
Abstract
Coronaviruses (CoVs) pose a huge threat to public health as emerging viruses. Bat-borne CoVs are especially unpredictable in their evolution due to some unique features of bat physiology boosting the rate of mutations in CoVs, which is already high by itself compared to other viruses. Among bats, a meta-analysis of overall CoVs epizootiology identified a nucleic acid observed prevalence of 9.8% (95% CI 8.7–10.9%). The main objectives of our study were to conduct a qPCR screening of CoVs’ prevalence in the insectivorous bat population of Fore-Caucasus and perform their characterization based on the metagenomic NGS of samples with detected CoV RNA. According to the qPCR screening, CoV RNA was detected in 5 samples, resulting in a 3.33% (95% CI 1.1–7.6%) prevalence of CoVs in bats from these studied locations. BetaCoVs reads were identified in raw metagenomic NGS data, however, detailed characterization was not possible due to relatively low RNA concentration in samples. Our results correspond to other studies, although a lower prevalence in qPCR studies was observed compared to other regions and countries. Further studies should require deeper metagenomic NGS investigation, as a supplementary method, which will allow detailed CoV characterization.
Similar content being viewed by others


Introduction
Coronaviruses (CoVs) have a high potential to adapt and evolve due to their fast replication rate, recombination in their RNAs and subsequent mutations1. It is hypothesized that this quality allows CoVs to overcome the interspecies barrier and adapt to new hosts2. In some cases, interspecies jumps could be a threat to public health and result in epidemic outbreaks of CoV infections in the human population. In recent years, we have witnessed outbreaks (MERS and SARS) and a pandemic (COVID-19) caused by animal-derived CoVs3. Animals such as bats, rodents, civets, cattle, swine, and camelids are recognized as hosts of CoVs diversity that have been involved in spillover and adaptation to new hosts including humans in the past, suggesting that favorable conditions may see the emergence of additional human CoVs4.
It has been suggested that the human CoVs HCoV-229E, HCoV-NL63, SARS-CoV, MERS-CoV, and SARS-CoV-2 may have emerged from progenitor CoVs present in bats5. Bats are not only hosts for CoVs, but also other emerging viruses such as Nipah, Hendra, Marburg, and Ebola viruses6. Several mechanisms are making these animals ideal "incubators" for emerging viruses. Limited antiviral immune reactions7 and inflammatory response in bats contribute to relatively long persistence of viruses, which gives emerging CoVs more time and space to get mutations for interspecies transmissions8. In addition to the unique antiviral immune response of bats, the ability to fly also contributes to developing new emerging viruses. Reactive oxygen species, formed as a result of active metabolism during the flight of these mammals, damage cell DNA and enzymes involved in viral replication, such as RNA-dependent RNA polymerase of CoVs9,10. Our previous study reveals pro-mutagenic properties of lactic acid producing bacteria within the gut microbiota of bats, which also can promote mutations in viruses of these animals11.
All the points mentioned above make bats the most important animals to be screened for the persistence of emerging viruses, especially CoVs. Before the COVID-19 outbreak, several papers were published on the detection of CoV in bats in Italy12,13, Germany14, France15, Spain16, United Kingdom17, Slovenia18, Netherlands19, Luxembourg20, Denmark21, Korea22, and China23,24. Based on this data, data from SARS-CoV and MERS-CoV outbreaks, and data on the ability of interspecies transmission of CoV, some scientists suggested that there would be a new CoV outbreak from bats25,26,27,28, which in the end indeed resulted in the COVID-19 pandemic. From today’s perspective, it is especially essential to conduct studies on the detection and characterization of CoVs in bats, as any obtained data can help predict the next CoV outbreaks. In this pilot study, we report the first data on qPCR screening of bats from several Fore-Caucasus regions of Russia for the presence of CoVs. We also conducted an in silico study to test the sensitivity and specificity of the pan-coronavirus assay used in this study and performed NGS of samples with detected CoV RNA.
Results
RNA concentration
The mean RNA concentration in liquid oropharyngeal probe samples was 2.67 ng/μL (2.58–2.77 ng/μL) and in liquid rectal samples was 2.71 ng/μL (2.59–2.82 ng/μL) There are no significant differences in RNA concentration in oropharyngeal and rectal swabs (p = 0.997) (Supplementary materials, File S1, Fig. S1).
qPCR screening
Positive signals on detection of CoV RNA were found in five samples from five different animals: samples from two Kuhl's pipistrelles, two serotine bats, and one lesser horseshoe bat. CoV RNA was detected in four oropharyngeal swab samples and one rectal swab sample. A detailed database of bats and CoV detection can be found in supplementary materials, File S2.
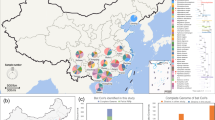
The mean proportion of detected CoVs in the studied population of bats is 3.33% (CI 1.1–7.6%). There is no significant effect of bat species (χ2 = 6.23, df = 6; p = 0.39), swab origin (χ2 = 1.83, df = 1; p = 0.18), sex (χ2 = 1.38, df = 1; p = 0.24), and sample collection site (χ2 = 2.9, df = 3; p = 0.4) on the detection of CoV RNA, likely also due to the low number of positive samples. There are no significant predictors for CoV RNA detection observed, the results of the simple logistic regression are given in the Table 1. The global positioning system (GPS) representing the locations of collection sites is shown in Fig. 1.

Global positioning system (GPS) showing (A) probe locations in this study, and (B) locations, from where probes with detected CoV RNA were taken. (This map was created by QGIS version 3.28, which can be accessed on https://qgis.org/en/site/).
