
Abstract
In this study, we aimed to comprehensively characterize the microbiomes of various samples from pregnant women and their neonates, and to explore the similarities and associations between mother-neonate pairs, sample collection sites, and obstetrical factors. We collected samples from vaginal discharge and amniotic fluid in pregnant women and umbilical cord blood, gastric liquid, and meconium from neonates. We identified 19,597,239 bacterial sequences from 641 samples of 141 pregnant women and 178 neonates. By applying rigorous filtering criteria to remove contaminants, we found evidence of microbial colonization in traditionally considered sterile intrauterine environments and the fetal gastrointestinal track. The microbiome distribution was strongly grouped by sample collection site, rather than the mother-neonate pairs. The distinct bacterial composition in meconium, the first stool passed by newborns, supports that microbial colonization occurs during normal pregnancy. The microbiome in neonatal gastric liquid was similar, but not identical, to that in maternal amnionic fluid, as expected since fetuses swallow amnionic fluid in utero and their urine returns to the fluid under normal physiological conditions. Establishing a microbiome library from various samples formed only during pregnancy is crucial for understanding human development and identifying microbiome modifications in obstetrical complications.
Similar content being viewed by others

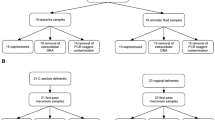

Introduction
The human microbiome potentially carries the answer to many secrets of the human body. It has been linked to maintaining homeostasis in health and is associated with numerous diseases1,2. Recent research has shifted to explore the microbiome in less-studied populations, such as infants or pregnant women, to better understand its role in human development. Microbiome development is likely to start from the in-utero environment and changes in a lifetime, continuously affecting the immune system and metabolism. Pregnancy has been shown to alter microbial populations within the maternal body and may impact future maternal, fetal, and neonatal health3. Pregnancy allows temporary immunotolerance to a foreign body, facilitating microbiome remodelling and potential adaptations to the immune system and metabolism4. Some microbiome studies in pregnancy have proposed that fetal environments, including placenta and amniotic fluid, traditionally known as sterile, contain several characteristic microbiotas not identified in routinely performed culture techniques5,6. However, the biomass of these microbiotas is small and the reliability of the sequencing methods and potential for contamination have been criticized. The association between those microbiota and specific obstetric conditions has not yet been proven and warrants further investigation.
The vagina is the most commonly studied site of bacteria in the female reproductive organ, as it is connected to the uterus through the cervix and is exposed to the skin. Microbiome research in pregnancy, however, has advanced slowly due to ethical concerns and difficulties in accessing samples. Aagaard et al. found that the vaginal microbiome changes during pregnancy based on gestational age and that Lactobacillus species play a role in preventing pathogenic bacterial growth7. More specifically, pregnancy leads to decreased diversity, increased proportion of Lactobacillus, and higher stability in the vaginal microbiome8,9. Some vaginal bacteria have been linked to preterm birth via intrauterine inflammation or infection10,11,12,13,14, yet there are no clinical guidelines for testing or monitoring these microbiota. Other sites that had been evaluated for microbiome in pregnancy are maternal15, oral cavity16, placenta5, amniotic fluid17,18, and neonatal gut19; but previous studies were fragmentary and more systematic research is needed.
In this study, we have comprehensively characterized the microbiome in vaginal discharge (VD) and amniotic fluid (AF) from pregnant women and in umbilical cord blood (CB), gastric liquid (GL), and meconium (M) from their neonates. The goal was to determine the relationships between these samples and various obstetric conditions.
Results
Description of the study populations and clinical characteristics
A total of 141 low-risk pregnant women were enrolled sequentially and 178 neonates were born from the study population with 37 cases being twin pregnancies. All women were of Asian ethnicity (Korean), and the median age was 34 (interquartile range 31–37) years (Table 1). The proportion of nulliparity was slightly over half of the population (67%), and the median values of height, weight, and body mass index (BMI) were 162 cm, 70 kg, and 27 kg/m2, respectively. About 30% were conceived by assisted reproductive technology (ART), including intrauterine insemination (IUI) and in vitro fertilization with embryo transfer (IVF-ET). As mentioned above, twin pregnancy was approximately one-fourth of the total population, and among them, 19% were monochorionic. The median gestational age at delivery was 37.7 weeks (interquartile range 36.9–38.6), and preterm birth before 37 weeks of gestation was 26.2% (37/141). The rate of cesarean section was 55% (77/141). Seven neonates had congenital structural anomalies (atrioventricular septal defect, absence of corpus callosum in the brain, achondroplasia, cleft lip, polydactyly, and syndactyly), which did not directly affect the neonatal survival. The frequencies of other obstetric complications or underlying maternal diseases are described in Table 1.
