TTK, and KIF20A, in the occurrence of various subtypes of breast cancer and even its progression toward the metastatic phase.
Bone-metastatic and skin-metastatic breast cancer: similarities and differences
As described before, appropriate samples of GSE56493 were selected to identify significant DEGs in skin-metastatic breast cancer. While the comparison between upregulated genes in skin-metastatic and bone-metastatic breast cancer revealed slight overlap among these two types of metastasis (overlap percentage: 5%), the overlap percentage jumped to 29.6% when the hub genes of both groups were compared. Figure 13 displays the Venn Diagram of this comparison schematically.

The Venn diagram of upregulated genes in the skin- and bone-metastatic breast cancer tumors (A); and hub genes in skin-metastatic versus common hub genes in primary and bone-metastatic breast cancer (B).
Once again, this analysis emphasized the importance of suggested upregulated hub genes as novel targets for drug discovery, inhibitor designing, and siRNA-based therapeutic agents. It also indicated that any potential inhibitor against the introduced hub genes in this study could target not only various types of primary breast cancer but also its possible metastasis to bene and skin vigorously.
Discussion
Nowadays, breast cancer is regarded to be one of the most critical health and medical issues due to its high prevalence among females worldwide. Epidemiological studies have suggested that in the United States, breast cancer holds the second place among all various diseases, accidents, etc., resulting in women’s death74. The high prevalence of breast cancer, the emergence of drug-resistant cancerous cells, and the possibility of metastasis, which causes the involvement of other organs, prove the urgent demand for apprehending the molecular basis of this disease75. Herein, the differential expresses genes in the breast cancer primary tumor cells and P7731 cell line (bone metastasis for breast cancer) was probed to detect the common DEGs between these cells to shed light on designing novel therapeutic agents targeting both of them efficiently.
To dive into details, the gene ontology analysis demonstrates that common upregulated genes are enriched in the crucial biological process, including the DNA-related pathways such as DNA replication and DNA metabolic process confirming the significance of such genes in cancer occurrence and cell proliferation. Similarly, previous studies have shown a strong relationship between gene ontology and mammary malignancies76. Furthermore, from a molecular function perspective, DNA replication, cadherin, and mRNA binding functions were the most relevant MFs to upregulated genes. The role of deregulation of cadherin and catenin in cancer progression, despite their primary role in mammary development, was investigated by Pamela Cowin et al.77. In addition, it is worth mentioning that although a low percentage of genes contribute to intracellular bounded and non-bounded organelles and nucleus-related components, these cellular components have the most robust connection with common upregulated genes. Investigating the gene ontology, particularly cellular components, is vital since their role in breast cancer pathways has been observed and proven repeatedly78,79. The analysis of common upregulated genes between the defined groups revealed that most of the top 10 pathways having the most meaningful association (based on Log adjusted p-value) are related to the cell cycle, apoptosis, and cancer-related pathways. For instance, 17.74% of 573 common upregulated genes that play critical roles in the cell cycle pathways have the most meaningful correlation with both primary and metastatic breast cancer types. The significance of cell cycle pathways was also investigated in other research designs80, highlighting the importance of the result of the present study.
Moreover, the obtained results also showed that some influential pathways in cancer (e.g., mRNA surveillance) are highly associated with the common downregulated genes. Thus, it is safe to say that such genes dangerously impact cell proliferation. The high complexity of the PPI network of common DEGs constructed using the STRING database and Cytoscape application indicated the importance of genes contributing to both primary and metastatic cancer types. Notably, the PPI analysis is noticeable since other studies have proven their functions in breast cancer72 and their relation with other types of cancers, such as colon cancer73.
After the detection of hub genes utilizing the PPI network of common upregulated genes, the top five hub genes, including AURKA, AURKB, KIF20A, MELK, and TTK that played the most critical role in both primary tumor cell and P7731 cell line, were selected to validate the study. Additionally, the investigation of these five genes by Human Atlas Protein, GEPIA, and Kaplan–Meier databases showed their correlation with tumorigenesis, so it validated the results. Aurora kinase A (AURKA) and Aurora kinase B (AURKA), which their overexpression is highly related to cancer emergence, have been found to play a crucial role in cell proliferation and division81,82. AURKA or STK61, a protein from the serine/threonine kinases family, is essential for the cell division during mitosis. It is also highlighted as one of the essential biomarkers in cancer prognosis, which its overexpression may activate deleterious phosphorylation pathways and induce cancer83. Moreover, AURKA has also a robust correlation with other genes contributing to Wnt and Ras-MAPK signaling pathways84. Due to the cogent effect of AURKA in various cancers, different inhibitors are designed and tested to halt cancer progress by suppressing this gene’s upregulation85,86. AURKB, which was detected as the second pivotal hub gene in this study, also belongs to serine/threonine kinases, and its amplification is clarified to result in tumorigenesis in diverse organs87. In fact, AURKB is proven to ameliorate the cell cycle by targeting different genes contributing to mitosis. To be more precise, AURKB diminishes the expression of p21 by inhibiting p53 activity; thereby, causing upregulation of CDK1, eventually leading to cell division and increasing the tumor cell survival88. KIF20A gene encodes a protein named as kinesin family member 20A, which is necessary for the spindle assembly and chromosome segregation during mitosis, particularly anaphase and cytokinesis89. This gene is also highly associated with other crucial genes in cell proliferation (e.g., MKLP1, PLK1, and RAB6). Furthermore, CDK1 is also affected by the proportion of KIF20A overexpression, which highlights the decisive role of KIF20A in the cell division and mitosis89. It is indicated that other types of cancer, like bladder cancer, are also caused by the upregulation of this gene resulting in more complicated tumor differentiation and a lower rate of survival in patients90.