In silico primer sensitivity and specificity
In silico analyses were conducted on the ability of pancoronavirus primers to detect RNA of various types of CoVs. As a result, it was established that the used degenerate primers from Vijgen et al.29 study could detect at least 65 CoV species from available reference genomes belonging to the Coronaviridae family. No hits were detected against other viruses, therefore, specificity could be assumed as 100% (Fig. 2). Most mismatches were observed in the forward primer—1 mismatch with 24.18% of published CoV genomes and 2 mismatches with 6.59% of CoV genomes. There were more than two mismatches in the genomes of 3 CoVs: Beluga Whale CoV SW1, unidentified human CoV isolate FT1407-6/2014, and Shrew-CoV/Tibet2014. Pairs of primers from Lelli et al.13 showed similar sensitivity. However, they also detected the Edwardsiella virus pEt-SU reference genome, and based on this we cannot assume 100% specificity for this pair of primers. Primers from Watanabe et al.30 study mostly had more than two mismatches in used reference CoV genomes, which is above the number of allowed mismatches. Detailed results of in silico analyses are shown in the supplementary file (File S3). The sensitivity of forward and reverse primers is presented in Table 2.

Phylogenetic tree built using 68 full reference genomes of CoV species and one reference genome of Edwardsiella virus pEt-SU (indicated by the NC-code from the NCBI RefSeq database). The rectangular heatmap represents the number of mismatches for forwards (F) and reverse (R) primers from Vijgen et al.29 (PrimeA), Lelli et al.13 (PrimeB), and Watanabe et al.30 (PrimeC) during in silico PCR testing.
Results NGS of samples with detected CoV RNA
After performing library preparation with NEBNext ARTIC SARS-CoV-2 Library Prep Kit (New England Biolabs, England) we did not observe amplified cDNA of SARS-like CoVs’ gRNA suitable for NGS. Results of electrophoresis of cDNA amplicons can be found in supplementary materials, File S4 (Fig. S2). Although, sequencing of target genome fragments of SARS-like CoVs was not successful due to the absence of amplicons, we obtained metagenomic data from samples with detected CoV RNA. According to the taxonomical identification of raw reads (FASTQ) with Kraken2, there were 30 reads of Coronaviridae genomes in data obtained from MiSeq runs and 54 reads of Coronaviridae genome in data obtained from NovaSeq runs. Results of taxonomical identification can be found in supplementary materials, File S5. Some assembled contigs had 89–100% identity with SARS-related CoVs and HcoV-OC43 records from the NCBI Virus database with 3–16% coverage. The length of these contigs is from 458 to 1236 bp. Gene annotation by VADR was not successful, however, part of the contigs was aligned to the SARS-CoV-2 reference genome used for this tool. Assembled contigs and outcomes of BLAST search of similar viral nucleotide sequences in the NCBI Virus database (ssRNA(+) records) can be found in supplementary materials, File S6.
Discussion
Compared to CoVs from other animals bat-borne CoVs are especially dangerous as some unique features of bat physiology boost the mutation rate in CoVs, which themselves have a high mutation rate already4,11. To predict future CoV outbreaks, more and improved surveillance is required to investigate CoV prevalence among bats and to characterize their genomic features. In this study, screening of CoV prevalence in bats from multiple locations in the European part of Southern Russia was performed using qPCR with pan-coronavirus primers.
The proportion of detected CoVs in the studied population of 150 bats is 3.33% (5/150; 95% CI 1.1–7.6%). In most previous studies CoV prevalence in bats is higher. Bonilla-Aldana et al. conducted a meta-analysis of 33 reports of PCR screening of bats and established that prevalence by RT-PCR in these studies (n = 14,295 bats) for CoV was 9.8% (95% CI 8.7–10.9%)31. However, there are some studies with a low rate of CoV RNA detection by PCR. Mendenhall et al. reported that they did not detect CoVs with PCR targeting polymerase gene in 431 pooled oral-rectal samples from six bat species and 1124 urine and faecal samples from cave nectar bat (Eonycteris spelaea)32. Although, the authors did detect CoV genomes with next-generation sequencing in pooled faecal material and pooled urine from E. spelaea32. Cappelle et al. also reported a relatively low proportion of positive PCR tests for CoVs in Cambodian bats—4.2% (24/573) in bats from Kampot, and 4.75% (22/463) in flying foxes from Kandal33, which overall corresponds to our results. Xu et al. detected CoV RNA in 5.3% (50/951 intestinal specimens) in bats of eight species from four provinces and the Tibet Autonomous Region of China by pan-coronavirus RT-PCR screening34, which is also below pooled average prevalence of CoVs in bats according to Bonilla-Aldana et al.31. Corman et al. conducted PCR screening of 1868 faecal, blood, and intestinal tissue specimens from 1560 bats in Costa Rica, Panama, Ecuador, and Brazil. They also used an assay targeting the RNA-dependent RNA-polymerase of CoVs and detected CoV RNA in 50 specimens from nine different bat species with overall CoV prevalence in 2.7 % of the total samples35. Corman et al. included more samples and animals in screening than in our study, which likely means that the smaller sample size did not have a negative impact on the overall results of PCR CoV screening. Some of the highest CoV prevalence in bats are reported in China by Ge et al.—50% (138/276 faecal samples)36, in Italy by Balboni et al.—42% (19/45 faeces samples)12, and in the Philippines by Tsuda et al.—29.6 % (53/179 faeces samples)37.
