
Abstract
Proteostasis regulates protein folding and degradation; its maintenance is essential for resistance to stress and aging. The loss of proteostasis is associated with many age-related diseases. Within the cell, molecular chaperones facilitate the refolding of misfolded proteins into their bioactive forms, thus preventing undesirable interactions and aggregation. Although the mechanisms of intracellular protein degradation pathways for intracellular misfolded proteins have been extensively studied, the protein degradation pathway for extracellular proteins remain poorly understood. In this study, we identified several misfolded proteins that are substrates for alpha 2-macroglobulin (α2M), an extracellular chaperone. We also established a lysosomal internalization assay for α2M, which revealed that α2M mediates the lysosomal degradation of extracellular misfolded proteins. Comparative analyses of α2M and clusterin, another extracellular chaperone, indicated that α2M preferentially targets aggregation-prone proteins. Thus, we present the degradation pathway of α2M, which interacts with aggregation-prone proteins for lysosomal degradation via selective internalization.
Similar content being viewed by others

Introduction
Protein misfolding can occur upon exposure to stresses such as heat, oxidation, and pH change. The exposure of hydrophobic sites on misfolded proteins causes non-specific interactions and induces the formation of protein aggregates1. Protein aggregates are associated with the onset of neurodegenerative diseases such as Alzheimer's disease2; the maintenance of proteostasis prevents loss of function and eliminates toxicity. In mammals, the mechanisms that maintain intracellular proteostasis are well-known. Molecular chaperones interact with misfolded proteins in cells, maintain solubility, and mediate refolding to prevent the aggregation of such proteins3. Major molecular chaperones (e.g., heat shock proteins) are upregulated and inhibit protein denaturation when cells are exposed to heat stress4,5. Intracellular protein degradation pathways are also essential for proteostasis. The proteasome degrades misfolded proteins that are ubiquitinated by specific E3 ubiquitin ligases6. Autophagy degrades damaged proteins and organelles via lysosomal degradation7. However, intracellular proteostasis cannot occur outside of the cell; moreover, intracellular chaperones could not mediate refolding if they were present in the extracellular environment because there is a considerably lower concentration of adenosine triphosphate (ATP) in extracellular fluids3,8. In addition, extracellular conditions are generally harsher than intracellular conditions9. Therefore, a distinct mechanism mediated by extracellular chaperones is required to maintain extracellular proteostasis and prevent aggregation in the extracellular environment. In the first step, an extracellular chaperone interacts with a substrate in the extracellular environment, ensuring the solubility of the substrate. In the second step, the extracellular chaperone–substrate complex is selectively internalized into the cell via endocytosis; it then undergoes lysosomal degradation10.
Extracellular chaperones including clusterin, Haptoglobin, alpha 2-macroglobulin (α2M) and casein prevent aggregate formation in the extracellular environment9,11,12,13,14,15,16. Clusterin has been characterized in terms of its chaperone activity and its role in the degradation pathway10,17,18; however, other extracellular chaperones have not been well characterized, and the degradation pathways of extracellular chaperones remain poorly understood. A clear understanding of the mechanisms by which extracellular chaperones mediate extracellular protein degradation will contribute to the development of novel therapies for various diseases that involve protein degeneration; such diseases include Alzheimer's disease19, which impacts approximately 50 million patients worldwide but has no effective treatment. We recently reported that a clusterin–misfolded protein complex is selectively delivered into cells through a cell surface receptor for lysosomal degradation10. α2M has also been shown to interact with stressed proteins and has been proposed to facilitate their disposal via receptors20; however, the degradation pathway of α2M remains uncharacterized. In the present study, we investigated α2M with the aim of determining its substrate specificity and its role in the lysosomal degradation of extracellular misfolded proteins.α2M is a 180-kDa protein that forms a homotetramer21; it is mainly produced in the liver and secreted into the blood22. α2M is the sixth most abundant plasma protein23 (2.1 mg/mL24); it is also present in cerebrospinal fluid at low levels (1.0–3.6 μg/mL25). The plasma concentration of α2M increases during inflammatory responses in mice26. The molecular mechanism underlying the protease inhibitor effects of α2M has been widely studied21. When the highly protease-sensitive bait region of α2M is cleaved by a protease, a structural change entraps the protease, thus inhibiting protease activity21. This change also exposes a receptor recognition site for low-density lipoprotein receptor-related protein (LRP1), which targets the α2M–protease complex for endocytic degradation20,27,28. Through this mechanism, α2M eliminates all classes of proteases from the extracellular environment29. α2M reportedly exhibits chaperone-like activity and inhibits the aggregation of heat-stressed proteins20. However, it remains unclear whether α2M participates in the lysosomal degradation of extracellular misfolded proteins. Therefore, in this study, we established a fluorescence internalization assay to measure α2M-mediated lysosomal degradation. Using green (GFP) and red fluorescence protein (RFP), the internalization assay quantitatively detects α2M on the plasma membrane and in lysosomes. We then conducted a comprehensive analysis to identify blood cell proteins that bind to α2M in a misfolding-dependent manner. The results of this study will provide a conceptual framework to explain how extracellular chaperones cooperatively contribute to extracellular proteostasis in the process that leads to lysosomal degradation of misfolded extracellular proteins.
Results
α2M and misfolded proteins are internalized into cells for lysosomal degradation
We recently discovered that clusterin mediates the lysosomal degradation of extracellular abnormal proteins10. To determine whether another extracellular chaperone, α2M, also participates in the lysosomal degradation pathway, we produced a recombinant protein of α2M fused with two fluorescent proteins (mCherry [an RFP] and superfolder GFP) and a His-tag sequence from mammalian cells. RFP is highly resistant to lysosomal proteases and is pH-insensitive, whereas GFP and α2M are not resistant to lysosomal proteases and acidic environments30. Therefore, when α2M–RFP–GFP–His protein is internalized into lysosomes, the fluorescence of RFP, but not GFP, is detected10 (Fig. 1A). To avoid potential structural interference of RFP–GFP (RG) tagging to α2M, we prepared two plasmids that expressed α2M–RG (–His) or (His–) RG–α2M. These plasmids were introduced into Flp-in T-Rex HEK293 cells using the FLP/FRT recombination system to generate stable cell lines. Immunoblotting demonstrated that α2M–RG was secreted in the culture supernatant, whereas RG–α2M was not (Fig. S1A), which indicated that α2M–RG–His was correctly folded. Therefore, we purified α2M–RG protein from the conditioned medium (Fig. S1B). We characterized the conformational state of α2M-RG using Native-polyacrylamide gel electrophoresis (Native-PAGE) and methylamine, which undergoes conformational collapse of α2M that is highly similar to its protease-cleaved conformation by aminolysis of its thiol ester31,32. Native-PAGE revealed that α2M–RG migrated to ~ 920 kDa, and treatment of methylamine results in faster migration of α2M–RG (collapsed a2M-RG) than untreated α2M–RG (Fig. S1C). An intact thiol ester of α2M apparent from formation of characteristic heat-induced autolysis products in sodium dodecyl sulfate (SDS)-PAGE31. Purified α2M–RG was boiled in SDS sample buffer and analyzed by SDS-PAGE. The result showed that the thiol-ester-dependent heat-fragmentation bands (TE120 for N-terminus and TE110 for C-terminus with RG) were generated, which were inhibited by methylamine (Fig. S1D). These results indicate that purified α2M–RG is a tetramer with an intact thiol ester.