Maternal and neonatal microbiome landscape during delivery
We identified 19,597,239 bacterial sequences and 22,412 unique amplicon sequence variants (ASVs) from 641 samples, including cervicovaginal discharge (n = 154), amniotic fluid (n = 40), gastric liquid (n = 100), umbilical cord blood (n = 125), meconium (n = 160), and negative controls (n = 62). The ASVs were taxonomically annotated, but we found evidence of batch effects in our sequencing data for all sample types except VD (Fig. 1 and Supplementary Fig. S1). The batch effects were likely introduced during library construction for next-generation sequencing (NGS) and not during sequencing itself (Supplementary Fig. S2). However, this was expected because our samples were collected from body sites with low-biomass specimens, making our samples prone to contamination20. Therefore, we expected to find many false positives and applied a series of filters, as outlined in Supplementary Fig. S3. Notably, we found and removed 203 ASVs that were statistically determined as contaminants because they were highly prevalent in negative controls (Supplementary Fig. S4) or they showed higher frequencies in low-concentration samples (Supplementary Figs. S5 and S6).

Batch effect detection in 16S rRNA amplicon sequencing data. Center log-ratio transformation was used to normalize the filtered ASV table before generating a hierarchically clustered heatmap based on correlation coefficients. AF, amniotic fluid; CB, umbilical cord blood; GL, gastric liquid; M, meconium; VD, cervicovaginal discharge; NC, negative control.
We measured the alpha diversity of the samples by calculating Shannon indices (Fig. 2A). The alpha diversity decreased in the following order: GL, AF, M, CB, and VD. The negative control group showed a slightly higher diversity compared to the VD sample, suggesting that negative controls for 16S amplicon sequencing can have microbiome diversity as rich as real biological specimens. Next, we estimated the beta diversity of our samples by computing the weighted UniFrac distances (Fig. 2B). The samples were moderately well separated by sample collection site when projected using principal coordinates analysis (PCoA).

Alpha and beta diversity of the Korean maternal and neonatal microbiome. (A) Alpha diversity: The filtered ASV table was rarefied before Shannon index was computed for each sample. The VD group exhibited the least amount of alpha diversity. AF, amniotic fluid; CB, umbilical cord blood; GL, gastric liquid; M, meconium; VD, cervicovaginal discharge; NC, negative control; (B) Beta diversity: The filtered ASV table was rarefied before the samples were projected into 2D-space with principal coordinates analysis using the weighted UniFrac distance.
To determine if mothers share microbes with their newborns, we compared the average number of ASVs shared between mother-neonate pairs and 1000 pairs randomly selected from different families. We conducted a one-tailed t-test and found that mother-neonate pairs did not have a higher number of shared ASVs compared to randomly selected pairs (N = 0.7 vs. N = 1.8, respectively; p = 1.0).
Clinical relevance of microbiome in pregnancy
To better understand the sources of variation seen in the beta diversity of our samples, we carried out the permutational multivariate analysis of variance (PERMANOVA) using different factors, including clinical information. As shown in Table 2, when all sample types were included in the analysis, the variable “Site” explained 17.2% of the variation (p-value = 0.001), and the variable “LibraryMonth,” was 7.4% (p-value = 0.002). This result indicates that the samples could still be separated well based on the microbiome pattern unique to their body site, despite the significant batch effects present within our dataset. When the analysis was restricted to each sample type, except for the sample VD group, the variable “LibraryMonth” was found to be significant for all sample types. The explanatory power increased to a range between 24.5 and 48.9%. These results align with the hypothesis that our samples are predominantly low-biomass specimens and prone to contamination.
Additionally, the variable “DeliveryMethod” was returned as significant for the VD group, the variables “PretermBirth37” and “AntibioticsUse” for the M group, and the variable “Weight” for the CB group (Fig. 3). We explored the significant variables in each group using PCoA with weighted UniFrac distance. Several ASVs of Lactobacillus and one ASV of Gardnerella were found in the VD group. In the M group Staphylococcus showed a strong association with preterm birth. Lastly, the lists of bacterial taxa were connected to the weights of neonates in the CB group. Table 3 shows the analysis of the composition of microbiomes (ANCOM) for various clinical data to study any statistically significant relevance with bacteria in multiple sample types.

Beta diversity results of the PERMANOVA analysis. Principal coordinates analysis using weighted UniFrac distance is shown for (A) the cervicovaginal discharge samples, (B) and (C) the meconium samples, and (D) the umbilical cord blood samples.
The resemblance of twin microbiome in delivery
To test the hypothesis that samples from twins, both monochorionic and dichorionic, have higher similarity in microbiome composition than randomly chosen samples, we compared the mean of weighted UniFrac distance between twin samples and randomly selected samples. More specifically, for each of the AF, CB, GL, and M groups, we performed bootstrapping hypothesis testing by randomly sampling pairwise distances with replacement from all samples 1000 times to build a 95% confidence interval with the means of the sampled distances. We rejected the null hypothesis that there was no difference between the twin samples and randomly selected samples for all four sample types because the mean pairwise distance for twin samples was below the confidence interval (Fig. 4 and Supplementary Fig. S7). Next, we divided the twins into monochorionic and dichorionic twins and repeated hypothesis testing. We found that we could still reject the null hypothesis for all four sample types for dichorionic twins. For monochorionic twins, however, only the CB and M groups passed the test.