We detected CoVs in P. kuhlii, E. serotinus, and R. hipposideros. Various types of CoVs were detected in these species before. For example, Lelli et al.13 detected alpha- and betaCoVs in P. kuhlii and betaCoVs in R. hipposideros in Italy. In the other study, which was conducted at the same time in Italy, betaCoV was also detected in E. serotinus38. CoVs have been detected previously in Asian populations of E. serotinus in Kazakhstan and Korea39,40.
To investigate pan-coronavirus primer efficiency, we performed in silico analyses of the ability of used primers and additional pairs from other studies to detect several CoV species. Vijgen et al. designed their primers based on a 251-bp fragment of the polymerase gene of CoVs known at the time and checked their sensitivity on 14 CoVs of human and animal origin29. We established that these primers could detect at least 65 CoV species, including SARS-like CoV (File S3). This is because Vijgen et al. designed their primers based on a target region that is highly resistant to mutations and variations, which occurred in the most of used CoV genomes for the in silico assay. In addition, we tested pairs of primers from Lelli et al.13 and Watanabe et al.30, that are also designed for a highly conserved region of the RNA-dependent RNA polymerase gene of CoVs. In silico, primers from Lelli et al. showed similar sensitivity to primers from Vijgen et al., however, the specificity was lower, as they detected the Edwardsiella virus pEt-SU genome in addition to CoVs. The primers from Watanabe et al.30 mostly detected bat CoVs, but the overall sensitivity was relatively low compared to the other pairs of primers. We suggest that the first two pairs of primers showed better sensitivity, as they are degenerate primers and can cover more possible nucleotide combinations of targeted polymerase gene and naturally detect more CoVs. The study by Watanabe et al.30 is primarily aimed at the detection of bat CoVs, which is why they probably mostly used bat CoVs genomes for primers design. We believe that this approach has some limitations, as degenerate pairs of primers are able to detect the target region of the CoV genome with various nucleotide sequence combinations, and most importantly they are more suited for the qPCR assay, which is more efficient for the initial screening. That is why we used degenerate pairs of primers for this first epizootiological study of CoVs in the bats population of Fore-Caucasus. Our results from in silico analyses support the statement that pan-coronavirus assays can be an effective tool for the management of emerging CoV diseases41,42.
We did not observe a significant effect of bat species, swab origin, sex, or sample collection site on the detection of CoV RNA by χ2 tests and simple logistic regression, due to the low prevalence detected. Also, there were no significant differences in RNA concentration in oropharyngeal and rectal swabs, which additionally states that swab origin did not contribute to the CoV RNA detection rate. But we should note that the mean RNA concentration was probably too low for NGS analysis sufficient for the effective assembly of CoV genomes and their annotation.
The overall CoV RNA detection rate is probably relatively small for the studied sample size (150 bats) for predictor calculations. As mentioned before, Cappelle et al. established a similar rate of CoV RNA detection in samples from bats in Cambodia and Kandal. The sample size in their study is larger, and they calculated that the rate of CoV detection is significantly higher in juveniles and immature individuals than in mature adults33. We suggest that there are two ways to define a significant predictor for CoV RNA detection in bats population of the Fore-Caucasus. The first, is to increase the sample size with a similar amount of sample collection sites to increase the sample size effect according to the basic principles of biostatistics43. The second is to reduce the amount of sample collection sites with the same sample size, as in studies with a lower amount of sample collection locations, the CoV detection rate in bats is relatively higher. For example, Alkhovsky et al. collected 120 oral swabs and 77 faeces samples from bats in 6 caves and 2 house attics in the Sochi National Park of Russia and detected an overall CoV prevalence of 14%. Most interesting is that the prevalence of SARS-like CoV Khosta-1 in greater horseshoe bats from Khosta 1 cave is 62.5% (15/24 faeces samples). However, the authors did not perform any statistical testing to investigate predictors for CoV detection rate44. Our study included many more collection sites (Fig. 1a), as we aimed to collect samples from different parts of four studied regions. This pilot study creates a foundation for future studies of CoVs in bats in the European part of Southern Russia. For example, five collection sites where we detected bat CoVs (Fig. 1b) can be included in the next studies with the inclusion of more animals from these locations. The detection rate of CoVs in bats’ populations strongly depends on the characteristics of colonies included in the study, particularly colony size, reproduction/hibernation status of the animals, number of species in the colony, and the location of the colony. A sampling for future studies of Fore-Caucasus bats’ populations should be done considering not only the results of this study but also these features of bats’ colonies.
NGS data from samples with detected CoV RNA was not sufficient for the detailed characterization of identified CoVs with qPCR. Probably, the concentration of extracted RNA was too low to obtain good quality genomic CoVs’ data suitable for gene annotation and following phylogenetic analysis. We performed three attempts for NGS. The first one included sequencing of shotgun metagenomic libraries consisting of 2 × 150 bp cDNA fragments with NovaSeq (Illumina, USA) generating 800 million paired reads per sample. Taxonomical analysis of raw reads from this run resulted in the identification of several reads similar to the SARS-related CoV records from the NCBI RefSeq database used by Kraken2. However, we did not assemble any contigs that were precisely recognized as any CoV by BLAST or VADR. Next, we tried to perform library preparation targeting SARS-related CoVs by RT-PCR with NEBNext ARTIC SARS-CoV-2 Library Prep Kit (New England Biolabs, England). We hypothesized that the amplification of SARS-related CoVs’ genomic fragments with primer pools from this kit could improve the outcome of obtaining at least a partial genome of detected CoVs, but amplification during library preparation was not successful. Then we decided to use longer cDNA fragments (2 × 300 bp) for the third attempt at a shotgun metagenomic library preparation and sequence it with the generation of 40 million paired reads per sample. Compared to the first run there were more identified reads similar to Coronaviridae records in the obtained raw reads. However, this did not result in the assembly of useful contigs. We should acknowledge that according to BLAST search in NCBI Viruses database there are some records of SARS-related CoV and HCoV-OC43 that share 89–100% identity with part of assembled contigs, but they also are similar to other even non-viral records as their length does not allow for proper alignment during BLAST search. According to the taxonomical identification of raw and assembled metagenomic data, we assume that we identified betaCoVs, which are not SARS-related, as otherwise parts of the SARS-like CoVs would be amplified during library preparation targeting their specific regions.