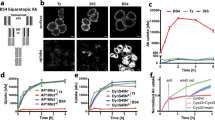
α2M induces lysosomal degradation of extracellular misfolded proteins. (A) Schematic representation of lysosomal degradation of α2M–RFP–GFP–His (–RG). Recombinant α2M–RG are internalized into lysosomes, leading to the accumulation of protease- and pH-resistant red fluorescent protein (RFP), but not α2M and green fluorescent protein (GFP). If α2M induces degradation of misfolded proteins, RFP should accumulate in the cell only in the presence of misfolded proteins. (B) α2M–RG internalization assay with red blood cell (RBC) lysate. RBC lysate and α2M–RG were heat-shocked in serum-free medium at 50 °C for 1 h. HeLa cells were cultured in the medium for 17 h at 37 °C, then analyzed using flow cytometry. Bar graph shows the relative fluorescence intensities of GFP and RFP in cells normalized to those intensities in untreated cells (n = 3). Data are means ± standard errors of the mean (SEMs). n.s., not significant; *P < 0.05 (two-tailed Student’s t-test). Small circles indicate each data point. (C) α2M–RG internalized into lysosomes. Cells were treated as described in (B), immunolabeled with the lysosomal marker LAMP1, and imaged using confocal microscopy. Dashed lines represent cell surface region. Scale bar, 10 μm. (D) RFP cleavage assay with α2M–RG and RBC lysate. Huh7 cells were treated as described in (B), and the cells were lysed for immunoblotting.
Haptoglobin captures hemoglobin leaking into plasma from intravascular hemolysis; this leads to lysosomal degradation of the hemoglobin. However, scavenger systems for other hemolysis-related intracellular proteins remain unclear. Therefore, we used red blood cell (RBC) lysate as a model substrate for α2M. To investigate whether α2M–RG is involved in the degradation of denatured proteins, we performed an α2M–RG internalization assay. RBC lysate and recombinant α2M–RG were heat-shocked in culture in advanced Dulbecco’s Modified Eagle Medium (DMEM) without serum at 50 °C for 1 h. HeLa cells were cultured in the resultant α2M–RG-containing media for 17 h at 37 °C. After incubation, the cells were collected; the fluorescence intensities of GFP and RFP were measured by flow cytometry (Fig. 1B). Incubation with α2M–RG alone or with RBC lysate did not strongly affect the RFP signal in the cells. However, heat-stressed lysate induced a statistically significant 1.3-fold increase in the RFP signal, compared with non-heat stress conditions. The fluorescence intensity of GFP tended to increase in cells treated with the lysosomal inhibitor bafilomycin A1 (Baf A), suggesting that α2M–RG is internalized into cells in the presence of misfolded proteins.
To observe the subcellular localization of α2M–RG, HeLa cells were subjected to identical treatment, then immunostained with an anti-LAMP1 antibody as a lysosomal marker (Fig. 1C). The number and intensity of RFP dots were elevated in the presence of heat-stressed RBC lysate; RFP dots were co-localized with LAMP1, indicating that α2M–RG was internalized into the lysosome. Importantly, treatment with Baf A enhanced RFP and GFP signals on both lysosomes and the cell membrane, suggesting that secondary inhibition of endocytosis by Baf A causes α2M–RG accumulation on the cell surface; this accumulated α2M–RG may be bound to an unknown cell surface receptor. Next, we confirmed lysosomal degradation of α2M–RG using an RFP cleavage assay33, which examines the release of free 25-kDa RFP that originates from the digestion of 250-kDa α2M–RG by lysosomal proteases. Free RFP was detected in cells treated with α2M–RG; the amount of free RFP was further increased in the presence of heat-stressed RBC lysate (Fig. 1D). Baf A inhibited the increase of free RFP. These cell biological and biochemical data demonstrate that α2M–RG in complexes with denatured RBC proteins is subjected to lysosomal degradation.
Identification of heat stress-dependent α2M-binding proteins
To identify RBC proteins that bound to α2M–RG in a misfolding-dependent manner, we performed co-immunoprecipitation using α2M fused to an ALFA-tag, a 13-amino acid tag that forms a stable α-helix34. A mixture of recombinant α2M–ALFA and RBC lysate was heated at 50 °C for 1 h, then subjected to co-immunoprecipitation. Interactions of α2M–RG with several proteins under heat stress were observed (Fig. 2A); we identified 142 proteins via mass spectrometry (Table S1). Spectra from the heat-stressed sample and non-heat-stressed control were graphically compared (Fig. 2B). We selected 12 candidate proteins for evaluation in pilot experiments (α2M–RG and clusterin–RG internalization assay with purified recombinant proteins), then extracted six proteins as candidate substrates for α2M: carbonic anhydrase 1 (CA1), carbonic anhydrase 2 (CA2), 26S proteasome non-ATPase regulatory subunit 5 (PSMD5), retinal dehydrogenase 1 (ALDH1A1), glutamate–cysteine ligase regulatory subunit (GCLM), and S-formylglutathione hydrolase (ESD). After purification of these recombinant proteins, including CA1, CA2, PSMD5, ALDH1A1, GCLM, and ESD, from E. coli (Fig. S2), an α2M–RG internalization assay using the purified recombinant proteins showed that the fluorescence intensity of RFP significantly increased in the presence of heat-stressed PSMD5, ALDH1A1, GCLM, and ESD (Fig. 2C). However, GCLM increased the fluorescence intensity of RFP regardless of heat stress, suggesting that its ability to bind α2M may be non-specific or different functions. In contrast, we detected no increase in the fluorescence intensity of RFP in an RFP–GFP internalization assay using the recombinant proteins (Fig. S3A); this finding indicated that α2M mediates internalization. α2M–RG internalization assays with CA1 and CA2 did not show changes in the fluorescence intensity of RFP. To determine whether α2M recognizes misfolded or aggregated proteins, substrate (PSMD5, ALDH1A1, or ESD) and α2M–RG were co-heated at 50 °C (Fig. 2D, left panel), substrates were pre-heated alone and then mixed with α2M–RG (Fig. 2D, right panel), or α2M–RG or substrate was pre-heated alone and then mixed with together (Fig. S3B); all mixtures were analyzed using an internalization assay. Flow cytometry analysis showed that pre-heated substrates or pre-heated α2M–RG did not induce α2M–RG internalization. Co-immunoprecipitation using recombinant proteins demonstrated that α2M–RG directly interacted with PSMD5, ALDH1A1, and ESD in a heat stress-dependent manner (Fig. 2E). Next, we explored whether the substrates were internalized into lysosomes by α2M–RG. Immunoblotting analysis confirmed that the amount of intracellular ESD increased in the presence of α2M–RG (Fig. 2F). Co-treatment of α2M–RG with the lysosomal inhibitor Baf A led to ALDH1A1 and ESD accumulation. These data indicate that α2M directly interacts with misfolded proteins and leads to the lysosomal degradation of the substrate.