Higher similarity of microbiome composition in twin samples than in randomly chosen samples. For each sample type, the means of weighted UniFrac distances are shown for the twin samples. A 95% confidence interval was constructed by randomly sampling pairwise distances with replacement from the samples for 1000 times.
Characterization of the vaginal health-related microbiome
Several pathogenic and commensal vaginal microbiota have been shown to have important consequences for a woman’s reproductive and general health. To establish reference ranges of vaginal microbiota with known clinical associations in generally healthy pregnant women, we searched for bacterial targets commonly tested for assessing vaginal health within VD samples. More specifically, we focused on 31 bacterial targets (15 genera and 16 species) that are tested by the “SmartJane” assay from uBiome Inc., including Lactobacillus, Sneathia, and Gardnerella21. Of the 31 bacterial taxa of clinical importance, 12 were identified in our samples (Fig. 5).

Relative abundance of bacteria associated with vaginal health. Only bacterial targets in uBiome’s SmartJane assay that are also present in the vaginal discharge samples are shown.
We observed a higher relative abundance of Lactobacillus at the genus level but lower abundances of Aerococcus, Fusobacterium, Gardnerella, Peptoniphilus, Porphyromonas, and Prevotella. Most of our patients did not have any severe pregnancy-related complications. In addition, the majority of preterm birth ranged in the late preterm period from 34 + 0 weeks to 36 + 6 weeks. Therefore, the “SmartJane” assay did not capture almost any pathogenic microbiome. The specification level was examined and is listed in Fig. 5. We found Lactobacillus iners and Lactobacillus jensenii from the assay lists, but Lactobacillus crispatus was not commonly found in the vaginal microbiome. This could be simply because the SILVA reference database we used omitted Lactobacillus crispatus. We confirmed that some of the ASVs from the Lactobacillus genus were indeed Lactobacillus crispatus using the National Center for Biotechnology Information (NCBI) database (data not shown).
Discussion
Controversies surrounding in utero colonization
Since contamination is a critical issue in microbiome research, we used several up-to-date methods to confirm the presence of bacteria and found evidence of in utero colonization. The distinct bacterial composition in meconium, the first stool passed by newborns, supports that microbial colonization occurs in the intrauterine environment during normal pregnancy22,23. The microbiome in neonatal gastric liquid was similar to that in maternal amnionic fluid, as expected since fetuses swallow amnionic fluid in utero and their urine returns to the fluid under normal physiological conditions. However, the microbiome in gastric liquid was not exactly the same as in amnionic fluid, indicating the existence of unknown mechanisms for flora formation in the fetal oral cavity or proximal gastrointestinal tract, such as esophagus, from the intrauterine environment.
Do different samples from mothers and newborns share the same microbiome?
The study aimed to determine whether samples from various body sites of pregnant women and their infants would have similar microbiomes, or if the maternal microbiome would be passed on to her fetus. Our results suggest that the microbiome primarily differed based on the body compartment where it was obtained, not the mother-fetus pair. That is, out of all factors, including various obstetric conditions, the sampling site was the most significant factor in determining microbiome similarity.
Establishing a representative microbiome library of various samples to understand the microbiomes of typical pregnant women, fetuses, and neonates
This study’s key strength is its study population, which comprised of pure Asians and reflected general, low-risk pregnancies. The maternal age range was between 20 and 45 years, which is considered typical for reproductive age. There were roughly equal numbers of nulliparous women, caesarean sections, and male and female neonates. Other than a small number of instances of fetal distress, such as low Apgar scores and meconium staining, newborns with extremely pathological conditions that could alter the microbiome, such as severe preterm birth and treatment in a neonatal intensive care unit (NICU), were excluded. As a result, the microbiome analyzed in this study population is likely to represent typical pregnancy. It is crucial to establish a microbiome library for low-risk pregnant women and their normal neonates as a basis for comparison with pathological conditions, to better understand the microbiome composition during pregnancy.
Association between microbiome and various pregnancy-related phenotypes
To identify the microbiomes associated with pregnancy-related conditions, such as delivery method, we conducted statistical analysis of differential abundance. Despite the challenges posed by the low microbial biomass and difficulties in controlling study subjects, which can result in false positive results, the bacteria listed in Table 3 seem to align with previous findings. For example, Finegoldia and Bifidobacterium have been previously linked to a healthier pregnancy, and our data confirms this association24,25. Other taxa listed in the table also have links to inflammation and pregnancy complications, such as gestational diabetes mellitus, preeclampsia, and preterm birth. The presence of Campylobacter and Lachnospiraceae in vaginal discharge, for example, is in line with previous research showing that these bacterial infections can lead to inflammation and preterm birth26,27.