The present results pronounce the importance of the Maternal embryonic leucine zipper kinase (MELK) gene as one of the most influential hub genes in both primary tumor cells and the P7731 cell line. More specifically, MELK is also a kinase that noticeably exerts its oncogenic impacts by interacting with cyclin B and cyclin D1 genes91. This gene is related to tumors’ aggressive growth and drug resistance emergence in cancerous cells92. In addition, it is acknowledged that inhibiting MELK, as a pivotal gene in breast cancer, can reduce cell division by suppressing the expression of cyclin B and D170 and increasing the sensitivity of breast cancer tumors to chemo- and radiotherapy92. Threonine and tyrosine kinase (TTK), the last important hub gene utilized to validate the outcomes of the present study, is part of the spindle assembly checkpoint and has an essential role in the chromosomal separation during mitosis93. In addition, the meaningful relation of TTK (which is known as a biomarker for the poor prognosis of various cancer, including breast94) with tumorigenesis, especially the advent of aneuploidy tumors, is already discovered95.
Various proteins and genes primarily having crucial roles in cell division are already mentioned as other important hub genes in multiple types and metastasis of breast cancer. On the other hand, uncontrolled cell division is an undeniable part of the “cancer” definition96. While CDC45 is indirectly required for the initiation of DNA replication due to its high connections with other cell proliferation genes like MCMs97, CDC20 directly activates anaphase-promoting complex (APC/C), resulting in chromatid separation and cell going through anaphase in mitosis98. Moreover, due to being one of the hub genes in bioinformatics analysis by Arulprakasam Ajucarmelprecilla et al.99, CDC45 is reportedly a crucial biomarker for gastric cancer as well. CDCA8 and CDCA5 are the other stated hub genes that regulate the cell division cycle by coding an essential protein complex for chromosomal migration called chromosome passenger complex (CPC)100 and chromatid cohesion in mitosis stabilizing101, respectively. Similarly, KIF20A is also involved in chromosomal transportation by CPC during mitosis102. As the role of other hub genes including MELK, AURKA and AURKB genes are explained before, it can be concluded that suppressing some of the pivotal genes (not all of them) in the cell division can efficiently pause the whole process of tumorigenesis and cell proliferation. Although most of the important genes in tumorigenesis seems to be explicitly liked to cell cycle, some other enzymes and proteins like TYMS and FN1 are also reported to be effective in breast adenocarcinoma by Jhansi Pandi et al.103. Finally, suppression of the genes that are statistically ranked as the more important genes is expected to be more effective in cancer treatment; however, the clinical research may be accompanied by other reasons due to the unknown biological functions and metabolisms in the cell.
The analysis of genes’ transcription in other molecular subtypes of primary breast cancer, including basal-like (the closest PAM50 subtype to triple-negative breast cancer), HER2-enriched, and Luminal B, proved the significance of the mentioned above hub genes. Moreover, some genes, such as TTK and MELK, were repeatedly observed in various PAM50 subtypes of primary mammary carcinoma and even in bone-metastatic and skin-metastatic breast cancer. Also, AURKB and KIF20A, the other two genes amongst five critical genes which are introduced as pivotal targets for prospective drug discovery and inhibitor designing, were indicated to play a crucial role in breast carcinoma metastasis to the skin. All in all, it appears that AURKB, AURKA, TTK, MELK, and KIF20A initiate the signaling pathways that eventually result in uncontrolled cell proliferation; therefore, inactivation and degradation of such genes’ proteins or mRNA illuminates a novel strategy to target the cancerous cells efficiently92,104,105. Based on the statistical analyses of the PPI network of upregulated genes, AURKA, AURKB, TTK, MELK, and KIF20A were also involved in module 1, indicating their importance both in primary tumorigenesis and cancer progression.
As pointed before, modules are defined as a group of highly-related genes in a gene regulatory (GR) or PPI network primarily affected by the same transcription factors106. Since the same TFs play an essential role in the transcription/expression of co-expressed genes, simultaneous transcription is usually observed for the genes that are categorized as one module in a GR or PPI network. In other words, modules are composed of a highly connected cluster of genes forming a subgraph in the leading network; such genes are involved in the same biological pathway or function, and targeting one may interrupt the whole module and even the entire network107. Clustering plug-ins such as MCODE follow a statistical approach to identify and rank the most critical modules in a PPI using mathematical parameters like K-Core. The top 3 modules usually reveal the vital genes involved in tumorigenesis and often include the hub genes of a particular network, increasing the validity of previous calculations. As can be seen, most of the 15 upregulated hub genes are also present in the module 1 which emphasizes their role in cancer-relevant pathways.
