
Abstract
Iron homeostasis and dyserythropoiesis are poorly investigated in pyruvate kinase deficiency (PKD), the most common glycolytic defect of erythrocytes. Herein, we studied the main regulators of iron balance and erythropoiesis, as soluble transferrin receptor (sTfR), hepcidin, erythroferrone (ERFE), and erythropoietin (EPO), in a cohort of 41 PKD patients, compared with 42 affected by congenital dyserythropoietic anemia type II (CDAII) and 50 with hereditary spherocytosis (HS). PKD patients showed intermediate values of hepcidin and ERFE between CDAII and HS, and clear negative correlations between log-transformed hepcidin and log-EPO (Person’s r correlation coefficient = − 0.34), log-hepcidin and log-ERFE (r = − 0.47), and log-hepcidin and sTfR (r = − 0.44). sTfR was significantly higher in PKD; EPO levels were similar in PKD and CDAII, both higher than in HS. Finally, genotype–phenotype correlation in PKD showed that more severe patients, carrying non-missense/non-missense genotypes, had lower hepcidin and increased ERFE, EPO, and sTFR compared with the others (missense/missense and missense/non-missense), suggesting a higher rate of ineffective erythropoiesis. We herein investigated the main regulators of systemic iron homeostasis in the largest cohort of PKD patients described so far, opening new perspectives on the molecular basis and therapeutic approaches of this disease.
Similar content being viewed by others


Introduction
Pyruvate kinase deficiency (PKD), due to bi-allelic mutations in PKLR gene (1q21), is the most common enzyme-related glycolytic defect that results in red blood cell (RBC) hemolysis1. Pyruvate kinase enzyme plays a key role in glycolysis, converting phosphoenolpyruvate to pyruvate, upon which RBC metabolism completely hinges. Lack of PK enzyme affects RBC ATP production, causing RBC damage, and the consequent trapping of defective cells by splenic and hepatic capillaries. Patients with PKD present with varying degrees of clinical manifestations, ranging from mild or asymptomatic well-compensated hemolysis to severe transfusion-dependent anemia from birth; complications include iron overload, pulmonary hypertension, endocrinopathies, liver failure, biliary disease, and extramedullary hematopoiesis2. The high phenotype variability reflects the molecular heterogeneity of the disease, with more than 400 pathogenic variants reported in PKLR gene3. A more severe phenotype, including rare complications, is usually observed in patients carrying non-missense mutations or missense mutations characterized by severely decreased protein stability4.
Intriguingly, PKD patients have been shown to develop iron overload even in absence of transfusions, suggesting dyserythropoietic features5. Moreover, recent studies showed that a quote of patients with clinical suspicion of congenital dyserythropoietic anemia (CDA) had a conclusive molecular diagnosis of chronic anemia arising from enzymatic defects, mainly PKD6. The presence of bone marrow abnormalities and ineffective erythropoiesis in these patients led to misdiagnosis, further supporting this evidence7.
Human PK comprises four isozymes (L, R, M1, M2). R-PK is the only one found in normal mature red cells. Hematopoietic stem cells and progenitor cells express M2-PK, which switches to R-PK in cells of the erythroid lineage, where it is constantly synthesized during erythroid cell maturation7. Thus, it was suggested that metabolic abnormalities in R-PK deficiency could alter the differentiation of erythroid progenitors into mature erythrocytes. Indeed, it was reported that hematopoiesis was enhanced in the spleen of PK-deficient patients8.
Evaluation of hepcidin, the master regulator of iron balance, has a key role in determining iron status due to ineffective erythropoiesis. It was demonstrated that the pathogenesis of iron overload in iron-loading anemias, such as CDA type II (CDAII) and beta-thalassemia, is related to the over-expression of the erythroblast-derived hormone erythroferrone (ERFE)9, leading to hepcidin suppression10,11,12. Dysregulation of the EPO-ERFE-hepcidin pathway has been recently reported in a group of patients with congenital hemolytic anemia including PKD13.