By cross-referencing with clinical databases, our analysis revealed several significant associations. First, the abundant presence of Lactobacillus and Gardnerella in vaginal discharge is a well-known indicator of the pregnancy microbiome. Lactobacillus plays a protective role in the maternal microbiome during pregnancy, while Gardnerella is considered a pathogen and is strongly associated with preterm birth or pregnancy complications11,13,26. The presence of Faecalibacterium in cord blood is noteworthy, as it has been shown to be depleted in gestational diabetes mellitus28, even though the number of cases in our study population was relatively small. Additionally, Staphylococcus was found to be strongly associated with preterm birth in meconium. This result coincides with previous findings that suggest Staphylococcus infections can lead to preterm birth29,30.
Regarding the effect of antibiotics, we analyzed the relationship between antibiotic use and meconium samples, but the results showed limited association due to the small sample size. As Tormo-Badia et al. reported, antibiotics can alter the gut microbiome of offspring in pregnant mice31. Given that the existence of a “healthy microbiome” during pregnancy is considered crucial for maintaining a normal pregnancy, it is easy to imagine the potential negative consequences of antibiotics administration during pregnancy. Since antibiotics are only given to pregnant women who have signs of infection or inflammation, specific diseases, or preterm premature rupture of membranes with the risk of ascending infection to the fetus, it is practically challenging to determine the effect of antibiotics on the modification of the birth-related microbiome.
The meconium samples showed the presence of microbiome taxa such as Lactobacillus, Staphylococcus, and Ureaplasma, which are collectively known as the vaginal flora11,32. We attempted to evaluate the relationship between delivery mode and the microbiome in meconium, but we did not find any statistically significant differences in composition or diversity. According to a study by Dominguez-Bello et al., there are differences in the bacterial communities in the guts of infants depending on the mode of delivery33. Neonates born vaginally have a microbiome resembling their mother's vaginal microbiota, dominated by Lactobacillus. Conversely, infants born via cesarean section have a microbiome dominated by Staphylococcus, Corynebacterium, and Propionibacterium, which are commonly found on their mothers’ skin surfaces.
Twin pregnancy and microbiome
Approximately a quarter of the pregnancies in our study were twin pregnancies (37/141). To the best of our knowledge, this is the first study to assess the microbiomes of twin newborns. Generally, our twin samples (AF, CB, GL, and M) showed a more similar composition compared to randomly selected samples, even for dichorionic twins who have separate intrauterine compartments. The only exception was CB and M samples from monochorionic twins, where randomly selected samples showed greater similarity, which is likely due to the small sample size of monochorionic twins.
Conclusion
Exploring the microbiologic features related to pregnancy has been a challenging and controversial task for many years. Microbial invasion of the gestational cavity such as amniotic fluid or placenta can lead to serious obstetric complications such as preterm birth and severe neonatal morbidities that may persist throughout life. Despite the importance of research on the microbiome in pregnancy, progress has been limited due to ethical and accessibility issues. We have collected various samples from pregnant women and their neonates using a standardized protocol and established a microbiome database, which can serve as a reference library for studying samples with other pregnancy-related or pathologic conditions.
Methods
Study design and sample collection
A prospective study was performed on live births delivered between March 2020 and January 2021. Samples were collected from women who had delivered at Seoul National University Bundang Hospital and their newborns. Women with unstable vital signs or those requiring urgent management such as transfusion and neonates admitted to the NICU or who had unstable vital signs after birth were excluded from the study. Samples for microbiome analysis included maternal VD, AF, CB, neonatal GL, and M. As a pregnant woman was hospitalized with expectancy of delivery, the VD sample was obtained using a polyester swab inserted into the posterior fornix of the vagina, assisted by sterile speculum examination. For those who had undergone cesarean section for delivery or amniocentesis for specific indications (i.e., for detection of intraamniotic inflammation/infection), approximately 10 cc of AF was obtained through a syringe for the study. During delivery, both cesarean section and vaginal delivery, approximately 20 cc of CB was taken through a syringe from the vein of the umbilical cord immediately after clamping. The syringe needle was directly inserted into the umbilical cord at the delivery site surrounded by sterile drapes to minimize surgical field contamination. Since removing amniotic fluid or other liquid from the newborn’s mouth and stomach after birth is a part of initial management to help the airway and to stimulate spontaneous breathing, most neonates received suctioning procedures, and the liquid collected in the suction bottle (approximately 15 ml) was carried into a conical tube for analysis of GL. The M sample, the newborn's very early stool, was carefully obtained within 24 h after birth using a polyester swab inserted into the anus as the neonate stabilized after initial management. We tried to collect all five different samples from each woman and neonate(s), nonetheless, a small part of samples from mother-neonate pairs were not obtained or missed for clinical circumstances. The primary outcome was the distribution and composition of the microbiome of the above samples from pregnant women and their neonates. To determine the association between the microbiome from different compartments and obstetric factors, medical records were collected and thoroughly reviewed. Data included maternal age, gestational age at delivery, delivery mode (vaginal delivery or cesarean section), the use of ART, other obstetric complications, and neonatal outcomes such as sex and birth weight.