Identification of misfolding-dependent substrates for α2M. (A) Several heat-stressed proteins directly interacted with α2M. Purified α2M–ALFA was mixed with or without RBC lysate, then pre-incubated at 4 °C or 50 °C (heat stress) for 1 h. Samples were subjected to co-immunoprecipitation with anti-ALFA sepharose and analyzed by ruby staining. (B) Mass spectrometric analysis of α2M-binding proteins (see also Table S1). Orange (gray) dots indicate proteins that bind α2M in a heat stress-dependent (independent) manner. (C) α2M–RG internalization assay with substrates. Six substrates (CA1, CA2, PSMD5, ALDH1A1, GCLM, and ESD) and α2M–RG were heat-shocked in serum-free medium at 50 °C for 1 h. HeLa cells were cultured in medium for 17 h at 37 °C, then analyzed using flow cytometry. Bar graph shows the relative fluorescence intensities of GFP and RFP in cells normalized to those intensities in untreated cells (n = 3). Data are means ± SEMs. Small circles indicate each data point. (D) α2M did not deliver aggregated proteins to lysosomes. Substrates (PSMD5, ALDH1A1, or ESD) were co-heated with α2M–RG at 50 °C (left panel) or pre-heated alone, then mixed with α2M–RG (right panel). HeLa cells were cultured in medium for 17 h at 37 °C, then analyzed using flow cytometry (n = 1). (E) α2M interacted directly with substrates. PSMD5, ALDH1A1, and ESD were each mixed with or without α2M–RG, then pre-incubated at 4° or 50 °C (heat stress) for 1 h. Samples were subjected to co-immunoprecipitation with anti-GFP sepharose. (F) α2M–RG cleavage assay with ALDH1A1 and ESD. Huh7 cells were treated as described in (C), and the cells were lysed for immunoblotting using anti-GFP, anti-ALFA RFP, and anti-β-tubulin. Quantitative analysis of band intensity using ImageJ. Data are presented as the ratio of Alfa-tag protein (α2M–RG or α2M–RG + Baf A treated sample/Baf A treated sample) after normalization to tubulin as mean of two independent experiments.
Lysosomal degradation of the α2M–misfolded protein complex is a universal system present in various tissue-derived cells
We conducted α2M–RG internalization assays using seven cell lines derived from different origins: HEK293 (embryonic kidney), HeLa (cervical cancer), A549 (lung cancer), U2OS (osteosarcoma), Huh7 (liver carcinoma), HCT116 (colon cancer), and T98G (glioblastoma). Most cell lines showed some degree of RFP signal enhancement in the presence of heat-stressed proteins (Fig. 3), suggesting that the α2M complex-degrading mechanism is a ubiquitous system.

Lysosomal degradation of α2M with substrate is ubiquitous in different cell lines. α2M–RG internalization assay with PSMD5, ALDH1A1, or ESD. In the absence or presence of substrate, α2M–RG was heat-shocked in serum-free medium at 50 °C for 1 h. Each cell line (HEK293, HeLa, A549, U2OS, Huh7, HCT116, or T98G) was cultured in medium for 17 h at 37 °C, then analyzed using flow cytometry (n = 1).
The bait region promotes α2M internalization
α2M sequesters proteases through a conformational change triggered by cleavage of the bait region, an unstructured 39-amino acid sequence. To investigate the involvement of the bait region in α2M–misfolded protein internalization, we generated α2M Δb–RG, a mutant in which the bait region is entirely replaced with a glycine–serine linker sequence that is not susceptible to protease cleavage31. The purified α2M wild type (WT) and Δb were incubated with trypsin to assess cleavage of the bait region. We detected a 120-kDa fragment of α2M–RG, which is a protease-cleaved conformation (collapsed α2M), in the α2M WT–RG condition, whereas no 120-kDa fragment was produced in the α2M Δb–RG condition (Fig. 4A). Trypsin treatment at a higher concentration resulted in random degradation. Thus, the protease-sensitive bait region of α2M Δb–RG was completely abolished.
α2M Δb–RG internalization assays showed that the increase in RFP intensity after treatment with α2M Δb–RG and ESD was 0.65-fold lower than the increase after treatment with α2M WT–RG (Fig. 4B). Conversely, there was a marginal difference after treatment with ALDH1A1; no effect was observed after treatment with PSMD5. These results suggest that the bait region is not essential for α2M internalization, although it promotes cellular uptake for specific substrates.

The bait region of α2M promotes lysosomal degradation. (A) The bait region of α2M Δb–RG is not cleaved by trypsin. Recombinant α2M WT–RG or α2M Δb–RG was treated with trypsin at 4 °C for 30 min, then analyzed by immunoblotting using anti-GFP. (B) α2M Δb–RG and α2M WT–RG internalization assays with substrates. Mixtures of substrate (PSMD5, ALDH1A1, or ESD) and α2M–RG (WT or Δb) were heat-shocked in serum-free medium at 50 °C for 1 h. HeLa cells were cultured in medium for 17 h at 37 °C, then analyzed using flow cytometry. Bar graph shows the relative fluorescence intensities of GFP and RFP in cells normalized to those intensities in untreated cells (n = 3). Data are means ± SEMs. n.s., not significant; ***P < 0.005 (two-tailed Student’s t-test). Small circles indicate each data point. (C) α2M–RG internalization assay with LRP1 KO cells. Substrate (ALDH1A1 or ESD) and α2M–RG were heat-shocked in serum-free medium at 50 °C for 1 h. HeLa WT and LRP1 KO cells were cultured in medium for 17 h at 37 °C, then analyzed using flow cytometry. Bar graph shows the relative fluorescence intensities of GFP and RFP in cells normalized to those intensities in untreated cells (n = 3). Data are means ± SEMs. n.s., not significant; ***P < 0.005 (two-tailed Student’s t-test). Small circles indicate each data point. KO, knockout; WT, wild type.
Because LRP1 is a cell-surface receptor for the α2M–protease complex35,36, we explored whether LRP1 mediates internalization of the α2M–misfolded protein complex. LRP1 knockout (KO) cell lines were generated using Cas9 and two different gRNAs for LRP1 (gLRP1 #8 or gLRP1 #38); LRP1 expression was completely undetectable in both cell lines (Fig. S4). However, α2M–RG in the presence of heated ALDH1A1 and ESD was internalized even in LRP1 KO cells; this finding suggested that LRP1 is not involved in the α2M–misfolded protein degradation pathway (Fig. 4C).