Herein, we investigated the pathophysiological basis of iron overload and ineffective erythropoiesis in a larger cohort of PKD patients, stratified by PKLR genotype, compared either with the model of a structural RBC membrane defect, such as hereditary spherocytosis (HS), or with the typical model of dyserythropoietic anemia with ineffective erythropoiesis, as CDAII.
Results
Analysis of erythropoiesis biomarkers in different inherited anemias
We examined 41 PKD, 42 CDAII, and 50 HS patients who differed for a number of characteristics (Table 1). In PKD patients, median age at the study was 26 years, hemoglobin (Hb) level was 9.1 g/dL, reticulocytes absolute number was 156 × 109/L, and ferritin level was 546 ng/mL. Twenty-nine PKD patients received at least three transfusions during their life, 14 of them were transfusion dependent (≥ 6 transfusions/year), 12 underwent splenectomy, and 15 patients required chelation therapy for iron overload developed also in absence of transfusions. Comparing with the other patient groups, PKD patients were younger, had Hb level similar to CDAII but lower than HS and showed intermediate median reticulocytes number between CDAII and HS patients (Table 1).
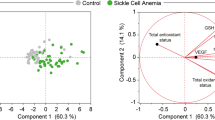
All patients showed ERFE, EPO, and sTfR levels clearly increased respect to normal controls. On the contrary, hepcidin levels were lower than normal in both PKD and CDAII (Table 2). Interestingly, the comparison among the three groups showed that PKD patients had intermediate values of hepcidin and ERFE between CDAII and HS. No differences were observed regarding EPO levels in PKD and CDAII patients, both higher than in HS. Finally, sTfR was significantly higher in PKD compared with the other hemolytic patients. These results were confirmed in multiple regression analysis adjusted for center (data not shown). The analysis of single data distribution, although showing an expected variability, highlighted that PK deficient patients have intermediate values between CDAII and HS, suggesting that PK deficiency displays ineffective erythropoiesis features as in CDAII, independently from transfusion status (Table 2, Fig. 1). In PKD and HS patients we observed positive correlations between EPO, ERFE and sTfR and a negative correlation between hepcidin and ERFE. A similar pattern, although weaker, was observed in CDAII patients (Fig. 2A–C).

Distribution of hepcidin, EPO, ERFE, and sTfR in CDAII, PKD, and HS patients. Median values and range are reported. Green lines represent median control’s values. Transfusion-dependent (TD—blue filled circle) and not transfusion-dependent (NTD—red filled circle) PKD patients are shown. Kruskal–Wallis test; P-values and reference ranges for each variable are reported in Table 2.

Pairwise correlations between selected variables in CDAII (A), PKD (B), and HS patients (C). Pearson’s r correlation coefficients are indicated in the figure. *P < 0.05 and **P < 0.001. Hepcidin, EPO, and ERFE were ln-transformed.
Iron homeostasis biomarkers and PKLR genotype
PKD patients were grouped according with their genotype on the basis of the different types of mutations: missense (M)/M, missense/non-missense (NM) or NM/NM. Twenty-four patients showed M/M, 8M/NM and 8 NM/NM genotype. In one case, PKD genotype was not available, and the patient was excluded by the analysis. NM/NM patients showed lower hepcidin level although not statistically significant (P = 0.07), increased ERFE (P = 0.004), EPO (P = 0.003) and sTfR (P = 0.007) than the other PKD patients suggesting a higher level of ineffective erythropoiesis in this group of patients (Table 3, Fig. 3). Comparable results were observed after considering in the NM/NM group the four patients carrying homozygous c.1529A > G, p.Arg510Gln variant, known to be characterized by severely decreased protein stability14.

Distribution of hepcidin, EPO, ERFE, and sTfR in PKD patients grouped according with their genotype. Green lines represent median control’s values. *P < 0.001, Mann–Whitney test. Reference ranges for each variable are reported in Table 2.