Ethics approval and consent to participate
This study was performed with the informed consent of appropriate participants in compliance with the Declaration of Helsinki. The study protocol was approved by the Institutional Review Board of the Seoul National University Bundang Hospital (B-1606/350-003).
Microbial DNA isolation
Microbial deoxyribonucleic acid (DNA) was extracted from the VD, GL, AF, and CB samples with the ZymoBIOMICS DNA Miniprep Kit (Zymo Research, Irvine, CA) and the sample M using the DNeasy PowerSoil Pro Kit (Qiagen, Germantown, MD) according to the manufacturer’s instructions. Briefly, samples were enzymatically and mechanically lysed by bead beating, followed by washing and filtering in the provided column. Extracted DNA concentrations were measured using a Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA). The total amounts of extracted DNA were varied based on sample types, such as 1–10 ug for VD, 3 μg for CB, 30–200 ng for M, and 50 ng for GL and AF. For each box of the DNA extraction kit used, no material was used as a negative control. The blanks were processed in the entire protocol and analyzed.
16S rRNA gene amplification
The 16S ribosomal ribonucleic acid (rRNA) gene was amplified using the two-step polymerase chain reaction (PCR) protocol in the 16S Metagenomic Sequencing Library Preparation (Illumina, San Diego, CA). In the first PCR step, the V3–V4 hypervariable region of the 16S rRNA gene was amplified using 10 ng of each sample, 10 µM of 341F/785R primers, and Herculase II fusion DNA polymerase (Agilent, Santa Clara, CA). In the below primer sequence, ‘N’ base is selected from any random base, ‘W’ base is A or T, ‘H’ base is A, C or T, and ‘V’ base is A, C, or G.
341F: 5′- TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCTACGGGNGGCWGCAG-3′
785R: 5′-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTACHVGGGTATCTAATCC-3′
PCR cycling was performed with an initial cycle at 95 °C for 3 min, followed by 25 cycles of 95 °C for 30 s, 55 °C for 30 s, 72 °C for 30 s, and a final extension cycle at 72 °C for 5 min. The amplicons were cleaned with AMPure XP beads (Beckman Coulter, Brea, CA, USA). In the second PCR, index primers from the Nextera DNA CD Index Kit (Illumina, San Diego, CA) were added to the ends of the amplicons generated in the first PCR. PCR cycling was performed with an initial cycle at 95 °C for 3 min, followed by ten cycles of 95 °C for 30 s, 55 °C for 30 s, 72 °C for 30 s, and a final extension cycle at 72 °C for 5 min. Each sample was cleaned with AMPure XP beads (Beckman Coulter, Brea, CA, USA) and eluted in UltraPure DNase/RNase-Free Water (Thermo Fisher Scientific, Waltham, MA). The amplified DNA was checked using a 2100 Bioanalyzer system using an Agilent DNA 1000 Kit (Agilent, Santa Clara, CA, USA). For each library production, no template was used as a negative control.
16S rRNA gene sequencing and analysis
Based on the DNA size and concentration, the amplicons were pooled in equimolar amounts and spiked with 30% PhiX (Illumina, San Diego, CA). These were then sequenced on the Illumina MiSeq platform using paired-end 250 cycle MiSeq Reagent Kit V2 (Illumina, San Diego, CA) and a 300 cycle MiSeq Reagent Kit V3 (Illumina, San Diego, CA). Negative controls from the DNA extraction and library were sequenced.
Sequencing data generation
We divided the samples into nine batches (Runs 1–9) and sequenced the V3-V4 region of the 16S rRNA gene using Illumina MiSeq machines with a target depth of 100,000 per sample (Supplementary Fig. S8). Sequencing was performed with 250 bp paired-end reads for all of the sequencing runs except for the last one (Run 9), where sequencing was performed with 300 bp paired-end reads for practical reasons. The read quality scores for each sequencing run are shown in Supplementary Fig. S9. The bcf2fastq program of Illumina was used to demultiplex raw sequencing data (BCL files) and output forward and reverse FASTQ files for each sample. Of note, some samples were sequenced more than once to assess the impact of batch effects. These included “sequencing duplicates” in which the identical NGS library of one sample was sequenced in separate runs and “library duplicates” in which multiple NGS libraries were prepared from the identical sample at different dates and then sequenced separately.