Five various databases to verify the trueness of the study hypothesis, analysis conduction, and integrity of detected hub genes indicated that the upregulation of detected common hub genes is already recorded in breast cancer patients. Also, it shows that the overexpression of upregulated hub genes causes tissue deformation, and it is correlated with less probability of survival in people diagnosed with breast carcinoma. On the other hand, the drawn heat map demonstrated that most of the marked genes are increasingly vital for breast cancer cell lines authorizing the results once again. The Kaplan–Meier analysis was selected as one of the validating approaches for detected hub genes in the PPI network of common upregulated genes showing a significant Hazard Ratio (HR). The HR is one of the most-used statistical parameters in clinical trials or survival analysis, indicating the possibility of a particular event like death/survival in two identical groups (test group vs. control group) which only have one distinct characteristic such as a specific expression of a gene over a period of time (month/year)108. As there is a downward slope in the survival of cancer patients, as displayed in Kaplan–Meier curves, HR determines the probability of death in the patients who have shown the higher expression rate for the inquired gene109. GEPIA analysis disclosed that breast cancer patients have significantly higher expression of AURKA (4.0 vs. 1.5), AURKB (4 vs. 1), TTK (2.5 vs. 0.5), MELK (3.0 vs. 0.5), KIF20A (3.5 vs. 0.5) genes compared to healthy people. On the other hand, Kaplan–Meier curves revealed the HR value to be 1.87, 1.43, 1.87, 1.81, 1.66 for AURKA, AURKB, TTK, MELK, and KIF20A, respectively, meaning the risk of death to be about 87%, 43%, 87%, 81% and 66% higher in patients whom these genes are upregulated in comparison to the patients with regular expression. To conclude, the conducted analyses showed that detected hub genes in this study are pretty suitable targets for future drug discovery research as they are vital not only in the various subtypes of primary breast carcinoma but also in both bone- and skin-metastatic mammary cancer.
Although there have been some recent endeavors for designing effective drugs against introduced hub genes in this study, more efficient research is required due to unmet achievements in this area. As the MELK gene is discovered to be responsible for Glioblastoma multiforme (GBM), OTSSP167, a MELK inhibitor recently in clinical trial phase I/II, has been synthesized for cancer treatment through MELK inhibition110. OTSSP167, an oral inhibitor of MELK, effectively speeds up the destabilization of MELK by stopping its autophosphorylation, which is necessarily demanded for MELK protein stability and function111,112. MELK-8a is another empirically used substance that has effectively shown selective inhibition of the MELK protein113. The valuable attempts for detecting and targeting the important genes in gastroenteropancreatic neuroendocrine tumors (GEP-NETs)114, prostate cancer115, etc., provided new inhibitors for Aurora genes (i.e., AURKA and AURKB) like ZM447439 and Hesperadin ultimately. Aurora inhibitors target the AURKB protein by acting as an ATP-competitive agent and suppressing DNA-related mechanisms116. While Hesperadin is typically used at a concentration of approximately 50–500 nM117, ZM447439 is administered around 2–20 μM118, determining the higher potency of Hesperadin for AURKB inhibition. Furthermore, Tozasertib (VX-680) and LY3295668 are other Aurora inhibitors actively targeting AURKA119. Since immature hematopoietic cells have proved to express KIF20A highly, there has been enormous research to find KIF20A to treat leukemia. DIACC2010 is a KIF20A inhibitor in the preclinical phase, which has passed in vitro step successfully with a median half inhibitory concentration (IC50) of 40 nM120. Due to the high importance of TTK in hepatocellular carcinoma (HCC), CFI-402257 has been designed to knock out the TTK gene121. As stated before, there are recently some other therapeutic agents for various cancers’ treatment and critical genes inhibition; however, more in silico, in vitro, and clinical trials are required for accurate tumor targeting.
To sum up, all of the abovementioned genes validated the results and were identified as the most critical hub genes in both primary and bone metastatic cancer cells. To extend the deduced results of this research and synthesize more accurate inhibitors or other therapeutic agents, other high throughput methods such as RNA-seq as well as other metastatic tumors can be used as models.
Conclusion
In conclusion, the most critical differentially expressed genes were detected by utilizing a comprehensive transcriptomic analysis. The usage of bioinformatics tolls resulted in the identification of AURKA, AURKB, TTK, MELK and KIF20A as top 5 hub genes involved in breast cancer occurrence and progression. Other bioinformatics databases and software were used to reveal the characterization of identified genes. Since we authorized the integrity of determined hub DEGs, it is now proven that such genes can now be counted as novel targets for breast carcinoma treatment by designing new drugs, inhibitors, and siRNA-based therapeutics. Furthermore, the high expression level of identified genes has made them advent biomarkers for mammary carcinoma diagnosis by simple evaluating methods. Moreover, the efficiency of cell lines as emerging cancer models is approved in this study once again. Although the significance of found hub genes in the occurrence and progression of diverse types of breast cancer from primary to advanced-metastatic stages was validated by applying various experimental-clinical databases, the importance of further experiments should not be ignored. As some other genes such as BUB1 and NCAPG were also reported to be influential in breast adenocarcinoma using bioinformatics tool103, it appears that a large-scale clinical research is highly required for detection of most critical genes. Ultimately, this study provides a robust basis to discover and highlight undisclosed signaling pathways in breast cancer by marking new genes, TFs, metabolites, kinases, miRNAs, and PP interactions.