Babiker et al. performed screening of SARS-CoV-2 and other respiratory viruses in human samples with a similar study design, including RT-PCR screening following metagenomic NGS of positive samples with MiSeq platform and similar sequencing depth (42 million reads per sample)45. In all cases, they detected the same viruses with metagenomic NGS as were detected with RT-PCR. The number of minimum SARS-CoV-2 reads from NGS data per one sample detected with KraenUniq was 27, which allowed Babiker et al. to assemble at least 6 genome regions from the sample with the least detected SARS-CoV-2 reads45. In our study, we detected reads of various CoV species (Supplementary File S5), and we suggest that the number of reads per sample from NovaSeq and MiSeq data was relatively low for the proper CoV genomes assembly. Crook et al. also performed a search of CoVs in bats, however, they only used metagenomic NGS of 2x150 cDNA fragments with MiSeq and longer cDNA fragments with Oxford nanopore GridION (Oxford Nanopore Technologies, Great Britain)46. The largest single contig identified as part of the CoV genome and assembled from MiSeq data was ~ 7 kb, while in our study the largest contig was ~ 1.2 kb. This also could confirm that the RNA concentration could affect the results of metagenomic NGS. However, we cannot compare the RNA concentration obtained in our work and in Crook et al. study, since the authors did not provide any relevant data on this. One of the possible ways for studying CoVs phylogenetics is amplifying long cDNA fragments with PCR following capillary sequencing, as described in Lelli et al.13 and Watanabe et al.30 studies. We should mention that this approach is also not the best option, as some CoVs can be not detected with PCR. For example, Kohl et al. reported that in their study, metagenomic NGS identified bat viruses that PCR did not detect47.
This study has some limitations, as we used only oropharyngeal and rectal swabs, which probably resulted in low RNA concentration and the impossibility of detected CoV characterization based on metagenomic NGS data due to this. Some other studies also include tissue, urine, faecal samples, and even exoparasites for CoV screening32,34,35,48. We did not collect tissue biopsies or urine from bats to prevent keeping animals in captivity for sampling. Faecal samples were not collected as it is impossible to predict bat species origin in some cases, contributing to some uncertainties during biostatistical analysis. However, future studies should include not only swab sampling, but also tissues and faeces to increase the RNA concentration needed for various sequencing studies. Also, our future studies should include not only samples of different origins, but also some other approaches, such as capillary sequencing of amplified CoV genes. Metagenomic NGS should be used as a main supplementary method to assist in characterizing previously unknown CoV genomes, that could not be done with PCR.
Conclusions
This study provides the first data on CoVs detection, with a mean overall prevalence of 3.33% (95% CI 1.1–7.6%). Although these results correspond to similar studies conducted in other regions and countries, further studies are required to investigate the reservoir of CoVs in bats and other animals, and to be able to predict a possible next pandemic. Metagenomic NGS did not provide sufficient data for detailed CoV characterization, but in raw NGS data of all qPCR-positive samples, betaCoV reads were detected with a taxonomical identification tool, which supports the results of the qPCR screening.
Methods
Sampling
A total of 300 probes from 150 bats of seven species (oropharynx and rectal swabs from each animal) were collected in Rostov region, Adygea Republic, Krasnodar Krai, and Stavropol Krai from March 2021 to June 2021. The following species of bats were included in the study: common noctule (Nyctalus noctula), Kuhl's pipistrelle (Pipistrellus kuhlii), serotine bat (Eptesicus serotinus), lesser horseshoe bat (Rhinolophus hipposideros), greater horseshoe bat (Rhinolophus ferrumequinum), Daubenton's myotis (Myotis daubentonii) and western barbastelle (Barbastella barbastellus). Identification of bat species was performed by a trained zoologist-taxonomist in the location site of sampling. The characterization of animals included in the study is represented in Table 3.
Sampling was performed with sterile swabs with the 2.5 mm diameter of the collecting part (Biomedical Innovations LLC, Russia). The probe was placed in a sterile 2 ml tube filled with a transport medium for virus-containing samples TVM (Biomedical Innovations LLC, Russia). After the procedure, each animal was released within the range they were caught to exclude the negative impact on the environment. Animals were not kept in captivity. Tubes were delivered to the laboratory within 24 h after sampling.
RNA extraction
RNA extraction from liquid probe samples was carried out using the QIAamp Viral RNA mini kit (Qiagen, Germany) according to the manufacturer's protocol. Extracted RNA was divided into aliquots for quality and concentration studies using fluorimetry using the Qubit RNA HS Assay Kit (Thermo-Fisher Scientific, USA), DNase processing and reverse transcription, and the preparation of negative reverse transcription controls. Reverse transcription was performed using the MMLV RT kit (Evrogen, Russia) according to the manufacturer's protocol for liquid virus-containing samples using random decanucleotide primers.
