
Abstract
Magnetic resonance imaging (MRI) and magnetic resonance spectroscopy (MRS) have made great successes in clinical diagnosis, medical research, and neurological science. MRI provides high resolution anatomical images of tissues/organs, and MRS provides information of the functional molecules related to a specific tissue/organ. However, it is difficult for classic MRI/MRS to selectively image/probe a specific metabolite molecule other than the water or fat in tissues/organs. This greatly limits their applications on the study of the molecular mechanism(s) of metabolism and disease. Herein, we report a series of molecularly targeted MRI/MRS methods to target specific molecules. The optimal control method was used to efficiently prepare the singlet spin orders of varied multi-spin systems and in turn greatly expand the choice of the targeted molecules in the molecularly targeted MRI/MRS. Several molecules, such as N-acetyl-l-aspartic acid (NAA), dopamine (DA), and a tripeptide (alanine-glycine-glycine, AGG), have been used as targeted molecules for molecularly targeted MRI and MRS. We show in vivo NAA-targeted 1H MRS spectrum of a human brain. The high-resolution signal of NAA suggests a promising way to study important issues in molecular biology at the molecular level, e.g., measuring the local pH value of tissue in vivo, demonstrating the high potential of such methods in medicine.
Similar content being viewed by others

Introduction
Magnetic resonance imaging (MRI) has been widely used in clinical diagnosis, biomedical research, and neurological science1,2,3. Most MRI techniques essentially map the spatial locations/distribution of 1H spins of water and fat in organs4. Although it can image anatomy in vivo with high spatial resolution, conventional MRI techniques lack “chemical resolution” in discriminating metabolites. That is, it is difficult for MRI to selectively image/probe a specific metabolite molecule other than water and fat1. Complementary to MRI, magnetic resonance spectroscopy (MRS) can directly probe different metabolites1,2,5. A routine MRS procedure includes the acquisition of MR images and the corresponding MR spectra. Here, MR images are used to indicate the spatial location of the MR spectra. MRS can help explain the molecular mechanism of metabolism and disease and in turn facilitate accurate diagnoses, recognition, and therapy6,7,8,9. In clinical applications, MRS can detect diseases much earlier than anatomic MRI because abnormal metabolism often occurs long before the resulting changes in tissue/organ morphology10,11. MRS signals are often difficult to analyze and quantify due to signal overlap and a corrupted baseline. This originates from the complexity of the composition of metabolites and the abundance of water in the body5,12. Thus, selective imaging/detection of endogenous molecules in MRI/MRS will greatly facilitate their applications in clinical environments and in brain research13,14,15,16,17.
Many efforts have been made to scan specific molecules in MRI and MRS. For example, exogenous biomarkers or agents have been used to image specific molecules16,17,18,19. By exploiting the chemical exchange process between hydroxyl groups of water and the active hydrogens of the biological substances, Balaban et al., developed chemical exchange dependent saturation transfer (CEST)20,21,22 to indirectly detect biological substances such as amino acids and sugars. In MRS, various techniques, such as MEGA editing by Mescher and Garwood et al.23, double quantum filtered (MQF) MRS23,24,25,26, and magnetic resonance spectroscopic imaging (MRSI) by Brown et al.27, can selectively probe signals from specific biomolecules. Although these techniques can probe the specific molecules, they fail to accurately select a specific molecule for molecularly targeted imaging/detection.
More recently, a nuclear singlet spin state/order28,29,30,31 has been used to select magnetic resonance signals from a specific group and/or molecule in vitro32,33,34,35. For example, DeVience et al.36 demonstrated a method known as ‘Suppression of Undesired Chemicals using Contrast-Enhancing Singlet States (SUCCESS) to create, preserve and measure the target singlet state produces strong contrast relative to undesired spectral components when the spectra of two molecules nearly overlap. Chen et al., propose a singlet-filtered spectroscopy method based on adiabatic passage singlet-order conversion (SFS-APSOC)33,37, which enables targeted detection of the coupled proton groups from extensive components in vitro. Simpson et al. reported a DREAMTIME-NMR technique which provides “a molecular window” to only refocus J-coupled magnetization while all other unwanted signals38. Glöggler et al., demonstrated a general coupling magnetization-to-singlet (gc-M2S) singlet-filtered method34 to distinctly observe the glutamate (Glu) in the hippocampus of a living mouse in vivo. Numerous π hard pulses in the gc-M2S sequence may inevitably bring about the RF power deposition with the risk of biological tissues heating in human brains in vivo.