Data availability
All data are available in the text of the article and Supplementary files. All data used an analyzed in this article are freely available in the mentioned databases e.g. Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/).
Abbreviations
- RAG-2:
-
Recombination activating gene 2
- CDK4/6:
-
Cyclin-dependent kinase 4 and 6
- BRCA-2:
-
Breast cancer gene type 2
- EGFR:
-
Epidermal growth factor receptor
- DEG:
-
Differentially expressed gene
- GEO:
-
Gene expression omnibus
- ChEA:
-
ChIP enrichment analysis
- FDR:
-
False discovery rate
- BP:
-
Biological process
- CC:
-
Cellular component
- miRNA:
-
Micro-ribonucleic acid
- NGS:
-
Next generation sequencing
- PPI:
-
Protein–protein interaction
- MCC:
-
Maximal clique centrality
- MAPK14:
-
Mitogen-activated protein kinase-14
- CK2ALPHA:
-
Casein kinase two alpha-1
- CDC2:
-
Cyclin-dependent kinase 2
- AURKB:
-
Aurora kinase B
- TTK:
-
Tyrosine-threonine kinase
- GRN:
-
Gene regulatory network
- FN1:
-
Fibronectin 1
- NCAPG:
-
Non-SMC condensin I complex subunit G
- BUB1:
-
Serine/threonine protein kinase
- TNBC:
-
Triple-negative breast cancer
- GEP-NET:
-
Gastro-entero-pancreatic neuroendocrine tumors
- HCC:
-
Hepatocellular carcinoma
- CPC:
-
Chromosome passenger complex
- IDC:
-
Invasive ductal carcinoma
- BRCA-1:
-
Breast cancer gene type 1
- HER2:
-
Human epidermal growth factor receptor 2
- Fc:
-
Fold change
- NCBI:
-
National center for biotechnology information
- TRE:
-
Trans regulatory elements
- TF:
-
Transcription factor
- GO:
-
Gene ontology
- MF:
-
Molecular function
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- MTI:
-
Micro RNA-target interaction
- HMDB:
-
Human metabolites database
- MCODE:
-
Molecular complex detection
- DNA-PK:
-
DNA-dependent protein kinase
- CCL:
-
Cancer cell line
- AURKA:
-
Aurora kinase A
- MELK:
-
Maternal embryonic leucine zipper kinase
- P-value:
-
Probability value
- IC50:
-
Half inhibitory concentration
- TYMS:
-
Thymidylate synthetase
- HR:
-
Hazard ratio
- GBM:
-
Glioblastoma multiforme
- APC/C:
-
Anaphase-promoting complex
- NP:
-
Nanoparticle
References
Hanahan, D. & Weinberg, R. A. The hallmarks of cancer. Cell 100(1), 57–70 (2000).
Mitra, S., Ganguli, S. & Chakrabarti, J. Chapter 1 - Introduction. In Cancer and Noncoding RNAs Vol. 1 (eds Chakrabarti, D. J. & Mitra, D. S.) 1–23 (Academic Press, 2018).
Dunn, G. P., Old, L. J. & Schreiber, R. D. The immunobiology of cancer immunosurveillance and immunoediting. Immunity 21(2), 137–148 (2004).
Jones, P. A. & Baylin, S. B. The epigenomics of cancer. Cell 128(4), 683–692 (2007).
Shankaran, V. et al. IFNγ and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature 410(6832), 1107–1111 (2001).
Shinkai, Y. et al. RAG-2-deficient mice lack mature lymphocytes owing to inability to initiate V (D) J rearrangement. Cell 68(5), 855–867 (1992).
Harbeck, N. et al. Breast cancer. Nat. Rev. Dis. Primers. 5(1), 1–31 (2019).
Organization, W. H. Breast cancer. https://www.who.int/news-room/fact-sheets/detail/breast-cancer (accessed 13 December 2021).
Sjöstedt, E. et al. An atlas of the protein-coding genes in the human, pig, and mouse brain. Science 367(6482), eaay5947 (2020).
Uhlen, M. et al. A pathology atlas of the human cancer transcriptome. Science 357(6352), eaan2507 (2017).
Vinay, K., Abbas, A. K., Fauston, N., Aster, J. Robbins and Cotran Pathologic Basis of Disease. In Robbins Pathology 10th edn, 628–636 (Elsevier, 2005).