Primers and qPCR
For qPCR screening we used pan-coronavirus primers with sequences: Cor-FW (5′-ACWCARHTVAAYYTNAARTAYGC-3′) and Cor-RV (5′-TCRCAYTTDGGRTARTCCCA-3′) as described by Vijgen et al.29. The synthesis of oligonucleotides, including degenerate primers, was carried out by Evrogen. qPCR reactions were performed using hot-start PCR mixture qPCRmix-HS and SYBR Green dye (Evrogen, Russia) on a QuantStudio 5 amplifier (Thermo-Fisher Scientific, USA). Optimization of the reaction parameters was carried out according to the following parameters: serial dilution of the matrix (reaction efficiency analysis), annealing temperature, annealing time, elongation temperature, elongation time. The optimal annealing temperature was selected using gradient PCR using the capabilities of the Veriflex block of a QuantStudio 5 cycler (Thermo-Fisher Scientific, USA). The melting curve analysis was carried out in the cases of all PCR settings. The PCR program was as follows: 1 cycle of 5 min at 95 °C, 54 cycles of 15 s at 94 °C, 30 s at 44 °C, 30 s at 60 °C, 1 cycle of melting assay − 0.15 °C increment starting at 60 °C and up to 95 °C. Complete negative control qPCR reactions were performed using DEPC water as a matrix with the main reactions. The synthesized sequence corresponding to 251 target nucleotides of entries EF584908.1 (Asian leopard cat coronavirus), DQ249235.1 (Bat coronavirus HKU2), and AF304460.1 (Human coronavirus 229E) were used as the reaction/primers optimization matrix and positive controls in the subsequent reactions. The sequence was synthesized by Evrogen and diluted to 1 ng/μL to serve as the template. The cutoff values were determined automatically by QuantStudio software. The analysis of PCR data was carried out in the Design and Analysis environment (Thermo-Fisher Scientific, USA). The qPCR screening was performed in three independent replicates.
Statistical analysis
Data for RNA concentration was not normally distributed according to the Kolmogorov–Smirnov test. To compare mean RNA concentration in probes of different swab origin unpaired U-test was performed. Pearson χ2 tests to explore the effect of (i) bat species, (ii) swab origin, (iii) sex, and (iv) sample collection site on the detection of CoV RNA were performed. For predictors calculations, simple logistic regression was performed. For calculation of confidence intervals for binomial proportions, the Clopper–Pearson interval was used. Data were presented as mean and 95% CI where ap-propriate. Differences were considered statistically significant when p < 0.05.
In silico analysis of pan-coronavirus assay
The sensitivity (% of target organisms detected) and specificity (% of non-target organisms detected) of primes were tested using in silico PCR approach49. From the degenerate primers pairs from Vijgen et al.29, Lelli et al.13, and Watanabe et al.30, all possible pairs of primers without ambiguous bases were generated using a custom R script (R v4.1.0, R Foundation for Statistical Computing, Vienna, Austria). Each generated primer pair was tested against a search database with the "primersearch" function from "EMBOSS" package50. The maximum number of allowed mismatches was set to two per primer. Two search databases were compiled from genome sequences available in the NCBI refSeq database (15 November 2022). Available genome sequences from the Coronaviridae family (68 in total) were used to test sensitivity, all available virus genomes (12026 in total including the Coronaviridae) were used to test specificity. Whole genomes from the Coronaviridae family were aligned with MAFFT51, and the phylogenetic tree was constructed using FastTree52 and visualized with "ggtree" package53.
NGS of samples with detected CoV RNA and bioinformatical analysis of obtained data
RNA extraction was carried out using the QIAamp Viral RNA mini kit (Qiagen, Germany) according to the manufacturer's protocol. Single-strand cDNA synthesis was performed with reverse transcriptase M-MuLV–RH (Biolabmix, Russia), and double-strand cDNA synthesis was carried out using Klenow fragment (SibEnzyme, Russia). Library preparation was performed using three different aliquots from one sample with NEBNext Ultra II DNA Library Prep Kit for Illumina (New England Biolabs, England), NEBNext ARTIC SARS-CoV-2 Library Prep Kit (New England Biolabs, England), and LIB Display kit (DNA Display, Russia) according to the manufacturers’ protocols. The concentration of cDNA in the fifth sample aliquot was not sufficient for library preparation with NEBNext Ultra II DNA Library Prep Kit (New England Biolabs, England). We used Illumina NovaSeq 6000 (Illumina, USA) for NGS with the generation of 800 million paired reads per sample of the library prepared with NEBNext Ultra II DNA Library Prep Kit and consisting of 2 × 150 bp cDNA fragments and Illumina MiSeq (Illumina, USA) for NGS with the generation of 40 million paired reads per sample of the library prepared with LIB Display kit consisting of 2 × 300 bp cDNA fragments. Graphical representation of molecular studies is shown in Fig. 3.

Flow-chart representing conducted molecular studies.
Quality control, filtering, and trimming of obtained FASTQ files was performed with fastp (version 0.23.2) software54. Taxonomical identification of raw reads was conducted with Kraken2 (version 2.1.2) using the NCBI RefSeq database as a reference55. Visualisation of taxonomical identification of metagenomics data was performed with Krona (version 2.8.1)56. SPAdes (version 3.15.4)57 and MEGAHIT (version 1.2.9)58 were used for de novo assembly of metagenomes. Binning of obtained contigs was performed with the abovementioned tools for taxonomical identification and seqtk tool (version 1.3)59. We used BLAST60 and NCBI Virus database61 for search of similar viral nucleotide sequences and VADR62 for validation and gene annotation of assembled contigs.