Data analysis and visualization
Unless stated otherwise, all analyses were carried out using the QIIME 2 platform, a powerful community-developed platform for microbiome bioinformatics34. For each sequencing run, FASTQ files were imported to QIIME 2 and the DADA2 plugin35 to identify ASVs by trimming low-quality parts of sequence reads, denoising trimmed reads, and then merging the forward and reverse reads (Supplementary Fig. S8). The observed ASVs from individual sequencing runs were then merged into one ASV table. To detect and remove potential contaminants, we ran the decontam program on our samples, which looked for ASVs per sequencing batch that appeared at higher frequencies in low-concentration samples and were repeatedly found in the negative control36. Taxonomy classification was performed using a naive Bayes classifier using the SILVA database37. To visualize the outputs from QIIME 2, we developed the Dokdo program (https://github.com/sbslee/dokdo), an open-source and MIT-licensed Python package for microbiome sequencing analysis using QIIME 2. Dokdo internally uses the application programming interface of QIIME 2 and therefore does not require any other dependencies. Dokdo can be used to perform a variety of secondary analyses or create publication-quality figures from QIIME 2 files/objects (e.g. a taxonomic bar plot or an alpha rarefaction plot).
Diversity analysis
We used the QIIME 2 command “qiime diversity core-metrics-phylogenetic” to compute the alpha and beta diversity metrics of our samples. When running the command, to normalize for the difference in read depth across the samples, we used the “-p-sampling-depth” option to rarefy our samples to 5,000 sequence reads and have an equal depth of coverage. We also ensured that all samples were sequenced to a sufficient depth of coverage for diversity analysis by creating rarefaction curves (Supplementary Fig. S10). Additionally, we used the “-i-phylogeny” option to provide a rooted phylogenetic tree of observed ASVs, which is required for performing PCoA based on the weighted UniFrac distance38.
Statistical analysis
To assess the differential abundance of the microbiome in the context of clinical information such as preterm birth, we used the QIIME 2 command “qiime composition ancom” to perform ANCOM, which compares the centered log-ratio (CLR) of relative abundance between two or more groups of samples39. To determine whether groups of samples are significantly different from one another in beta diversity, we carried out PERMANOVA using the QIIME 2 command “qiime diversity adonis” which fits linear model assumptions to a distance matrix (e.g., weighted UniFrac) with the chosen variables. We performed bootstrapping hypothesis testing by building a 95% confidence interval with the “scipy.stats.t.interval” method in the scipy package to compare similarities in microbiome composition between twins and randomly chosen samples40.
Data availability
The sequencing data generated from this study has been deposited in the INSDC databases through the European Nucleotide Archive (ENA) under accession number PRJEB52455. The ENA URL is https://www.ebi.ac.uk/ena/browser/view/PRJEB52455. The data generated during the current study are available from the corresponding author on reasonable written request.
Change history
02 November 2023
A Correction to this paper has been published: https://doi.org/10.1038/s41598-023-46309-3
Abbreviations
- AF:
-
Amniotic fluid
- ANCOM:
-
Analysis of the composition of microbiomes
- ART:
-
Assisted reproductive technology
- ASVs:
-
Amplicon sequence variants
- BMI:
-
Body mass index
- CB:
-
Umbilical cord blood
- CLR:
-
Centered log-ratio
- DNA:
-
Deoxyribonucleic acid
- GL:
-
Gastric liquid
- IUI:
-
Intrauterine insemination
- IVF-ET:
-
In vitro fertilization and embryo transfer
- M:
-
Meconium
- NCBI:
-
National Center for Biotechnology Information
- NGS:
-
Next-generation sequencing
- NICU:
-
Neonatal intensive care unit
- PCoA:
-
Principal coordinates analysis
- PCR:
-
Polymerase chain reaction
- PERMANOVA:
-
Permutational multivariate analysis of variance
- RNA:
-
Ribosomal ribonucleic acid
- VD:
-
Cervicovaginal discharge
References
Mesa, M. D. et al. The evolving microbiome from pregnancy to early infancy: A comprehensive review. Nutrients https://doi.org/10.3390/nu12010133 (2020).
Knight, R. et al. Best practices for analysing microbiomes. Nat. Rev. Microbiol. 16, 410–422. https://doi.org/10.1038/s41579-018-0029-9 (2018).
Fox, C. & Eichelberger, K. Maternal microbiome and pregnancy outcomes. Fertil. Steril. 104, 1358–1363. https://doi.org/10.1016/j.fertnstert.2015.09.037 (2015).
Chen, X., Liu, S., Tan, Q., Shoenfeld, Y. & Zeng, Y. Microbiome, autoimmunity, allergy, and helminth infection: The importance of the pregnancy period. Am. J. Reprod. Immunol. https://doi.org/10.1111/aji.12654 (2017).
Aagaard, K. et al. The placenta harbors a unique microbiome. Sci. Transl. Med. 6, 237ra265. https://doi.org/10.1126/scitranslmed.3008599 (2014).