Eheman, C. R. et al. The changing incidence of in situ and invasive ductal and lobular breast carcinomas: United States, 1999–2004. Cancer Epidemiol. Prev. Biomark. 18(6), 1763–1769 (2009).
Walker, S., Hyde, C. & Hamilton, W. Risk of breast cancer in symptomatic women in primary care: A case–control study using electronic records. Br. J. Gen. Pract. 64(629), e788–e793 (2014).
Koo, M. M. et al. Typical and atypical presenting symptoms of breast cancer and their associations with diagnostic intervals: Evidence from a national audit of cancer diagnosis. Cancer Epidemiol. 48, 140–146 (2017).
Redaniel, M. T., Martin, R. M., Ridd, M. J., Wade, J. & Jeffreys, M. Diagnostic intervals and its association with breast, prostate, lung and colorectal cancer survival in England: historical cohort study using the clinical practice research datalink. PLoS ONE 10(5), e0126608 (2015).
Webber, C., Jiang, L., Grunfeld, E. & Groome, P. A. Identifying predictors of delayed diagnoses in symptomatic breast cancer: A scoping review. Eur. J. Cancer Care 26(2), e12483 (2017).
Waks, A. G. & Winer, E. P. Breast cancer treatment: A review. JAMA 321(3), 288–300 (2019).
Hassan, M., Ansari, J., Spooner, D. & Hussain, S. Chemotherapy for breast cancer. Oncol. Rep. 24(5), 1121–1131 (2010).
Senkus-Konefka, E. & Jassem, J. Complications of breast-cancer radiotherapy. Clin. Oncol. 18(3), 229–235 (2006).
Im, S.-A. et al. Overall survival with ribociclib plus endocrine therapy in breast cancer. N. Engl. J. Med. 381(4), 307–316 (2019).
Sun, Y.-S. et al. Risk factors and preventions of breast cancer. Int. J. Biol. Sci. 13(11), 1387 (2017).
Deng, C.-X. BRCA1: Cell cycle checkpoint, genetic instability, DNA damage response and cancer evolution. Nucl. Acids Res. 34(5), 1416–1426 (2006).
Dine, J. & Deng, C.-X. Mouse models of BRCA1 and their application to breast cancer research. Cancer Metastasis Rev. 32(1), 25–37 (2013).
Wang, Z., Gerstein, M. & Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 10(1), 57–63 (2009).
Stekel, D. Microarray Bioinformatics (Cambridge University Press, 2003).
Talkhabi, M., Razavi, S. M. & Salari, A. Global transcriptomic analysis of induced cardiomyocytes predicts novel regulators for direct cardiac reprogramming. J. Cell Commun. Signal. 11(2), 193–204 (2017).
Barrett, T. et al. NCBI GEO: Archive for functional genomics data sets—Update. Nucl. Acids Res. 41(D1), D991–D995 (2012).
Savci-Heijink, C. D., Halfwerk, H., Koster, J. & van de Vijver, M. J. A novel gene expression signature for bone metastasis in breast carcinomas. Breast Cancer Res. Treat. 156(2), 249–259 (2016).
Davis, S. & Meltzer, P. S. GEOquery: A bridge between the gene expression omnibus (GEO) and bioConductor. Bioinformatics 23(14), 1846–1847 (2007).
Zhao, Z., Yang, H., Ji, G., Su, S., Fan, Y., Wang, M., Gu, S., Identification of hub genes for early detection of bone metastasis in breast cancer. Front. Endocrinol. 13, 1–14. https://doi.org/10.3389/fendo.2022.1018639 (2022).
Oliveros, J. C. VENNY. An interactive tool for comparing lists with Venn's diagrams. https://bioinfogp.cnb.csic.es/tools/venny/index.html (accessed 12 November 2021).
Wittkopp, P. J., Haerum, B. K. & Clark, A. G. Evolutionary changes in cis and trans gene regulation. Nature 430(6995), 85–88 (2004).
Yusuf, D. et al. The transcription factor encyclopedia. Genome Biol. 13(3), 1–25 (2012).
Lachmann, A. et al. ChEA: transcription factor regulation inferred from integrating genome-wide ChIP-X experiments. Bioinformatics 26(19), 2438–2444 (2010).
Clarke, D. J. B. et al. eXpression2Kinases (X2K) Web: Linking expression signatures to upstream cell signaling networks. Nucl. Acids Res. 46(W1), W171–W179 (2018).
Lambert, S. A. et al. The human transcription factors. Cell 172(4), 650–665 (2018).
Ashburner, M. et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 25(1), 25–29 (2000).
Kuleshov, M. V. et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucl. Acids Res. 44(W1), W90–W97 (2016).
Yip, H. Y. K. & Papa, A. Signaling pathways in cancer: Therapeutic targets, combinatorial treatments, and new developments. Cells 10(3), 659 (2021).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucl. Acids Res. 28(1), 27–30 (2000).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 28(11), 1947–1951 (2019).
Kanehisa, M., Furumichi, M., Sato, Y., Kawashima, M. & Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucl. Acids Res. 51(D1), D587–D592 (2023).