α2M and clusterin have distinct specificities as extracellular chaperones
To compare substrate selectivity between α2M and clusterin, we used the internalization assay to evaluate the degradation efficiencies of recombinant clusterin–RFP–GFP (Clu-RG) and α2M–RG with five substrates. Three substrates (PSMD5, ALDH1A1, and ESD) promoted the lysosomal degradation of both chaperones in a similar manner (Fig. 5A). In contrast, after treatment with Clu-RG, RFP intensity was efficiently elevated twofold by the addition of CA1 or CA2, which did not promote the degradation of α2M–RG (Fig. 5A). These results suggest that α2M and clusterin have distinct substrate selectivities. Co-immunoprecipitation analysis also showed that binding to clusterin increased in a heat stress-dependent manner for all substrates (Fig. S5).

Degradation activity of α2M differed from degradation activity of clusterin. (A) α2M–RG and Clu-RG internalization assays. Substrates (CA1, CA2, PSMD5, ALDH1A1, and ESD) were each mixed with α2M–RG or Clu-RG; mixtures were heat-shocked in serum-free medium at 50 °C for 1 h. HeLa cells were cultured in medium for 17 h at 37 °C, then analyzed using flow cytometry. (B) α2M–RG and Clu-RG internalization assays with EXT1 KO cells. Substrates (ALDH1A1 and ESD) were each mixed with α2M–RG or Clu-RG; mixtures were heat-shocked in serum-free medium at 50 °C for 1 h. HeLa WT and EXT1 KO cells were cultured in medium for 17 h at 37 °C, then analyzed using flow cytometry. Bar graph shows the relative fluorescence intensities of GFP and RFP in cells normalized to those intensities in untreated cells (n = 3). Data are means ± SEMs. *P < 0.05; **P < 0.01; ***P < 0.005 (two-tailed Student’s t-test). Small circles indicate each data point.
Because our previous work suggests that the clusterin–misfolded protein complex binds to the heparan sulfate receptor10, we explored whether heparan sulfate is required for α2M. We performed an internalization assay of α2M–RG and Clu-RG using cells that lacked EXT1 (an essential synthase for heparan sulfate; i.e., EXT1 KO cells). As expected, increased uptake of Clu-RG was substantially inhibited in EXT1 KO cells, compared to control cells. In contrast, EXT1 depletion only marginally inhibited the uptake of α2M compared with clusterin (Fig. 5B), indicating that α2M uses a cell surface receptor different to that used by clusterin.
α2M had an affinity for aggregation-prone proteins
CA1 and CA2 underwent efficient clusterin-mediated lysosomal degradation, whereas they did not undergo α2M-mediated lysosomal degradation (Fig. 5A). To examine the properties of substrates targeted by α2M or clusterin, we performed a turbidity assay at 50 °C and categorized the heating results into three groups. In the first group, heating of CA2, PSMD5, and ALDH1A1 resulted in a large increase in turbidity. Heating of ESD produced the greatest protein precipitation among the five tested substrates (i.e., increased absorbance at 360 nm; Fig. 6A), suggesting that aggregation-prone proteins had strong affinity for both chaperones. In contrast, heating of CA1 led to a small increase in turbidity. In the second group, heating of CA2 led to a rapid increase in turbidity within 60 min, whereas other substrates exhibited progressive increases in turbidity over 240 min. In the third group, heating of CA1, CA2, and PSMD5 revealed a modest formation of aggregates at a low concentration (5 μM). Overall, these results indicate that α2M preferentially targets aggregate-prone misfolded proteins that are produced by gradual denaturing, whereas clusterin targets a broad range of denatured proteins.

CA1 and CA2 exhibit low and rapid aggregation, respectively. Substrates (CA1, CA2, PSMD5, ALDH1A1, or ESD) were incubated in phosphate-buffered saline (PBS) at 50 °C for 4 h (n = 3). Turbidity (A360) was measured at 3-min intervals at 5 μM (green), 10 μM (yellow), and 20 μM (orange). Relative turbidity is calculated as the ratio of turbidity at each time point to the initial turbidity. Data are means ± SEMs.
Discussion
Extracellular chaperones have been evaluated for their importance in extracellular proteostasis. The chaperone activity of extracellular chaperones against stressed proteins has been characterized relatively well16,17. In contrast, less is known about the degradation pathways of extracellular chaperones except that of clusterin10,18. In this study, we found that α2M mediates lysosomal degradation of extracellular misfolded proteins, thus maintaining extracellular proteostasis. Our data indicate that although the effects of α2M overlap with the effects of clusterin, α2M recognizes misfolded proteins depending on their level of aggregation. An α2M internalization assay showed that α2M also mediates the cellular internalization of extracellular misfolded proteins. α2M is a protease inhibitor and might inhibits lysosomal proteases after transport into lysosomes. However, lysosomes contain approximately 10 proteases, including serine-, cysteine-, and aspartate-type acidic proteases37. α2M would be eventually degraded by various lysosomal proteases. Thus, a key function of α2M is the removal of misfolded proteins generated by exposure to stress in the harsh extracellular environment.
Thus far, no quantitative method has been available to detect the lysosomal degradation of extracellular proteins; the clearance of stressed protein by α2M has been poorly understood. Although a conventional method for the detection of internalization involves fluorescence labeling of extracellular proteins, this method cannot distinguish between cell surface interaction and lysosomal internalization. To overcome this problem, we designed the recombinant protein α2M–RG, which is composed of two fluorescent proteins, RFP and GFP. Using α2M–RG, we quantitatively detected an increase in RFP, but not GFP, during the cellular internalization of α2M–RG. Biochemical and cell biological experiments revealed that α2M is involved in the lysosomal degradation of heat-stressed proteins (Fig. 1). Since the RFP-GFP tag is a large protein, it may affect the functions of tagged α2M. However, RFP-GFP does not facilitate the degradation of misfolded proteins (Fig. S2); therefore, facilitation of the degradation of misfolded proteins of α2M–RG is due to α2M.
Mass spectrometry revealed that PSMD5, ALDH1A1, and ESD were α2M-interacting proteins in RBC lysate; these proteins formed a complex with α2M under heat stress (Fig. 2E) and were internalized (Fig. 2F). Our findings suggest that these substrates are internalized into the cell together with α2M, where they undergo lysosomal degradation. Intravascular hemolysis causes autoimmune diseases38, infection39, and mechanical stress40 (e.g., running41), which might be associated with accumulation of normally intracellular proteins. Because more than half of all cells in the human body are erythrocytes, sufficient hemolysis would expose numerous intracellular proteins to the extracellular environment. Therefore, RBC-derived intracellular proteins in the blood may produce misfolded proteins in the harsh extracellular environment and might contribute to disease. In addition, α2M is abundant in blood23, and α2M internalization was confirmed in most of the cultured cells tested in this study (Fig. 3). Based on the present findings, we propose that α2M protects extracellular proteostasis from the effects of hemolysis.α2M functions as a protease inhibitor that traps and inactivates proteases via conformational changes caused by protease-induced cleavage of the bait region21. To investigate the relationship between the bait region and lysosomal internalization, we used the bait region mutant α2M Δb–RG31, in which the bait region was completely replaced with a glycine–serine linker sequence. α2M Δb–RG reduced internalization compared to WT (Fig. 4B). Although the bait region was not essential for α2M degradation in the presence of misfolded protein, our data suggest that the bait region facilitates degradation. Since the culture medium used during the internalization assay did not contain a protease (we used advanced DMEM/F12 without serum), the bait region might contribute to another role without cleavage. Although a cell surface receptor might be involved in the recognition of α2M–misfolded protein complexes via the bait region, LRP1 was not essential for lysosomal degradation of the α2M–misfolded protein complexes under our conditions (Fig. 4C). LRP1 may have another role, such as mediating α2M recycling via the recycling endosome42. For lysosomal degradation of the α2M–misfolded protein complex, other α2M receptors, such as Grp78, might be involved43.