Ethical approval
The study was approved by the ethics committee of the Don State Technical University, Rostov-on-Don, Russia (protocol number 67-43-4). Experimental procedures for this report did not include any in vivo studies. ARRIVE guidelines: not applicable63. The collection of probes was carried out according to the Sanitary and Epidemiological Regulations SP 3.2.1288-03. All studies were conducted according to the protocols recommended by the manufacturers of used kits, devices and software. All procedures were performed in accordance with relevant guidelines' in the manuscript.
Data availability
All reference data used for this study are retrieved from NCBI open database and are in open access. Supplementary File S1 contains a figure representing RNA concentration in oropharynx and rectal swabs from bats. Supplementary File S2 contains a database of studied bat species, bat sex, regions where sampling was done, and CoV RNA detection rate. Supplementary File S3 contains reference names of used CoV genomes. Supplementary File S4 contains the results of gel electrophoresis of cDNA fragments prepared with ARTIC SARS-CoV-2 Library Prep Kit. Supplementary File S5 contains the results of the taxonomical identification of raw reads. Supplementary File S6 contains assembled contigs and results of BLAST search in the NCBI Virus database (ssRNA(–) records). Raw metagenomic sequencing data are available in Sequence Read Archive (SRA) under accession numbers SRR22460268, SRR22460267, SRR22460266, SRR22460265, SRR22460264, SRR22460263, SRR22460271, SRR22460270, SRR22460269.
References
Ji, W., Wang, W., Zhao, X., Zai, J. & Li, X. Cross-species transmission of the newly identified coronavirus 2019-nCoV. J. Med. Virol. 92, 433–440. https://doi.org/10.1002/jmv.25682 (2020).
Lu, R. et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 395, 565–574. https://doi.org/10.1016/s0140-6736(20)30251-8 (2020).
Mohamadian, M. et al. COVID-19: Virology, biology and novel laboratory diagnosis. J. Gen. Med. 23, e3303. https://doi.org/10.1002/jgm.3303 (2021).
Donnik, I. M. et al. Coronavirus infections of animals: Future risks to humans. Biol. Bull. 48, 26–37. https://doi.org/10.1134/s1062359021010052 (2021).
Ye, Z.-W. et al. Zoonotic origins of human coronaviruses. Int. J. Biol. Sci. 16, 1686–1697. https://doi.org/10.7150/ijbs.45472 (2020).
Han, H. J. et al. Bats as reservoirs of severe emerging infectious diseases. Vir. Res. 205, 1–6. https://doi.org/10.1016/j.virusres.2015.05.006 (2015).
Serra-Cobo, J. & López-Roig, M. Bats and emerging infections: An ecological and virological puzzle. Adv. Exp. Med. Biol. 972, 35–48. https://doi.org/10.1007/5584_2016_131 (2017).
Banerjee, A. et al. Novel insights into immune systems of bats. Front. Immunol. 11, 26. https://doi.org/10.3389/fimmu.2020.00026 (2020).
Zhang, G. et al. Comparative analysis of bat genomes provides insight into the evolution of flight and immunity. Science 339, 456–460. https://doi.org/10.1126/science.1230835 (2013).
Subudhi, S., Rapin, N. & Misra, V. Immune system modulation and viral persistence in bats: Understanding viral spillover. Viruses 11, 192. https://doi.org/10.3390/v11020192 (2019).
Popov, I. V. et al. Gut microbiota of bats: Pro-mutagenic properties and possible frontiers in preventing emerging disease. Sci. Rep. 11, 21075. https://doi.org/10.1038/s41598-021-00604-z (2021).
Balboni, A., Gallina, L., Palladini, A., Prosperi, S. & Battilani, M. A real-time PCR assay for bat SARS-like coronavirus detection and its application to italian greater horseshoe bat faecal sample surveys. Sci. World J. 2012, 1–8. https://doi.org/10.1100/2012/989514 (2012).
Lelli, D. et al. Detection of coronaviruses in bats of various species in Italy. Viruses 5, 2679–2689. https://doi.org/10.3390/v5112679 (2013).
Fischer, K. et al. Insectivorous bats carry host specific astroviruses and coronaviruses across different regions in Germany. Infect. Genet. Evol. 37, 108–116. https://doi.org/10.1016/j.meegid.2015.11.010 (2016).
Goffard, A. et al. Alphacoronaviruses detected in french bats are phylogeographically linked to coronaviruses of European bats. Viruses 7, 6279–6290. https://doi.org/10.3390/v7122937 (2015).
Ar Gouilh, M. et al. SARS-CoV related Betacoronavirus and diverse Alphacoronavirus members found in western old-world. Virology 517, 88–97. https://doi.org/10.1016/j.virol.2018.01.014 (2018).
August, T. A., Mathews, F. & Nunn, M. A. Alphacoronavirus detected in bats in the United Kingdom. Vector Borne Zoonotic Dis. 12, 530–533. https://doi.org/10.1089/vbz.2011.0829 (2012).
Rihtarič, D., Hostnik, P., Steyer, A., Grom, J. & Toplak, I. Identification of SARS-like coronaviruses in horseshoe bats (Rhinolophus hipposideros) in Slovenia. Arch. Virol. 155, 507–514. https://doi.org/10.1007/s00705-010-0612-5 (2010).
Reusken, C. B. et al. Circulation of group 2 coronaviruses in a bat species common to urban areas in Western Europe. Vector Borne Zoonotic Dis. 10, 785–791. https://doi.org/10.1089/vbz.2009.0173 (2010).