Schirle, N. T., Sheu-Gruttadauria, J. & MacRae, I. J. Structural basis for microRNA targeting. Science 346(6209), 608–613 (2014).
Huang, H.-Y. et al. miRTarBase 2020: Updates to the experimentally validated microRNA–target interaction database. Nucl. Acids Res. 48(D1), D148–D154 (2020).
Chou, C.-H. et al. miRTarBase update 2018: a resource for experimentally validated microRNA-target interactions. Nucl. Acids Res. 46(D1), D296–D302 (2018).
Pan, Y.-C. et al. CEBPD reverses RB/E2F1-mediated gene repression and participates in HMDB-induced apoptosis of cancer cells. Clin. Cancer Res. 16(23), 5770–5780 (2010).
Mandal, R., Chamot, D. & Wishart, D. S. The role of the Human Metabolome Database in inborn errors of metabolism. J. Inherit. Metab. Dis. 41(3), 329–336 (2018).
Wishart, D. S. et al. HMDB 4.0: The human metabolome database for 2018. Nucl. Acids Res. 46(D1), D608–D617 (2018).
Fry, D. C. & Vassilev, L. T. Targeting protein–protein interactions for cancer therapy. J. Mol. Med. 83(12), 955–963 (2005).
Szklarczyk, D. et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucl. Acids Res. 47(D1), D607–D613 (2019).
Lin, C.-Y. et al. Module organization and variance in protein-protein interaction networks. Sci. Rep. 5(1), 1–12 (2015).
Bader, G. D. & Hogue, C. W. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 4(1), 1–27 (2003).
Chin, C.-H. et al. cytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 8(4), 1–7 (2014).
Zwyea, S.; Naji, L.; Almansouri, S. Kaplan-Meier plotter data analysis model in early prognosis of pancreatic cancer, in Journal of Physics: Conference Series, 012033 (IOP Publishing, 2021).
Tang, Z. et al. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucl. Acids Res. 45(W1), W98–W102 (2017).
Uhlén, M. et al. A human protein atlas for normal and cancer tissues based on antibody proteomics. Mol. Cell. Proteom. 4(12), 1920–1932 (2005).
Shimada, K., Bachman, J. A., Muhlich, J. L. & Mitchison, T. J. shinyDepMap, a tool to identify targetable cancer genes and their functional connections from cancer dependency map data. Elife 10, e57116 (2021).
Tsherniak, A. et al. Defining a cancer dependency map. Cell 170(3), 564–576 (2017).
Meyers, R. M. et al. Computational correction of copy number effect improves specificity of CRISPR–Cas9 essentiality screens in cancer cells. Nat. Genet. 49(12), 1779–1784 (2017).
Weinstein, J. N. et al. An information-intensive approach to the molecular pharmacology of cancer. Science 275(5298), 343–349 (1997).
Weinstein, J. et al. Predictive statistics and artificial intelligence in the US National Cancer Institute’s drug discovery program for cancer and AIDS. Stem cells 12(1), 13–22 (1994).
Piñero, J. et al. The DisGeNET knowledge platform for disease genomics: 2019 update. Nucl. Acids Res. 48(D1), D845–D855 (2020).
Information-GEO, N. C. F. B. Expression profiling of breast cancer samples from Institut Curie (Maire cohort). https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE65216 (accessed 28 April 2023).
Information-GEO, N. C. F. B. Expression data from Breast cancer subtypes. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE45827 (accessed 28 April 2023).
Parker, J. S. et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol. 27(8), 1160 (2009).
Kensler, K. H. et al. PAM50 molecular intrinsic subtypes in the nurses’ health study CohortsPAM50 in the NHS/NHSII. Cancer Epidemiol. Biomark. Prev. 28(4), 798–806 (2019).
Troester, M. A. et al. Racial differences in PAM50 subtypes in the Carolina Breast Cancer Study. JNCI J. Natl. Cancer Inst. 110(2), 176–182 (2018).
Latchman, D. S. Transcription factors: An overview. Int. J. Biochem. Cell Biol. 29(12), 1305–1312 (1997).
Hsia, C. C. & McGinnis, W. Evolution of transcription factor function. Curr. Opin. Genet. Dev. 13(2), 199–206 (2003).
Han, H. et al. TRRUST v2: An expanded reference database of human and mouse transcriptional regulatory interactions. Nucl. Acids Res. 46(D1), D380–D386 (2018).
Lin, A. & Sheltzer, J. M. Discovering and validating cancer genetic dependencies: Approaches and pitfalls. Nat. Rev. Genet. 21(11), 671–682 (2020).
Mirabelli, P., Coppola, L. & Salvatore, M. Cancer cell lines are useful model systems for medical research. Cancers 11(8), 1098 (2019).
Piñero, J., Bravo, À., Queralt-Rosinach, N., Gutiérrez-Sacristán, A., Deu-Pons, J., Centeno, E., García-García, J., Sanz, F., Furlong, L. I. (2016) DisGeNET: A comprehensive platform integrating information on human disease-associated genes and variants. Nucl. Acids Res. gkw943.