To investigate the physiological significance of the presence of multiple extracellular chaperones, we compared the substrate specificities of α2M and clusterin. Internalization assays showed that misfolded CA1 increased the degradation of clusterin, but did not affect the degradation of α2M, suggesting that α2M has comparatively greater substrate selectivity. The turbidity assay revealed that the aggregation growth phase of substrates for α2M and clusterin, including PSMD5, ALDH1A1, and ESD, slowly increased the turbidity (Fig. 6). In contrast, CA1 and CA2, both substrates for clusterin (Fig. 5), quickly reached a plateau. Since α2M did not induce the internalization of aggregated proteins (i.e., preheated proteins) (Fig. 2D), these results suggest that clusterin has higher binding kinetics to misfolding proteins than does α2M. Thus, the binding kinetics and other currently unknown differences in chaperone action may contribute to the different misfolded client protein specificities seen for clusterin and α2M. From this perspective, α2M-mediated lysosomal degradation and clusterin-mediated lysosomal degradation are not totally redundant pathways. Therefore, we propose that the cooperation of multiple types of extracellular chaperones is effective for protecting extracellular proteostasis from diverse misfolded proteins in harsh extracellular conditions (Fig. 7).

Model of the extracellular misfolded protein degradation pathway by extracellular chaperones. Clusterin targets a broad range of misfolded proteins than α2M. The binding kinetics of the extracellular chaperone might be one determinant of the substrate recognition of an extracellular chaperone. The α2M–misfolded protein complex is internalized via a putative receptor, leading to degradation of the α2M–misfolded protein complex.
This study provides evidence that different receptors mediate lysosomal degradation of clusterin or α2M-misfolded protein complexes. Because clusterin and α2M are both associated with protein deposition diseases such as Alzheimer's disease, prion disease, and atherosclerosis16,44, both chaperones have similar physiological roles. On the other hand, clusterin is conserved only among vertebrates45, whereas α2M is evolutionarily conserved from bacteria to vertebrates46, suggesting that α2M is a universal factor under diverse conditions. Our internalization assay of α2M–RG suggested cell specificity of the lysosomal degradation of α2M–misfolded protein complexes (Fig. 3). Identifying the tissue specificity of the degradation pathway will provide important information about the quality control system of extracellular proteostasis in vivo.
Materials and methods
Plasmids
We amplified α2M cDNA from HepG2 total cDNA via polymerase chain reaction and inserted it into a pcDNA5 FRT TO vector along with mCherry, sfGFP, His-tag, and signal sequence (ss) of prolactin (only RG–α2M) to generate the fusion proteins α2M–mCherry–sfGFP–His (α2M–RG) and ss–His–mCherry–sfGFP–α2M (RG–α2M). For the mutated bait region, the bait region (117 bp) of pcDNA5 FRT TO a2M–RG–His was replaced by a GGS repeat sequence via mutagenesis. For bacterial expression, CA1 from mouse lung total cDNA, CA2 from HEK293 total cDNA, ESD from HEK293 total cDNA, PSMD5 from HeLa total cDNA, ALDH1A1 from HEK293 total cDNA, and GCLM from HEK293 total cDNA were amplified and inserted into a pRSET-A vector along with ALFA tag and His-tag. The resultant plasmids were as follows: pcDNA5 FRT TO α2M–mCherry–sfGFP–His, pcDNA5 FRT TO ss–His–mCherry–sfGFP–α2M, pcDNA5 FRT TO α2M–ALFA–His, pcDNA5 FRT TO α2M(Δbait)–mCherry–sfGFP–His, pRSET-A CA1–ALFA–His, pRSET-A CA2–ALFA–His, pRSET-A His–ALFA–3C–PSMD5, pRSET-A His–ALFA–3C–ALDH1A1, pRSET-A His–ALFA–3C–GCLM, and pRSET-A His–ALFA–3C–ESD. pOG44 was used in the Flp-in system. pCMV-VSVG (Addgene plasmid #8454) and psPAX2 (Addgene plasmid #12260) were used for lentivirus production. The pcDNA5 FRT TO FLAG–Cas9 vector was previously described (Itakura et al., 2016). pLenti gEXT1a was previously described (Itakura et al. 2019).
Antibodies
Rabbit polyclonal anti-LAMP1 antibodies were gifted from Y. Tanaka (Kyushu University, Fukuoka, Japan). Mouse monoclonal anti-GFP (clone no. mFX75, cat no. 012-22541) antibody was purchased from Wako. Mouse monoclonal anti-RFP (clone no. 1G9, cat no. M204-3) antibody was purchased from MBL. Rabbit monoclonal anti-LRP1 (clone no. EPR3724, cat. no. ab92544) antibody was purchased from Abcam. Polyclonal anti-ALFA tag antibody was raised in rabbits by immunization with the ALFA peptide; this antibody generation was performed by Eurofins. GFP-nanobody and ALFA tag-nanobody sepharose were generated by conjugating GFP-nanobody protein purified from pOPINE GFP nanobody (Addgene plasmid #49172) and ALFA tag-nanobody protein34 purified from pRSET-A ALFA–His to N-hydroxy succinimide-activated Sepharose 4 Fast Flow (GE).
Cell culture
Flp-in T-Rex HEK293 (Thermo Fisher Scientific), HeLa (RIKEN BRC), Huh7 (RIKEN BRC), A549 (RIKEN BRC), HCT116 (RIKEN BRC), HEK293FT (Thermo Fisher Scientific), and U2OS cells were cultured in DMEM (Nacalai Tesque); T98G cells were cultured in Eagle’s Minimum Essential Medium (Nacalai Tesque). Each medium was supplemented with 10% fetal bovine serum (Biosera) and 50 µg/mL penicillin/streptomycin (regular medium) in a humidified 5% CO2 incubator at 37 °C. Flp-in T-Rex HEK293 cells were maintained in the presence of 100 μg/mL zeocin and 15 μg/mL blasticidin. To generate stable doxycycline (dox)-inducible secreted proteins or FLAG–Cas9-expressing cells, plasmids encoding the respective proteins were co-transfected with pOG44, encoding the FLP recombinase, into Flp-in T-Rex HEK293 cells. Transfected cells were selected by adding 100 μg/mL hygromycin, then maintained in the presence of 15 μg/mL blasticidin and 100 μg/mL hygromycin. Dox (100 ng/mL) was used to induce the integrated gene at the FRT site. Lentivirus-infected HeLa cells harboring Lenti Cas9 Blast were used to generate stable FLAG–Cas9-expressing cells.