Pauly, M. et al. Novel alphacoronaviruses and paramyxoviruses cocirculate with type 1 and severe acute respiratory system (SARS)-related betacoronaviruses in synanthropic bats of Luxembourg. Appl. Environ. Microbiol. 83, e01326-e1417. https://doi.org/10.1128/aem.01326-17 (2017).
Lazov, C. M. et al. Detection and characterization of distinct alphacoronaviruses in five different bat species in Denmark. Viruses 10, 486. https://doi.org/10.3390/v10090486 (2018).
Lo, V. T. et al. Long-term surveillance of bat coronaviruses in Korea: Diversity and distribution pattern. Transbound. Emerg. Dis. 67, 2839–2848. https://doi.org/10.1111/tbed.13653 (2020).
Li, W. et al. Bats are natural reservoirs of SARS-like coronaviruses. Science 310, 676–679. https://doi.org/10.1126/science.1118391 (2005).
Han, H.-J. et al. Novel coronaviruses, astroviruses, adenoviruses and circoviruses in insectivorous bats from northern China. Zoonoses Public Health. 64, 636–646. https://doi.org/10.1111/zph.12358 (2017).
Zhou, P. et al. Fatal swine acute diarrhoea syndrome caused by an HKU2-related coronavirus of bat origin. Nature 556, 255–258. https://doi.org/10.1038/s41586-018-0010-9 (2018).
Ng, O.-W. & Tan, Y.-J. Understanding bat SARS-like coronaviruses for the preparation of future coronavirus outbreaks—Implications for coronavirus vaccine development. Hum. Vaccin. Immunother. 13, 186–189. https://doi.org/10.1080/21645515.2016.1228500 (2017).
Reusken, C. B., Raj, V. S., Koopmans, M. P. & Haagmans, B. L. Cross host transmission in the emergence of MERS coronavirus. Curr. Opin. Virol. 16, 55–62. https://doi.org/10.1016/j.coviro.2016.01.004 (2016).
Hu, B., Ge, X., Wang, L.-F. & Shi, Z. Bat origin of human coronaviruses. Vir. J. 12, 221. https://doi.org/10.1186/s12985-015-0422-1 (2015).
Vijgen, L., Moës, E., Keyaerts, E., Li, S. & Van Ranst, M. A pancoronavirus RT-PCR assay for detection of all known coronaviruses. Methods Mol. Biol. 454, 3–12. https://doi.org/10.1007/978-1-59745-181-9_1 (2008).
Watanabe, S. et al. Bat coronaviruses and experimental infection of bats, the Philippines. Emerg. Infect. Dis. 8, 1217–1223. https://doi.org/10.3201/eid1608.100208 (2010).
Bonilla-Aldana, D. K. et al. Molecular and serological prevalence of Coronavirus in Chiropterans: A systematic review with meta-analysis. Infez. Med. 29, 181–190 (2021).
Mendenhall, I. H. et al. Identification of a Lineage D Betacoronavirus in Cave Nectar Bats (Eonycteris spelaea) in Singapore and an overview of Lineage D Reservoir Ecology in SE Asian Bats. Transbound. Emerg. Dis. 64, 1790–1800. https://doi.org/10.1111/tbed.12568 (2017).
Cappelle, J. et al. Longitudinal monitoring in Cambodia suggests higher circulation of alpha and betacoronaviruses in juvenile and immature bats of three species. Sci. Rep. 11, 24145. https://doi.org/10.1038/s41598-021-03169-z (2021).
Xu, L. et al. Detection and characterization of diverse alpha- and betacoronaviruses from bats in China. Virol. Sin. 2016(31), 69–77. https://doi.org/10.1007/s12250-016-3727-3 (2016).
Corman, V. M. et al. Highly diversified coronaviruses in neotropical bats. J. Gen. Virol. 94, 1984–1994. https://doi.org/10.1099/vir.0.054841-0 (2013).
Ge, X. Y. et al. Coexistence of multiple coronaviruses in several bat colonies in an abandoned mineshaft. Virol. Sin. 31, 31–40. https://doi.org/10.1007/s12250-016-3713-9 (2016).
Tsuda, S. et al. Genomic and serological detection of bat coronavirus from bats in the Philippines. Arch. Virol. 157, 2349–2355. https://doi.org/10.1007/s00705-012-1410-z (2012).
De Benedictis, P. et al. Alpha and lineage C betaCoV infections in Italian bats. Virus Genes 48, 366–371. https://doi.org/10.1007/s11262-013-1008-x (2014).
Karagulov, A. I. et al. The prevalence of viral pathogens among bats in Kazakhstan. Viruses 14, 2743. https://doi.org/10.3390/v14122743 (2022).
Do, H. Q. et al. Genomic characterization of a novel alphacoronavirus isolated from bats, Korea, 2020. Viruses 13, 2041. https://doi.org/10.3390/v13102041 (2021).
Erlichster, M., Chana, G., Zantomio, D., Goudey, B. & Skafidas, E. Pan-family assays for rapid viral screening: Reducing delays in public health responses during pandemics. Clin. Infect. Dis. 73, e3047–e3052. https://doi.org/10.1093/cid/ciaa1028 (2021).
Uhteg, K., Carroll, K. C. & Mostafa, H. H. Coronavirus detection in the clinical microbiology laboratory: Are we ready for identifying and diagnosing a novel virus?. Clin. Lab. Med. 40, 459–472. https://doi.org/10.1016/j.cll.2020.08.004 (2020).
Petrie, A. & Sabin, C. Medical Statistics at a Glance, 4th ed. (ed. Petrie, A. & Sabin, C.) 47–49 (Wiley-Blackwell, 2019).