In this work, we report a series of molecularly targeted MRI/MRS methods based on optimal control (OC) of spin singlet. The OC method33,39 provides the high efficiency and flexibility for systematically imposing desirable constraints and fully exploits the experimentally available high degree of freedom in pulse sequences to design experiments with specific properties which are typically not covered by traditional pulse design procedures. Thus, the OC method is suitable for weakly, strongly, and multiple spin systems. Besides, an optimized control pulse is composed of hundreds of pulse units, which avoids RF power deposition from π hard pulses. Taking advantage of the precise control and high efficient preparation of singlet order by using OC pulses, we demonstrate that it is possible to target the MR signals of the specific molecules in humans in vivo. Several molecules, such as N-acetyl-l-aspartic acid (NAA), dopamine (DA), and a tripeptide (alanine-glycine-glycine), have been used as target molecules in molecularly targeted MRI and MRS. Moreover, the analysis of the in vivo NAA targeted 1H MRS spectrum of a human brain suggests a promising way to study important issues of molecular biology at the molecular level, e.g., measuring the local pH value of tissue in vivo, which is important for one to understand the molecular mechanism of biological processes35,40,41.
Experiments
Materials
All reagents were used as received without further purification including: N-acetyl aspartate (NAA, 99%, Sigma-Aldrich), Dopamine hydrochloride (DA, 98%, Aladdin), a tripeptide (alanine-glycine-glycine, AGG, 98.89%, Nanjing Peptide Biotech Ltd), deuterium oxide (D2O, 99.9%).
Sample preparation
-
1.
Sample 1: The AGG, NAA, and DA aqueous solutions (concentration of each solution sample, 0.08 mol per liter) were prepared by dissolving the solutes into D2O as follows: First, four capillary tubes (1 mm diameter, ~ 0.1 mm thickness), filled with water (80% D2O and 20% H2O), NAA, AGG, and DA aqueous solution, were prepared, respectively. Then the four capillary tubes were carefully inserted into a 5 mm NMR tube that already contains about 500 micro-liter volume water (60% D2O and 40% H2O).
-
2.
Sample 2: Seven samples of the NAA with different pH (the pH values are 4.6, 5.4, 5.8, 6.4, 6.7, 7.4, and 7.6, respectively) were prepared. For each sample, pH was adjusted to the desired value with a calibrated pH meter (Jenway-3510, England) by adding the concentrated NaOH solution (0.05 mol per liter) and/or HCl (0.05 mol per liter).
Subjects
Three healthy volunteers including one 25-year-old female (weight 50 kg, height 150 cm), one 24-year-old male (weight 65 kg, height 177 cm) and one 25-year-old male (Weight 63 kg, height 175 cm), were scanned for the molecularly targeted MRS. All methods were performed according to the guidelines of the Declaration of Helsinki and the current ethical guidelines. This study was approved by the Institutional Ethics Committee of East China Normal University (Shanghai, People’s Republic of China). Written informed consent was obtained from all the study participants prior to their enrolment. Informed consents were obtained from all subjects. All experiments were carried out on a 3T Siemens MAGNETOM Prisma scanner.
Results and discussion
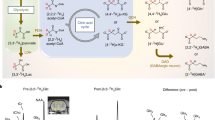
Figure 1a illustrates the scheme of the molecularly targeted MRS/MRI pulse sequences. In the pulse sequence, the Singlet order Filtering method based on an Optimal Control pulse (SFOC) is used to selectively probe the signals of endogenous molecules in vivo. The sequences consist of two parts: One for molecularly targeted signal selection and one for MRI/MRS acquisition. The molecular selectivity of the pulse sequences originates from the preparation of the singlet spin order (SSO). Sequence parameters used in SSO preparation were calculated using chemical shifts and J-couplings of a spin system in the targeted molecule. In principle, any molecule with a spin system suitable for SSO preparation can be used as the target. The efficiency of SSO preparation is related to the design of the pulse sequence. Here, the OC strategy is exploited to prepare SSO. Figure 1b and c show the amplitude modulation and the phase modulation of an OC pulse as an example. Both the amplitude modulation and the phase modulation consist of a series of modulated discrete units, which are optimized to ensure the highest efficiency of SSO preparation. Because SSO is not a single quantum coherence and is not directly observable42, the OC pulse II is required in the pulse sequence in Fig. 1a to transfer the SSO back into longitudinal polarization. MR signal is acquired by the following MRI/MRS sequence block. Figure 1d shows, as an illustration, the spin evolution of a three-spin NAA system (Ha, Hb, and Hb') during the OC pulse I. the chemical shift and J couplings of NAA could be found in Supporting Information Fig. S1. 4000 individual short pulses (total duration: 40 ms) make up the OC pulse. The longitudinal magnetization (the black line) is transferred to the spin order (I1z + 2I2xI3x + 2I2yI3y) with the highest efficiency. Comparing with the reported singlet-filtered methods for signal selection, including gM2S43, SUCCESS36, SISTEM35, the main advantage of the OC pulses lies on the accurate control of the spin dynamics of a multiple spin system, resulting in the high preparation efficiency of the singlet order of the multiple spin system (see Fig. S2). Herein, some important biochemicals, such as AGG, NAA, and DA, have been successfully targeted in the molecularly targeted MRI/MRS. More details about the spin evolution of specific pulse sequence can be found in Supporting Information.