Coughlin, S. S., Epidemiology of breast cancer in women. Breast Cancer Metastasis Drug Resist. 9–29 (2019).
Russo, J. & Russo, I. H. Molecular Basis of Breast Cancer: Prevention and Treatment (Springer, 2004).
Arciero, C. et al. Functional relationship and gene ontology classification of breast cancer biomarkers. Int. J. Biol. Markers 18(4), 241–272 (2003).
Cowin, P., Rowlands, T. M. & Hatsell, S. J. Cadherins and catenins in breast cancer. Curr. Opin. Cell Biol. 17(5), 499–508 (2005).
Blain, S. W. & Massague, J. Breast cancer banishes p27 from nucleus. Nat. Med. 8(10), 1076–1078 (2002).
Sutherland, R. L. & Musgrove, E. A. Cyclins and breast cancer. J. Mammary Gland Biol. Neoplasia 9(1), 95–104 (2004).
Caldon, C. E., Daly, R. J., Sutherland, R. L. & Musgrove, E. A. Cell cycle control in breast cancer cells. J. Cell. Biochem. 97(2), 261–274 (2006).
Umene, K. et al. Aurora kinase inhibitors: Potential molecular-targeted drugs for gynecologic malignant tumors (Review) Corrigendum in/10.3892/br. 2019.1249. Biomed. Rep. 1(3), 335–340 (2013).
Kivinummi, K. et al. The expression of AURKA is androgen regulated in castration-resistant prostate cancer. Sci. Rep. 7(1), 1–11 (2017).
Du, R., Huang, C., Liu, K., Li, X. & Dong, Z. Targeting AURKA in cancer: Molecular mechanisms and opportunities for Cancer therapy. Mol. Cancer 20(1), 1–27 (2021).
Jacobsen, A. et al. Aurora kinase A (AURKA) interaction with Wnt and Ras-MAPK signalling pathways in colorectal cancer. Sci. Rep. 8(1), 1–11 (2018).
Yang, Y. et al. Silencing of AURKA augments the antitumor efficacy of the AURKA inhibitor MLN8237 on neuroblastoma cells. Cancer Cell Int. 20(1), 1–16 (2020).
Donnella, H. J. et al. Kinome rewiring reveals AURKA limits PI3K-pathway inhibitor efficacy in breast cancer. Nat. Chem. Biol. 14(8), 768–777 (2018).
Tang, A. et al. Aurora kinases: novel therapy targets in cancers. Oncotarget 8(14), 23937 (2017).
González-Loyola, A. et al. Aurora B overexpression causes aneuploidy and p21Cip1 repression during tumor development. Mol. Cell. Biol. 35(20), 3566–3578 (2015).
Wu, W.-D., Yu, K.-W., Zhong, N., Xiao, Y. & She, Z.-Y. Roles and mechanisms of Kinesin-6 KIF20A in spindle organization during cell division. Eur. J. Cell Biol. 98(2–4), 74–80 (2019).
Shen, T. et al. KIF20A affects the prognosis of bladder cancer by promoting the proliferation and metastasis of bladder cancer cells. Dis. Markers https://doi.org/10.1155/2019/4863182 (2019).
Chlenski, A. et al. Maternal embryonic leucine zipper kinase (MELK), a potential therapeutic target for neuroblastoma. Mol. Cancer Ther. 18(3), 507–516 (2019).
Li, G., Yang, M., Zuo, L. & Wang, M. X. MELK as a potential target to control cell proliferation in triple-negative breast cancer MDA-MB-231 cells. Oncol. Lett. 15(6), 9934–9940 (2018).
Siddik, Z. H. Cisplatin: Mode of cytotoxic action and molecular basis of resistance. Oncogene 22(47), 7265–7279 (2003).
Maia, A. R. et al. Inhibition of the spindle assembly checkpoint kinase TTK enhances the efficacy of docetaxel in a triple-negative breast cancer model. Ann. Oncol. 26(10), 2180–2192 (2015).
Liu, Y. et al. TTK is a potential therapeutic target for cisplatin-resistant ovarian cancer. J. Ovarian Res. 14(1), 1–10 (2021).
Preston-Martin, S., Pike, M. C., Ross, R. K., Jones, P. A. & Henderson, B. E. Increased cell division as a cause of human cancer. Can. Res. 50(23), 7415–7421 (1990).
Broderick, R. & Nasheuer, H.-P. Regulation of Cdc45 in the cell cycle and after DNA damage. Biochem. Soc. Trans. 37(4), 926–930 (2009).
Kapanidou, M., Curtis, N. L. & Bolanos-Garcia, V. M. Cdc20: at the crossroads between chromosome segregation and mitotic exit. Trends Biochem. Sci. 42(3), 193–205 (2017).
Ajucarmelprecilla, A. et al. In silico identification of hub genes as observing biomarkers for gastric cancer metastasis. Evid.-Based Complement. Altern. Med. https://doi.org/10.1155/2022/6316158 (2022).