Generation of KO cells using clustered regularly interspaced short palindromic repeats (CRISPR)
SgRNA sequences for KO (gLRP1-a: GCCAAACGAGCATAACTGCC, gLRP1-b: CATTGTGTCCCCA-CACTCGA) were designed using CHOPCHOP and cloned into pLentiGuide-puro (Addgene plasmid #52963). HEK293 cells and HeLa cells stably expressing FLAG–Cas9 were infected with lentivirus harboring pLentiGuide-puro gEXT1 (HEK293), pLentiGuide-puro gLRP1-a, and pLentiGuide-puro gLRP1-b (HeLa), respectively. As a control, HEK293 cells and HeLa cells were infected with lentivirus harboring pLentiGuide-puro gControl (CGCAGTCATTCGATAGGAAT). After 24 h of transduction, cells were cultured with 1 μg/mL puromycin (HEK293 and HeLa) and 100 ng/mL dox (only HEK293) for 7 days; they were then used as KO cell lines.
Preparation of cell lysate and immunoblotting
Cells were washed with cold phosphate-buffered saline (PBS) and lysed in lysis buffer (1% Triton X-100, 50 mM Tris/HCl, pH 7.5, 1 mM ethylenediaminetetraacetic acid [EDTA], and 150 mM NaCl) supplemented with protease inhibitor cocktail (EDTA-free; Nacalai Tesque) and 1 mM phenylmethanesulfonyl fluoride for 15 min at 4 °C. The lysates were clarified by centrifugation at 20,630 × g for 5 min at 4 °C, then mixed with sodium dodecyl sulfate (SDS) sample buffer. Samples were boiled at 95 °C for 5 min prior to SDS–polyacrylamide gel electrophoresis (SDS-PAGE). We separated 10 μg of protein per lane via SDS–PAGE; proteins were then transferred to polyvinylidene difluoride membranes (Millipore). Immunoblotting analysis was performed using the indicated antibodies and immunoreactive proteins were visualized using the ImmunoStar Zeta reagent (Wako).
Flow cytometry
Cells were detached from dishes with trypsin and EDTA for collection, then passed through a 70-μm cell strainer and resuspended in 5% newborn calf serum plus 1 μg/mL 4′,6-diamidino-2-phenylindole (DAPI) in PBS. Flow cytometry was performed using a CytoFLEX S flow cytometer equipped with NUV 375-nm (DAPI), 488-nm (GFP), and 561-nm (mCherry) lasers (Beckman Coulter). Dead cells were detected by DAPI staining. In each sample, more than 10,000 cells were acquired.
Immunocytochemistry and fluorescence microscopy
Cells were plated on coverslips and fixed in 3.7% formaldehyde in PBS for 15 min. For immunostaining, fixed cells were permeabilized with 50 μg/mL digitonin in PBS for 5 min, blocked with 10% newborn calf serum in PBS for 30 min, and incubated with primary antibodies for 1 h. After a washing step, the cells were incubated with Alexa Fluor 647-conjugated goat anti-rabbit IgG secondary antibodies (Thermo Fisher Scientific) for 1 h. The stained cells were observed under a confocal laser microscope (FV1000 IX81; Olympus) using a 100 × oil immersion objective lens with a numerical aperture of 1.40.
Co-immunoprecipitation
Recombinant extracellular chaperones (Fig. 2A: 0.14 μM, Fig. 2E, S4: 0.1 μM) were mixed with protein substrates (e.g., BCL or CA1) that had been incubated at 50 °C for 1 h. For BCL, tissue lysate was centrifuged at 2290×g for 2 min to remove debris after heat shock. GFP-nanobody or ALFA tag-nanobody sepharoses were added to the mixture and incubated for 2 h at 4 °C. The sepharoses were washed four times with PBS and then transferred to fresh tubes; subsequently, they were subjected to elution with SDS sample buffer.
Preparation of blood cell lysate
Blood samples were collected from C57BL/6 mice and centrifuged at 2290×g for 2 min to separate plasma and blood cells. The cells were washed with PBS, resuspended in an equal volume of homogenization buffer (20 mM HEPES, pH 7.4, 1 mM EDTA, 1 mM phenylmethanesulfonyl fluoride, and protease inhibitor cocktail), and homogenized using a 1-mL syringe with a 27-G needle. The homogenized cells were centrifuged at 20,620×g for 10 min to remove cell debris. The supernatant was used as blood cell lysate (BCL). The concentration of BCL total proteins was determined by the Bradford method; it was typically 136 μg/μL. For mass spectrometry, BCL was cleared by ultra-centrifugation at 572,000×g (Hitachi S110AT) for 30 min, followed by immunoprecipitation.
Protein purification
Purification of secreted protein from conditioned medium was performed as previously described33. Briefly, cells expressing α2M–RG, RG–α2M, α2M–ALFA, α2M(Δbait)–RG, or Clu-RG were cultured with doxycycline in advanced DMEM medium for 4 days. The conditioned medium was collected and centrifuged at 780×g for 20 min at 4 °C to remove dead cells and debris. Secreted proteins were purified from the conditioned medium via Ni–NTA affinity chromatography. Purified proteins were stored in PBS with 10% glycerol at − 80 °C.
Human CA1, CA2, PSMD5, ALDH1A1, GCLM, and ESD were tagged with ALFA tag and 6xHis-tag, then cloned into pRSET-A. The plasmids were introduced into the BL21(DE3) LOBSTR strain of Escherichia coli47, then induced with 0.1 mM isopropyl β-D-1-thiogalactopyranoside at 18 °C. The cells were disrupted by sonication. After ultracentrifugation, recombinant protein was purified from the soluble fraction using HisPur Cobalt Resin (Thermo Fisher Scientific). Purity was assessed by SDS-PAGE with coomassie brilliant blue staining (Fig. S2).
Internalization assay
Conditioned advanced DMEM containing secreted α2M–RG (in which the extracellular chaperone concentration was diluted to 70 nM with advanced DMEM) was mixed with protein substrates (1.09 μg/μL BCL) (2 μM PSMD5, 0.5 μM ALDH1A1, 1 μM ESD in Figs. 2D, 3, 4B, C, 5B) (2 μM CA1, 2 μM Ca2, 2 μM PSMD5, 2 μM ALDH1A, 2 μM GCLM, 2 μM ESD in Figs. 2C and 5A) then heat-treated at 50 °C or incubated at 4 °C for 1 h. The medium was added to cells in 24-well plates and cultured at 37 °C for 20 h. Cells were then collected and analyzed via flow cytometry or immunoblotting.