Alkhovsky, S. et al. SARS-like coronaviruses in horseshoe bats (Rhinolophus spp.) in Russia, 2020. Viruses 14, 113. https://doi.org/10.3390/v14010113 (2022).
Babiker, A. et al. Metagenomic sequencing to detect respiratory viruses in persons under investigation for COVID-19. J. Clin. Microbiol. 59, e02142-e2220. https://doi.org/10.1128/JCM.02142-20 (2020).
Crook, J. M. et al. Metagenomic identification of a new sarbecovirus from horseshoe bats in Europe. Sci. Rep. 11(1), 14723. https://doi.org/10.1038/s41598-021-94011-z (2021).
Kohl, C. et al. The virome of German bats: Comparing virus discovery approaches. Sci. Rep. 11, 7430. https://doi.org/10.1038/s41598-021-86435-4 (2021).
Speranskaya, A. S. et al. Identification and genetic characterization of MERS-related coronavirus isolated from Nathusius’ pipistrelle (Pipistrellus nathusii) near Zvenigorod (Moscow region, Russia). bioRxiv. https://doi.org/10.1101/2022.06.09.495421 (2022).
Henriques, A., Cereija, T., Machado, A. & Cerca, N. In silico vs in vitro analysis of primer specificity for the detection of Gardnerella vaginalis, Atopobium vaginae and Lactobacillus spp. BMC Res. Notes. 5, 637. https://doi.org/10.1186/1756-0500-5-637 (2012).
Rice, P., Longden, I. & Bleasby, A. EMBOSS: The European molecular biology open software suite. Trends Genet. 16, 276–277. https://doi.org/10.1016/s0168-9525(00)02024-2 (2000).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. https://doi.org/10.1093/molbev/mst010 (2013).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree: Computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 26, 1641–1650. https://doi.org/10.1093/molbev/msp077 (2009).
Yu, G. Using ggtree to visualize data on tree-like structures. Curr. Protoc. Bioinform. 69, e96. https://doi.org/10.1002/cpbi.96 (2020).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. https://doi.org/10.1093/bioinformatics/bty560 (2018).
Wood, D. E., Lu, J. & Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 20, 257 (2019).
Ondov, B. D., Bergman, N. H. & Phillippy, A. M. Interactive metagenomic visualization in a Web browser. BMC Bioinform. 12, 385. https://doi.org/10.1186/1471-2105-12-385 (2011).
Antipov, D., Raiko, M., Lapidus, A. & Pevzner, P. A. Metaviral SPAdes: Assembly of viruses from metagenomic data. Bioinformatics 36, 4126–4129. https://doi.org/10.1093/bioinformatics/btaa490 (2020).
Li, D., Liu, C. M., Luo, R., Sadakane, K. & Lam, T. W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676. https://doi.org/10.1093/bioinformatics/btv033 (2015).
Li, H. Seqtk: "A toolkit for processing sequences in FASTA/Q formats". https://github.com/lh3/seqtk (GitHub, year 2022).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410. https://doi.org/10.1016/S0022-2836(05)80360-2 (1990).
Hatcher, E. L. et al. Virus variation resource—Improved response to emergent viral outbreaks. Nucleic Acids Res. 45, D482–D490. https://doi.org/10.1093/nar/gkw1065 (2017).
Schäffer, A. A. et al. VADR: Validation and annotation of virus sequence submissions to GenBank. BMC Bioinform. 21, 211. https://doi.org/10.1186/s12859-020-3537-3 (2020).
Percie du Sert, N. et al. The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research. PLoS Biol. 18, e3000410. https://doi.org/10.1371/journal.pbio.3000410 (2020).
Acknowledgements
Ig.V.P., P.V.Z., A.V.M., M.L.C., and A.M.E. were supported by Russian Foundation for Basic Research (research project № 20-04-60263). This research has been made possible with the support of the Dutch Province of Limburg with a grant to the Centre for Healthy Eating & Food Innovation (HEFI) of Maastricht University—campus Venlo. In silico and statistical analyses were conducted during the Ig.V.P. internship in Maastricht University—Campus Venlo started on 7 November 2021 under the supervision of K.V. and A.U. O.V.O was supported by the Ministry of Science and Higher Education of the Russian Federation (agreement №075-15-2019-1665). A.Y.A. was supported by the Russian Science Foundation (Grant № 22-24-00617).
Author information
Authors and Affiliations
Contributions
Ig.V.P., M.L.C., and A.M.E. planned the research and contributed to the initial study design. A.V.M. obtained swab samples. P.V.Z., A.A.B., and A.K.L. conducted qPCR studies. Il.V.P., T.A.L., and V.N.B. performed the statistical analysis. A.U., Ig.V.P., and K.V. conducted in silico study. I.M.D., A.Y.A., and A.M.S. planned the NGS experiments. K.A.S. and N.A.D. performed library preparations. O.V.O. and I.A.S. conducted NGS. I.V.P. and A.U. performed the bioinformatical analysis of metagenomic NGS data. A.M.E. and K.V. acquired funding. Ig.V.P., P.V.L., and A.U. wrote the draft of the manuscript. A.M.E., M.L.C., and K.V. revised the manuscript. All authors reviewed the final variant of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Popov, I.V., Ohlopkova, O.V., Donnik, I.M. et al. Detection of coronaviruses in insectivorous bats of Fore-Caucasus, 2021. Sci Rep 13, 2306 (2023). https://doi.org/10.1038/s41598-023-29099-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-29099-6
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