Cui, X. H. et al. Cell division cycle associated 8: A novel diagnostic and prognostic biomarker for hepatocellular carcinoma. J. Cell Mol. Med. 25(24), 11097–11112 (2021).
Huang, X. et al. Loss of cell division cycle-associated 5 promotes cell apoptosis by activating DNA damage response in clear cell renal cell carcinoma. Int. J. Oncol. 61(1), 1–17 (2022).
Jin, Z., Peng, F., Zhang, C., Tao, S., Xu, D., Zhu, Z., Expression, regulating mechanism and therapeutic target of KIF20A in multiple cancer. Heliyon 9(2), 1–16 (2023).
Pandi, J. et al. Biomarkers for breast adenocarcinoma using in silico approaches. Evid.-Based Complement. Altern. Med. https://doi.org/10.1155/2022/7825272 (2022).
Ozawa, H. et al. Targeting AURKA in treatment of peritoneal tumor dissemination in gastrointestinal cancer. Transl. Oncol. 16, 101307 (2022).
Yao, W., Jiang, M., Zhang, M., Zhang, H. & Liang, X. TTK: A promising target in malignant tumors. J. Cell. Signal. 2(3), 212–220 (2021).
Bar-Joseph, Z. et al. Computational discovery of gene modules and regulatory networks. Nat. Biotechnol. 21(11), 1337–1342 (2003).
Roy, S., Bhattacharyya, D. K. & Kalita, J. K. Reconstruction of gene co-expression network from microarray data using local expression patterns. BMC Bioinform. 15(7), 1–14 (2014).
Dudley, W. N., Wickham, R. & Coombs, N. An introduction to survival statistics: Kaplan-Meier analysis. J. Adv. Pract. Oncol. 7(1), 91 (2016).
Wei, Y. & Royston, P. Reconstructing time-to-event data from published Kaplan-Meier curves. Stand. Genom. Sci. 17(4), 786–802 (2017).
Zhang, X. et al. MELK inhibition effectively suppresses growth of glioblastoma and cancer stem-like cells by blocking AKT and FOXM1 pathways. Front. Oncol. 10, 608082 (2021).
Cho, Y.-S., Kang, Y., Kim, K., Cha, Y.-J. & Cho, H.-S. The crystal structure of MPK38 in complex with OTSSP167, an orally administrative MELK selective inhibitor. Biochem. Biophys. Res. Commun. 447(1), 7–11 (2014).
Simon, M., Mesmar, F., Helguero, L. & Williams, C. Genome-wide effects of MELK-inhibitor in triple-negative breast cancer cells indicate context-dependent response with p53 as a key determinant. PLoS ONE 12(2), e0172832 (2017).
McDonald, I. M. et al. Mass spectrometry–based selectivity profiling identifies a highly selective inhibitor of the kinase MELK that delays mitotic entry in cancer cells. J. Biol. Chem. 295(8), 2359–2374 (2020).
Georgieva, I. et al. ZM447439, a novel promising aurora kinase inhibitor, provokes antiproliferative and proapoptotic effects alone and in combination with bio-and chemotherapeutic agents in gastroenteropancreatic neuroendocrine tumor cell lines. Neuroendocrinology 91(2), 121–130 (2010).
Taylor, W. R. & Grabovich, A. Targeting the Cell Cycle to Kill Cancer Cells 429–453 (Elsevier, 2009).
Teusel, F., Henschke, L. & Mayer, T. U. Small molecule tools in mitosis research. In Methods in Cell Biology Vol. 144 137–155 (Elsevier, 2018).
Hauf, S. et al. The small molecule Hesperadin reveals a role for Aurora B in correcting kinetochore–microtubule attachment and in maintaining the spindle assembly checkpoint. J. Cell Biol. 161(2), 281–294 (2003).
Ditchfield, C. et al. Aurora B couples chromosome alignment with anaphase by targeting BubR1, Mad2, and Cenp-E to kinetochores. J. Cell Biol. 161(2), 267–280 (2003).
Chu, Q.S.-C. et al. Aurora kinase A inhibitor, LY3295668 erbumine: A phase 1 monotherapy safety study in patients with locally advanced or metastatic solid tumors. Invest. New Drugs 39, 1001–1010 (2021).
Collette, Y. et al. DIACC2010, a selective inhibitor of KIF20A. Cancer Res. 82(12_Supplement), 1813–1813 (2022).
Chan, C.Y.-K. et al. CFI-402257, a TTK inhibitor, effectively suppresses hepatocellular carcinoma. Proc. Natl. Acad. Sci. 119(32), e2119514119 (2022).
Author information
Authors and Affiliations
Contributions
F.R. contributed to the investigations, and did writing-original draft, visualization and figures preparation, data analysis, and literature search. All authorship and correspondence goes to F.R. as the only author.
Corresponding author
Ethics declarations
Competing interests
The author declares no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rommasi, F. Identification, characterization, and prognosis investigation of pivotal genes shared in different stages of breast cancer. Sci Rep 13, 8447 (2023). https://doi.org/10.1038/s41598-023-35318-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-35318-x
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