SYPRO Ruby staining
After gel electrophoresis, gels were fixed with 7% acetic acid and 50% methanol solution for 30 min. Fixed gels were incubated with SYPRO Ruby solution (Thermo Fisher Scientific) overnight at room temperature with continuous gentle agitation. The gels were destained during 30 min in 7% acetic acid and 10% methanol solution and then rinsed in deionized water. The gels were visualized using a iBright FL1500 Imaging System (Thermo Fisher Scientific).
Native-PAGE
Proteins were diluted in sample buffer (100 mM Tris pH8.6, 10% glycerol, 0.0025% bromophenol blue). Native-PAGE was performed on NuPAGE Novex 3 to 8% Tris–acetate gels (Thermo Fisher Scientific) and Tris–glycine running buffer (25 mM Tris, 192 mM glycine, pH8.3) (Fujifilm), with a constant voltage 125 V. Gels were stained using coomassie brilliant blue or SYPRO Ruby staining.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Wang, W., Nema, S. & Teagarden, D. Protein aggregation: Pathways and influencing factors. Int. J. Pharm. 390, 89–99. https://doi.org/10.1016/j.ijpharm.2010.02.025 (2010).
Aguzzi, A. & O’Connor, T. Protein aggregation diseases: Pathogenicity and therapeutic perspectives. Nat. Rev. Drug Discov. 9, 237–248. https://doi.org/10.1038/nrd3050 (2010).
Klaips, C. L., Jayaraj, G. G. & Hartl, F. U. Pathways of cellular proteostasis in aging and disease. J. Cell Biol. 217, 51–63. https://doi.org/10.1083/jcb.201709072 (2018).
Langer, T. et al. Successive action of DnaK, DnaJ and GroEL along the pathway of chaperone-mediated protein folding. Nature 356, 683–689. https://doi.org/10.1038/356683a0 (1992).
Frydman, J., Nimmesgern, E., Ohtsuka, K. & Hartl, F. U. Folding of nascent polypeptide chains in a high molecular mass assembly with molecular chaperones. Nature 370, 111–117. https://doi.org/10.1038/370111a0 (1994).
Schwartz, A. L. & Ciechanover, A. The ubiquitin-proteasome pathway and pathogenesis of human diseases. Annu. Rev. Med. 50, 57–74. https://doi.org/10.1146/annurev.med.50.1.57 (1999).
Lamark, T. & Johansen, T. Aggrephagy: Selective disposal of protein aggregates by macroautophagy. Int. J. Cell Biol. 2012, 736905. https://doi.org/10.1155/2012/736905 (2012).
Poon, S., Easterbrook-Smith, S. B., Rybchyn, M. S., Carver, J. A. & Wilson, M. R. Clusterin is an ATP-independent chaperone with very broad substrate specificity that stabilizes stressed proteins in a folding-competent state. Biochemistry 39, 15953–15960 (2000).
Mesgarzadeh, J. S., Buxbaum, J. N. & Wiseman, R. L. Stress-responsive regulation of extracellular proteostasis. J. Cell Biol. https://doi.org/10.1083/jcb.202112104 (2022).
Itakura, E., Chiba, M., Murata, T. & Matsuura, A. Heparan sulfate is a clearance receptor for aberrant extracellular proteins. J. Cell Biol. https://doi.org/10.1083/jcb.201911126 (2020).
Powers, J. M., Schlaepfer, W. W., Willingham, M. C. & Hall, B. J. An immunoperoxidase study of senile cerebral amyloidosis with pathogenetic considerations. J. Neuropathol. Exp. Neurol. 40, 592–612. https://doi.org/10.1097/00005072-198111000-00002 (1981).
Tomino, Y. et al. Immunofluorescent studies on acute phase reactants in patients with various types of chronic glomerulonephritis. Tokai J. Exp. Clin. Med. 6, 435–441 (1981).
Yerbury, J. J. et al. The extracellular chaperone clusterin influences amyloid formation and toxicity by interacting with prefibrillar structures. FASEB J. 21, 2312–2322. https://doi.org/10.1096/fj.06-7986com (2007).
Niewold, T. A., Murphy, C. L., Hulskamp-Koch, C. A., Tooten, P. C. & Gruys, E. Casein related amyloid, characterization of a new and unique amyloid protein isolated from bovine corpora amylacea. Amyloid 6, 244–249. https://doi.org/10.3109/13506129909007335 (1999).
Rosenberg, M. E. & Silkensen, J. Clusterin: Physiologic and pathophysiologic considerations. Int. J. Biochem. Cell Biol. 27, 633–645. https://doi.org/10.1016/1357-2725(95)00027-m (1995).
Wyatt, A. R., Yerbury, J. J., Ecroyd, H. & Wilson, M. R. Extracellular chaperones and proteostasis. Annu. Rev. Biochem. 82, 295–322. https://doi.org/10.1146/annurev-biochem-072711-163904 (2013).
Yerbury, J. J., Stewart, E. M., Wyatt, A. R. & Wilson, M. R. Quality control of protein folding in extracellular space. EMBO Rep. 6, 1131–1136. https://doi.org/10.1038/sj.embor.7400586 (2005).
Wyatt, A. R. et al. Clusterin facilitates in vivo clearance of extracellular misfolded proteins. Cell Mol. Life Sci. 68, 3919–3931. https://doi.org/10.1007/s00018-011-0684-8 (2011).
Hung, S. Y. & Fu, W. M. Drug candidates in clinical trials for Alzheimer’s disease. J. Biomed. Sci. 24, 47. https://doi.org/10.1186/s12929-017-0355-7 (2017).
French, K., Yerbury, J. J. & Wilson, M. R. Protease activation of alpha2-macroglobulin modulates a chaperone-like action with broad specificity. Biochemistry 47, 1176–1185. https://doi.org/10.1021/bi701976f (2008).
Barrett, A. J. & Starkey, P. M. The interaction of alpha 2-macroglobulin with proteinases. Characteristics and specificity of the reaction, and a hypothesis concerning its molecular mechanism. Biochem. J. 133, 709–724. https://doi.org/10.1042/bj1330709 (1973).
Bauer, J. et al. Interleukin-6 and alpha-2-macroglobulin indicate an acute-phase state in Alzheimer’s disease cortices. FEBS Lett. 285, 111–114. https://doi.org/10.1016/0014-5793(91)80737-n (1991).
Tirumalai, R. S. et al. Characterization of the low molecular weight human serum proteome. Mol. Cell Proteomics 2, 1096–1103. https://doi.org/10.1074/mcp.M300031-MCP200 (2003).
Nedić, O., Šunderić, M., Gligorijević, N., Malenković, V. & Miljuš, G. Analysis of four circulating complexes of insulin-like growth factor binding proteins in human blood during aging. Biochemistry (Mosc) 82, 1200–1206. https://doi.org/10.1134/S0006297917100133 (2017).