Discussion
Pyruvate kinase deficiency is the most common enzyme-related glycolytic defect that results in red cell hemolysis. This disorder is characterized by clinical heterogeneity, which results in a variable degree of hemolytic, non-spherocytic anemia. Manifestations occur from the neonatal period through adult life, causing countless complications5,15. The variety of unspecific and overlapping phenotypes with other congenital anemias, particularly between PKD and HS in the mildest forms, and hypo-regenerative anemias such as CDAII, often hampers a correct clinical management of the patients16. Presence of inadequate reticulocytosis, erythroblasts in peripheral blood, or of immature forms at bone marrow examination, are not so rare in patients with severe PKD5.
Several studies suggested that metabolic abnormalities in PKD could alter the differentiation of erythroid progenitors. Of note, studies on PK-1slc deficient mice showed an increased number of BFU-E, indicating enhanced erythropoiesis. In addition, the presence of apoptotic cells of erythroid lineage in the splenic red pulp suggested that the metabolic disturbance in PKD alters not only the survival of erythrocytes but also the maturation of erythroid progenitors, resulting in ineffective erythropoiesis17.
To demonstrate the occurrence of this condition in PKD, we measured the levels of different markers in our cohort of patients, and we compared the results with two different models of congenital hemolytic anemia, CDAII with overt dyserythropoiesis, and HS, characterized by normal erythroid production but increased red cell fragility.
Although most of the analyzed parameters were altered in all the hemolytic patients studied including HS, as expected we found higher level of ineffective erythropoiesis in PKD and CDAII. Indeed, significantly increased levels of EPO were found in PKD patients compared to reference values of healthy subjects. Of note, even with a slight difference, EPO levels were also significantly increased when compared to those of CDAII patients, probably due to a greater hemolytic component and the higher degree of anemia in PKD than CDAII.
It is well known that the concentration of sTfR reflects erythropoiesis rate18. Expansion of erythroid precursors to compensate a loss of RBCs leads to an augmented level of sTfR1 due to an increase of highly expressing TfR1+ cells19. We observed increased levels of sTfR in PKD patients compared not only to CDAII, but also to HS patients. This finding may be partially justified either by the higher ferritin levels found in the PKD subjects respect to the other patients analyzed in this series or by the increased reticulocyte number in both HS and PKD, particularly high after splenectomy in the latter. Of note, sTfR levels in CDAII patients were similar to those previously reported for these patients12,20. The increased sTfR levels in HS (in some cases also higher than CDAII patients) are in line with previous reports on different series of HS: a direct correlation between clinical severity and sTfR levels was in fact reported in a large case series of 82 HS21. Moreover, increased EPO and sTfR was reported in another case series of 32 HS, also with compensated hemolysis, suggesting that inappropriately high EPO and sTfR may represent a biological characteristic of the disease22.
It was already demonstrated that ERFE expression, which is produced by erythroblasts in response to EPO, is increased in conditions characterized by ineffective erythropoiesis, such as CDAII and beta-thalassemia12,13, resulting in hepcidin suppression23. All our PKD patients showed increased levels of ERFE compared to controls, and slightly decreased values compared to those of CDAII patients. Interestingly, they showed decreased hepcidin values, although to a lesser extent to those observed in CDAII cases, corroborating the observation that ERFE is not the only erythroid regulator that contributes to hepcidin inhibition23. Our results agree with a recent study investigating the EPO-ERFE-hepcidin pathway in PKD, HS, and beta-thalassemia patients13. In addition, given the extreme phenotypic variability in PKD24, we also considered PKLR genotype since nonsense PKLR variants are known to be associated with severe phenotype4, and because of genotype is an independent factor from differences in management approaches, age, or period of the study. Although the limited number of cases due to the rarity of this disease, and the low proportion of NM/NM cases (8/40, 20% of PKD cases), we observed higher level of ineffective erythropoiesis in patients with disruptive variants.