Perez-Munoz, M. E., Arrieta, M. C., Ramer-Tait, A. E. & Walter, J. A critical assessment of the “sterile womb” and “in utero colonization” hypotheses: Implications for research on the pioneer infant microbiome. Microbiome 5, 48. https://doi.org/10.1186/s40168-017-0268-4 (2017).
Aagaard, K. et al. A metagenomic approach to characterization of the vaginal microbiome signature in pregnancy. PLoS ONE 7, e36466. https://doi.org/10.1371/journal.pone.0036466 (2012).
Neuman, H. & Koren, O. The pregnancy microbiome. Nestle Nutr. Inst. Workshop Ser. 88, 1–9. https://doi.org/10.1159/000455207 (2017).
Nuriel-Ohayon, M., Neuman, H. & Koren, O. Microbial changes during pregnancy, birth, and infancy. Front. Microbiol. 7, 1031. https://doi.org/10.3389/fmicb.2016.01031 (2016).
Staude, B. et al. The microbiome and preterm birth: A change in paradigm with profound implications for pathophysiologic concepts and novel therapeutic strategies. Biomed. Res. Int. 2018, 7218187. https://doi.org/10.1155/2018/7218187 (2018).
DiGiulio, D. B. et al. Temporal and spatial variation of the human microbiota during pregnancy. Proc. Natl. Acad. Sci. USA 112, 11060–11065. https://doi.org/10.1073/pnas.1502875112 (2015).
Stout, M. J. et al. Early pregnancy vaginal microbiome trends and preterm birth. Am. J. Obstet. Gynecol. 217, 356.e351-356.e318. https://doi.org/10.1016/j.ajog.2017.05.030 (2017).
Callahan, B. J. et al. Replication and refinement of a vaginal microbial signature of preterm birth in two racially distinct cohorts of US women. Proc. Natl. Acad. Sci. USA 114, 9966–9971. https://doi.org/10.1073/pnas.1705899114 (2017).
Brown, R. G. et al. Vaginal dysbiosis increases risk of preterm fetal membrane rupture, neonatal sepsis and is exacerbated by erythromycin. BMC Med. 16, 9. https://doi.org/10.1186/s12916-017-0999-x (2018).
Kuperman, A. A. & Koren, O. Antibiotic use during pregnancy: How bad is it?. BMC Med. 14, 91. https://doi.org/10.1186/s12916-016-0636-0 (2016).
Fujiwara, N. et al. Significant increase of oral bacteria in the early pregnancy period in Japanese women. J. Investig. Clin. Dent. https://doi.org/10.1111/jicd.12189 (2017).
Collado, M. C., Rautava, S., Aakko, J., Isolauri, E. & Salminen, S. Human gut colonisation may be initiated in utero by distinct microbial communities in the placenta and amniotic fluid. Sci. Rep. 6, 23129. https://doi.org/10.1038/srep23129 (2016).
DiGiulio, D. B. et al. Prevalence and diversity of microbes in the amniotic fluid, the fetal inflammatory response, and pregnancy outcome in women with preterm pre-labor rupture of membranes. Am. J. Reprod. Immunol. 64, 38–57. https://doi.org/10.1111/j.1600-0897.2010.00830.x (2010).
Akagawa, S. et al. Effect of delivery mode and nutrition on gut microbiota in neonates. Ann. Nutr. Metab. 74, 132–139. https://doi.org/10.1159/000496427 (2019).
Moreno, I. & Simon, C. Relevance of assessing the uterine microbiota in infertility. Fertil. Steril. 110, 337–343. https://doi.org/10.1016/j.fertnstert.2018.04.041 (2018).
Bik, E. M. et al. A novel sequencing-based vaginal health assay combining self-sampling, HPV detection and genotyping, STI detection, and vaginal microbiome analysis. PLoS ONE 14, e0215945. https://doi.org/10.1371/journal.pone.0215945 (2019).
Hansen, R. et al. First-pass meconium samples from healthy term vaginally-delivered neonates: An analysis of the microbiota. PLoS ONE 10, e0133320. https://doi.org/10.1371/journal.pone.0133320 (2015).
Martin, R. et al. Early-life events, including mode of delivery and type of feeding, siblings and gender, shape the developing gut microbiota. PLoS ONE 11, e0158498. https://doi.org/10.1371/journal.pone.0158498 (2016).
Kumar, M. et al. Vaginal microbiota and cytokine levels predict preterm delivery in Asian women. Front. Cell Infect. Microbiol. 11, 639665. https://doi.org/10.3389/fcimb.2021.639665 (2021).
Nuriel-Ohayon, M. et al. Progesterone increases bifidobacterium relative abundance during late pregnancy. Cell Rep. 27, 730-736.e733. https://doi.org/10.1016/j.celrep.2019.03.075 (2019).