Biringer, R. G. et al. Enhanced sequence coverage of proteins in human cerebrospinal fluid using multiple enzymatic digestion and linear ion trap LC-MS/MS. Brief Funct. Genom. Proteomic 5, 144–153. https://doi.org/10.1093/bfgp/ell026 (2006).
Gehring, M. R. et al. Sequence of rat liver alpha 2-macroglobulin and acute phase control of its messenger RNA. J. Biol. Chem. 262, 446–454 (1987).
Van Leuven, F., Marynen, P., Sottrup-Jensen, L., Cassiman, J. J. & Van den Berghe, H. The receptor-binding domain of human alpha 2-macroglobulin Isolation after limited proteolysis with a bacterial proteinase. J. Biol. Chem. 261, 11369–11373 (1986).
Odom, A. R., Misra, U. K. & Pizzo, S. V. Nickel inhibits binding of alpha2-macroglobulin-methylamine to the low-density lipoprotein receptor-related protein/alpha2-macroglobulin receptor but not the alpha2-macroglobulin signaling receptor. Biochemistry 36, 12395–12399. https://doi.org/10.1021/bi970806k (1997).
Sottrup-Jensen, L., Sand, O., Kristensen, L. & Fey, G. H. The alpha-macroglobulin bait region. Sequence diversity and localization of cleavage sites for proteinases in five mammalian alpha-macroglobulins. J. Biol. Chem. 264, 15781–15789 (1989).
Katayama, H., Yamamoto, A., Mizushima, N., Yoshimori, T. & Miyawaki, A. GFP-like proteins stably accumulate in lysosomes. Cell Struct. Funct. 33, 1–12 (2008).
Harwood, S. L. et al. Development of selective protease inhibitors via engineering of the bait region of human α(2)-macroglobulin. J. Biol. Chem. 297, 100879. https://doi.org/10.1016/j.jbc.2021.100879 (2021).
Luque, D. et al. Cryo-EM structures show the mechanistic basis of pan-peptidase inhibition by human α. Proc. Natl. Acad. Sci. USA 119, e2200102119. https://doi.org/10.1073/pnas.2200102119 (2022).
Tomihari, A., Chiba, M., Matsuura, A. & Itakura, E. Protocol for quantification of the lysosomal degradation of extracellular proteins into mammalian cells. STAR Protoc. 2, 100975. https://doi.org/10.1016/j.xpro.2021.100975 (2021).
Götzke, H. et al. The ALFA-tag is a highly versatile tool for nanobody-based bioscience applications. Nat. Commun. 10, 4403. https://doi.org/10.1038/s41467-019-12301-7 (2019).
Strickland, D. K. et al. Sequence identity between the alpha 2-macroglobulin receptor and low density lipoprotein receptor-related protein suggests that this molecule is a multifunctional receptor. J. Biol. Chem. 265, 17401–17404 (1990).
Kristensen, T. et al. Evidence that the newly cloned low-density-lipoprotein receptor related protein (LRP) is the alpha 2-macroglobulin receptor. FEBS Lett. 276, 151–155. https://doi.org/10.1016/0014-5793(90)80530-v (1990).
Lübke, T., Lobel, P. & Sleat, D. E. Proteomics of the lysosome. Biochim. Biophys. Acta 1793, 625–635. https://doi.org/10.1016/j.bbamcr.2008.09.018 (2009).
Gehrs, B. C. & Friedberg, R. C. Autoimmune hemolytic anemia. Am. J. Hematol. 69, 258–271. https://doi.org/10.1002/ajh.10062 (2002).
Orf, K. & Cunnington, A. J. Infection-related hemolysis and susceptibility to Gram-negative bacterial co-infection. Front. Microbiol. 6, 666. https://doi.org/10.3389/fmicb.2015.00666 (2015).
Lee, I. H. et al. Renal hemosiderosis secondary to intravascular hemolysis after mitral valve repair: A case report. Medicine (Baltimore) 99, e18798. https://doi.org/10.1097/MD.0000000000018798 (2020).
Weight, L. M., Byrne, M. J. & Jacobs, P. Haemolytic effects of exercise. Clin. Sci. (Lond.) 81, 147–152. https://doi.org/10.1042/cs0810147 (1991).
Laatsch, A. et al. Low density lipoprotein receptor-related protein 1 dependent endosomal trapping and recycling of apolipoprotein E. PLoS ONE 7, e29385. https://doi.org/10.1371/journal.pone.0029385 (2012).
Misra, U. K. et al. The role of Grp 78 in alpha 2-macroglobulin-induced signal transduction. Evidence from RNA interference that the low density lipoprotein receptor-related protein is associated with, but not necessary for, GRP 78-mediated signal transduction. J. Biol. Chem. 277, 42082–42087. https://doi.org/10.1074/jbc.M206174200 (2002).
Thal, D. R., Schober, R. & Birkenmeier, G. The subunits of alpha2-macroglobulin receptor/low density lipoprotein receptor-related protein, native and transformed alpha2-macroglobulin and interleukin 6 in Alzheimer’s disease. Brain Res. 777, 223–227. https://doi.org/10.1016/s0006-8993(97)01021-4 (1997).
Jiao, S. et al. The conserved clusterin gene is expressed in the developing choroid plexus under the regulation of notch but not IGF signaling in zebrafish. Endocrinology 152, 1860–1871. https://doi.org/10.1210/en.2010-1183 (2011).
Wong, S. G. & Dessen, A. Structure of a bacterial α2-macroglobulin reveals mimicry of eukaryotic innate immunity. Nat. Commun. 5, 4917. https://doi.org/10.1038/ncomms5917 (2014).
Andersen, K. R., Leksa, N. C., Schwartz, T. U. & Optimized, E. coli expression strain LOBSTR eliminates common contaminants from His-tag purification. Proteins 81, 1857–1861. https://doi.org/10.1002/prot.24364 (2013).
Acknowledgements
We thank Prof. Yoshitaka Tanaka (University of Kyusyu) for anti-LAMP1 antibodies. This work was supported by Japan Society for the Promotion of Science (JSPS) KAKENHI Grants (Nos. 20H03249, 20H05312, and 22H04634 to E.I.), a Japan Science and Technology Agency (JST) Fusion Oriented REsearch for disruptive Science and Technology (FOREST) Grant (No. JPMJFR204N to E.I.), and the Takeda Science Foundation (to E.I.).
Author information
Authors and Affiliations
Contributions
A.T. and M.K. performed the experiments. A.T. and E.I. proposed the experiments, interpreted the data, and wrote the manuscript. A.M. contributed to manuscript writing and data interpretation. All authors discussed the results and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tomihari, A., Kiyota, M., Matsuura, A. et al. Alpha 2-macroglobulin acts as a clearance factor in the lysosomal degradation of extracellular misfolded proteins. Sci Rep 13, 4680 (2023). https://doi.org/10.1038/s41598-023-31104-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-31104-x
This article is cited by
-
Human platelet lysate stimulates neurotrophic properties of human adipose-derived stem cells better than Schwann cell-like cells
Stem Cell Research & Therapy (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