Although the parameter herein analyzed are well-known and widely used markers of ineffective erythropoiesis, they represent only a limited part of the many different direct and indirect players involved in this process25,26. Moreover, also iron deposits (as measured with LIC MRI) may put in light dyserythropoietic bases of some disorders27,28.
The clinical heterogeneity of the investigated patients in terms of clinical severity, different management approaches, different age at diagnosis, together with selection of the analyzed parameters might represent a bias that accounts for the variability of the results. However, unraveling mechanisms of iron loading and ineffective erythropoiesis in PKD and related disorders by identifying easy-to-access laboratory markers may be useful in differential diagnosis and for new therapeutic perspectives29.
Materials and methods
Patients, plasma collection, and ELISA assay
Overall, 133 patients with different types of hereditary hemolytic anemias were retrospectively included in the study after obtaining informed consent. For patients under the age of 18, written informed consent was obtained from the parents. The procedures followed were in accordance with relevant guidelines and regulations and with the Helsinki international ethical standards on human experimentation. Clinical diagnosis was based on history, clinical findings, laboratory data, morphological analysis of peripheral blood, and genetic testing30,31. Local institution ethical committees approved both the plasma sampling and the collection of patients’ data from Medical Genetics Ambulatory in Naples (University Federico II, DAIMedLab) and Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico of Milan (Milano, Area 2). Plasma samples were collected during routine laboratory investigation from peripheral blood of patients and healthy controls.
Plasma levels of human ERFE (Intrinsic Erythroferrone IE; Intrinsic Lifesciences, La Jolla, California, USA), erythropoietin (EPO) (Quantikine IVD ELISA Human Erythropoietin), and soluble transferrin receptor (sTfR) (Quantikine IVD ELISA Human sTfR, R&D System Minneapolis, Minnesota, USA) were quantified using ELISA kits. Hepcidin evaluation was performed with Intrinsic HEPCIDIN IDxTM (Intrinsic Lifesciences, CA, USA) and Hepcidin-25 ELISA (DRG Instruments GmbH, Germany). In 20/42 CDA patients iron status parameters were previously reported32. Prior to the analysis of plasma samples from patients, the reference intervals of both hepcidin assays were evaluated on reference samples for the harmonization of the results obtained by the two different kits used in Naples and Milan respectively. The concentration of each parameter in each sample was determined through the fitting of a four-parameter logistic curve, according to the manufacturer protocols.
Clinical severity and PKLR genotype
For the analysis of the correlation of iron homeostasis markers and clinical severity in PKD, we stratified patients according to transfusions, splenectomy, or iron chelation need. Transfusion needs were classified into the following categories: (a) regularly transfused ≥ 6/year during life; (b) not regularly transfused or not transfused. Intermittent chelation therapy was considered in case of transfusion dependency and/or ferritin levels > 500 ng/mL.
Based on previous observations in literature of the existence of genotype–phenotype correlation in PKD4, patients were categorized according to their genotype as follows: (a) missense/missense genotype (M/M); (b) missense/non-missense genotype (M/NM); or (c) non-missense/non-missense genotype (NM/NM). Non-missense pathogenic variants included stop gain, frameshift, in-frame small ins/del/indels, large deletions, and splicing variants; missense mutations also affecting splicing were categorized as non-missense (Supplemental Table 1). For statistical analysis, M/M and M/MN patients were grouped together, given similar clinical phenotype.
Statistical analysis
Demographic and hematologic data has been expressed as median and range and compared across the three diseases (CDAII, PK deficiency, and HS) by calculating chi-squared and Kruskal–Wallis tests (for categorical or quantitative variables, respectively).
Comparison of quantitative variables across the three diseases were analyzed with Mann–Whitney and Kruskal–Wallis test. Additional multiple regression analyses were performed after adjusting for center. We did not adjust for other variables because they are not confounders (by definition they do not affect inherited disorders)33. We calculated Pearson’s r correlation coefficients between selected hematological variables. In regression and correlation analyses hepcidin, EPO and ERFE were ln-transformed. Statistical analyses were performed with Stata 17 (StataCorp. 2021).