Serrano, M. G. et al. Racioethnic diversity in the dynamics of the vaginal microbiome during pregnancy. Nat. Med. 25, 1001–1011. https://doi.org/10.1038/s41591-019-0465-8 (2019).
Smith, J. L. Campylobacter jejuni infection during pregnancy: Long-term consequences of associated bacteremia, Guillain-Barre syndrome, and reactive arthritist. J. Food Prot. 65, 696–708. https://doi.org/10.4315/0362-028x-65.4.696 (2002).
Ferrocino, I. et al. Changes in the gut microbiota composition during pregnancy in patients with gestational diabetes mellitus (GDM). Sci. Rep. 8, 12216. https://doi.org/10.1038/s41598-018-30735-9 (2018).
Kriebs, J. M. Staphylococcus infections in pregnancy: Maternal and neonatal risks. J. Perinat. Neonatal. Nurs. 30, 115–123. https://doi.org/10.1097/JPN.0000000000000165 (2016).
Tang, Y., Yu, F., Hu, Z., Peng, L. & Jiang, Y. Characterization of aerobic vaginitis in late pregnancy in a Chinese population: A STROBE-compliant study. Medicine 99, e20732. https://doi.org/10.1097/MD.0000000000020732 (2020).
Tormo-Badia, N. et al. Antibiotic treatment of pregnant non-obese diabetic mice leads to altered gut microbiota and intestinal immunological changes in the offspring. Scand. J. Immunol. 80, 250–260. https://doi.org/10.1111/sji.12205 (2014).
Peretz, A. et al. Mycoplasma and Ureaplasma carriage in pregnant women: The prevalence of transmission from mother to newborn. BMC Pregnancy Childbirth 20, 456. https://doi.org/10.1186/s12884-020-03147-9 (2020).
Dominguez-Bello, M. G. et al. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc. Natl. Acad. Sci. USA 107, 11971–11975. https://doi.org/10.1073/pnas.1002601107 (2010).
Bolyen, E. et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857. https://doi.org/10.1038/s41587-019-0209-9 (2019).
Callahan, B. J. et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. https://doi.org/10.1038/nmeth.3869 (2016).
Davis, N. M., Proctor, D. M., Holmes, S. P., Relman, D. A. & Callahan, B. J. Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome 6, 226. https://doi.org/10.1186/s40168-018-0605-2 (2018).
Quast, C. et al. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 41, D590-596. https://doi.org/10.1093/nar/gks1219 (2013).
Lozupone, C. & Knight, R. UniFrac: A new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 71, 8228–8235. https://doi.org/10.1128/AEM.71.12.8228-8235.2005 (2005).
Mandal, S. et al. Analysis of composition of microbiomes: A novel method for studying microbial composition. Microb. Ecol. Health Dis. 26, 27663. https://doi.org/10.3402/mehd.v26.27663 (2015).
Virtanen, P. et al. SciPy 1.0: Fundamental algorithms for scientific computing in Python. Nat. Methods 17, 261–272. https://doi.org/10.1038/s41592-019-0686-2 (2020).
Acknowledgements
This study was supported by Seoul National University Bundang Hospital Research Fund (18-2018-002) and World-class 300 project by Ministry of SMEs and Start-ups.
Author information
Authors and Affiliations
Contributions
The final manuscript has been seen and approved by all authors and we have taken due care to ensure the integrity of the work as in the following; Guarantor of integrity of entire study: J.Y.P., H.Y., J.S.P., J.S.; study concepts/study design: J.Y.P., H.Y., S.L. H.J.K. ;data acquisition or data analysis/interpretation: J.Y.P., H.Y., Y.H.J., C.W.C., J.S.; manuscript drafting or manuscript revision for important intellectual content: J.Y.P., H.Y., J.S., J.S.P., J.S.; approval of final version of submitted manuscript: J.Y.P., H.Y., J.S.P., J.S.; agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved; J.Y.P., H.Y., S.L. H.J.K., Y.H.J., C.W.C., J.S.; literature research: J.Y.P., H.Y., S.L. H.J.K.; statistical analysis; J.Y.P., H.Y., J.S.; manuscript editing: J.Y.P., H.Y., J.S.P., J.S.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this Article was revised: The original version of this Article contained an error in the Acknowledgements section. “This study was supported by Seoul National University Bundang Hospital Research Fund (14-2017-026) and World-class 300 project by Ministry of SMEs and Start-ups.” now reads: “This study was supported by Seoul National University Bundang Hospital Research Fund (18-2018-002) and World-class 300 project by Ministry of SMEs and Start-ups.”
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Park, J.Y., Yun, H., Lee, Sb. et al. Comprehensive characterization of maternal, fetal, and neonatal microbiomes supports prenatal colonization of the gastrointestinal tract. Sci Rep 13, 4652 (2023). https://doi.org/10.1038/s41598-023-31049-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-31049-1
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