Data availability
All data generated or analysed during this study are included in this published article (and its Supplementary Information files).
References
Secrest, M. H. et al. Prevalence of pyruvate kinase deficiency: A systematic literature review. Eur. J. Haematol. 105, 173–184 (2020).
Grace, R. F. et al. Clinical spectrum of pyruvate kinase deficiency: Data from the Pyruvate Kinase Deficiency Natural History Study. Blood 131, 2183–2192 (2018).
Bianchi, P. & Fermo, E. Molecular heterogeneity of pyruvate kinase deficiency. Haematologica 105, 2218–2228 (2020).
Bianchi, P. et al. Genotype-phenotype correlation and molecular heterogeneity in pyruvate kinase deficiency. Am. J. Hematol. 95, 472–482 (2020).
Al-Samkari, H. et al. The variable manifestations of disease in pyruvate kinase deficiency and their management. Haematologica 105, 2229–2239 (2020).
Roy, N. B. A. et al. The use of next-generation sequencing in the diagnosis of rare inherited anaemias: A joint BSH/EHA good practice paper. Hemasphere 6, e739 (2022).
Takegawa, S., Fujii, H. & Miwa, S. Change of pyruvate kinase isozymes from M2- to L-type during development of the red cell. Br. J. Haematol. 54, 467–474 (1983).
Aizawa, S. et al. Ineffective erythropoiesis in the spleen of a patient with pyruvate kinase deficiency. Am. J. Hematol. 74, 68–72 (2003).
Coffey, R. et al. Erythroferrone is not required for the glucoregulatory and hematologic effects of chronic erythropoietin treatment in mice. Physiol. Rep. 6, e13890 (2018).
Andolfo, I. et al. The BMP-SMAD pathway mediates the impaired hepatic iron metabolism associated with the ERFE-A260S variant. Am. J. Hematol. 94, 1227–1235 (2019).
Kautz, L. et al. Erythroferrone contributes to hepcidin suppression and iron overload in a mouse model of β-thalassemia. Blood 126, 2031–2037 (2015).
Russo, R. et al. Increased levels of ERFE-encoding FAM132B in patients with congenital dyserythropoietic anemia type II. Blood 128, 1899–1902 (2016).
van Vuren, A. J. et al. A comprehensive analysis of the erythropoietin-erythroferrone-hepcidin pathway in hereditary hemolytic anemias. Hemasphere 5, e627 (2021).
Wang, C. et al. Human erythrocyte pyruvate kinase: Characterization of the recombinant enzyme and a mutant form (R510Q) causing nonspherocytic hemolytic anemia. Blood 98, 3113–3120 (2001).
Boscoe, A. N. et al. Comorbidities and complications in adults with pyruvate kinase deficiency. Eur. J. Haematol. 106, 484–492 (2021).
Russo, R., Marra, R., Rosato, B. E., Iolascon, A. & Andolfo, I. Genetics and genomics approaches for diagnosis and research into hereditary anemias. Front. Physiol. 11, 613559 (2020).
Aizawa, S. et al. Ineffective erythropoiesis in mutant mice with deficient pyruvate kinase activity. Exp. Hematol. 33, 1292–1298 (2005).
Kohgo, Y., Torimoto, Y. & Kato, J. Transferrin receptor in tissue and serum: Updated clinical significance of soluble receptor. Int. J. Hematol. 76, 213–218 (2002).
Richard, C. & Verdier, F. Transferrin receptors in erythropoiesis. Int. J. Mol. Sci. 21, 9713 (2020).
Russo, R. et al. Retrospective cohort study of 205 cases with congenital dyserythropoietic anemia type II: Definition of clinical and molecular spectrum and identification of new diagnostic scores. Am. J. Hematol. 89, E169–E175 (2014).
Rocha, S. et al. Complementary markers for the clinical severity classification of hereditary spherocytosis in unsplenectomized patients. Blood Cell Mol. Dis. 46, 166–170 (2011).
Guarnone, R., Centenara, E., Zappa, M., Zanella, A. & Barosi, G. Erythropoietin production and erythropoiesis in compensated and anaemic states of hereditary spherocytosis. Br. J. Haematol. 92, 150–154 (1996).
Rosato, B. E. et al. SEC23B loss-of-function suppresses hepcidin expression by impairing glycosylation pathway in human hepatic cells. Int. J. Mol. Sci. 23, 1304 (2022).
Grace, R. F., Layton, M. D. & Barcellini, W. How we manage patients with pyruvate kinase deficiency. Br. J. Haematol. 184, 721–734 (2019).
van Vuren, A. J. et al. Interplay of erythropoietin, fibroblast growth factor 23, and erythroferrone in patients with hereditary hemolytic anemia. Blood Adv. 28, 1678–1682 (2020).
Yan, H. et al. Comprehensive phenotyping of erythropoiesis in human bone marrow: Evaluation of normal and ineffective erythropoiesis. Am. J. Hematol. 96, 1064–1076 (2021).
Barcellini, W. et al. Iron overload in congenital haemolytic anaemias: Role of hepcidin and cytokines and predictive value of ferritin and transferrin saturation. Br. J. Haematol. 185, 523–531 (2019).
van Straaten, S. et al. Iron overload in patients with rare hereditary hemolytic anemia: Evidence-based suggestion on whom and how to screen. Am. J. Hematol. 93, E374–E376 (2018).
Matte, A. The pyruvate kinase activator mitapivat reduces hemolysis and improves anemia in a β-thalassemia mouse model. J. Clin. Investig. 131, e144206 (2021).
Fermo, E. et al. Targeted next generation sequencing and diagnosis of congenital hemolytic anemias: A three years experience monocentric study. Front. Physiol. 12, 684569 (2021).
Andolfo, I. et al. Complex modes of inheritance in hereditary red blood cell disorders: A case series study of 155 patients. Genes 23, 958 (2021).
Andolfo, I. et al. Gain-of-function mutations in PIEZO1 directly impair hepatic iron metabolism via the inhibition of the BMP/SMADs pathway. Am. J. Hematol. 95, 188–197 (2020).
Digitale, J. C., Marin, J. N. & Glymour, M. M. Tutorial on directed acyclic graphs. J. Clin. Epidemiol. 142, 264–267 (2022).
Acknowledgements
This research was funded by an EHA Junior Research Grant to Immacolata Andolfo (3978026), by Bando Star Linea 1—Junior Principal Investigator Grants—COINOR, Università degli Studi di Napoli ‘Federico II’ to Roberta Russo, by Italian Ministry of Health—PRIN, Italian Ministry of Research to Achille Iolascon (2020Z22PM7) an by Italian Ministry of Health—Current research IRCCS, Fondazione IRCCS Ca’ Granda Policlinico Milano, project no. RC 175/05. This work is generated within the European Reference Network on Rare Hematological Diseases (ERN-EuroBloodNet). FPA 739541. AI, RR, IA, WB, PB, EF are EuroBloodNet members.
Author information
Authors and Affiliations
Contributions
R.R. and P.B. designed the study and prepared the manuscript; A.Z. and R.M. performed ELISA assays and prepared the initial draft of the manuscript; I.A., B.E.R., and E.F. performed genetic testing; A.P.M., C.V. performed laboratory diagnosis of patients; D.C. performed statistical analysis; W.B. and A.I. cared for the patients. All the authors provided a critical review of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
PB, WB Agios Pharmaceuticals: advisory board member and consultancy; All the other authors declare no conflicts of interest.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zaninoni, A., Marra, R., Fermo, E. et al. Evaluation of the main regulators of systemic iron homeostasis in pyruvate kinase deficiency. Sci Rep 13, 4395 (2023). https://doi.org/10.1038/s41598-023-31571-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-31571-2
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