(a) The pulse sequence scheme of molecularly targeted MRI/MRS, namely SFOC-MRS or SFOC-MRI. Here, the solid blue rectangles represent the optimal control pulses (OC pulse I and II) and the decoupling block, respectively. The solid black rectangles, g1 and g2, are the gradient field pulses used for coherence dephasing. The grey rectangle represents the pulse sequence block for MRS/MRI. (b) The amplitude modulation and (c) the phase modulation an OC pulse as an example. (d) The evolution trajectory of a three-spin system under OC pulse I. The spin evolutions of the different spin systems and the specific pulse sequence examples can be found in Supplementary Materials.
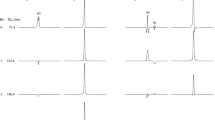
To demonstrate the signal targeting by using the developed pulse sequences, a phantom, i.e., Sample 1 in Fig. 2a, was prepared. Figure 2b shows the routine 1H MRI image of Sample 1 that was acquired with a spin echo imaging sequence. The pulse sequence is referred to Fig. S3. In the routine 1H MRI image of Sample 1, one can see one big grey disk containing four small disks, each surrounded by a black circle representing the wall of the capillary wall. The big grey disk is from the water inside the 5 mm glass tube, whereas the smaller disks are from the capillaries containing HDO water, the NAA, AGG, and DA aqueous solutions, respectively. The difference in signal intensity can be attributed to the difference in 1H concentrations in different solutions. Figure 2c–e show the molecularly targeted 1H MRI image of NAA, AGG, and DA, respectively. These were acquired using SFOC-MRI-11.7T (see Fig. S4). The capillary containing the targeted molecule can be selectively imaged, thus demonstrating the excellent molecular selectivity of SFOC-MRI-11.7T.

(a) A photo of the Sample 1. This sample is a 5 mm NMR glass tube containing water (40% D2O and 60% H2O). Four capillary tubes (1 mm diameter) containing D2O and the NAA, AGG, and DA aqueous solution were carefully set into the 5 mm tube. The (b) spin echo, (c) NAA-targeted, (d) AGG-targeted, and (e) DA-targeted 1H MRI image of the sample. The (f) conventional, (g) NAA-targeted, (h) AGG-targeted, and (i) DA-targeted 1H MRS spectra of the same sample. For the NAA-targeted MRI and MRS, the signals of the CH2 group of NAA (marked by red in the NAA molecular structure in c) were selected. For the AGG-targeted MRI and MRS, the signals of the CH2 group of AGG (marked by red in the AGG molecular structure in d) were selected. For the DA-targeted MRI and MRS, the signals of the two CH groups of DA (marked by red in the DA molecular structure in e) were selected. A 1H spin echo imaging sequence (see Fig. S3) was used to acquire the image in (b). The molecular targeted MRI images of the sample were acquired using SFOC-MRI-11.7T (see Fig. S4). A conventional MRS spectrum was acquired using the spin echo sequence (see Fig. S5). The molecularly targeted MRS spectra of the sample were acquired using SFOC-MRS-11.7T (see Fig. S6). The amplitude and phase of the OC pulses of AGG, NAA and DA used in the experiments are shown in the Figs. S7–S9. More experimental details can be found in Supporting Information. The nutation frequency of Continuous Wave (CW) decoupling pulse in SFOC-MRS-11.7T and SFOC-MRI-11.7T sequences is about 2 kHz (0.1 W for Bruker Triple resonance Broadband Inverse probe) and the duration is 200 ms. The experiment was done at room temperature. All experiments were carried out on a 500 MHz Bruker AVANCE III spectrometer.
Figure 2f–i shows the MRS spectra of the Sample 1. Figure 2f is the conventional 1H MRS spectrum, and Fig. 2g–i are the molecularly targeted MRS spectra acquired using SFOC-MRS-11.7T. The pulse sequence used to acquire conventional 1H MRS spectra can be found in Fig. S5. In the conventional 1H MRS spectrum in Fig. 2f, the signals from the HDO, NAA, AGG, and DA are clearly observable. In the molecularly targeted MRS spectra, i.e., Fig. 2g–i, acquired using SFOC-MRS-11.7T (see Fig. S6), the signals of the targeted molecules are dominant in the spectra. The other signals including the water signal are significantly suppressed, indicating the excellent molecular selectivity of the method. The life-time Ts of the singlet orders for the molecules was experimentally measured, which is much longer than the corresponding spin–lattice relaxation time T1 (see Fig. S10 for the details).
We have also used SFOC-MRS-3T (see Fig. S11) to probe the NAA molecule in human brains in vivo. The OC pulses used in the SFOC-MRS-3T are shown in Fig. S12. Figure 3a and b are the MRI image and a traditional 1H MRS of a human brain, respectively. The region of interest (ROI) where 1H MRS was acquired is marked with a white box in Fig. 3a. The MRS spectrum is featured with severe signal overlapping which can be attributed to the complex biochemical environment in the human brain. The red line in Fig. 3b presents the in vivo NAA-targeted 1H MRS acquired using SFOC-MRS-3T. The monitored spin system of NAA consists of Ha, Hb, and Hb’ (see cartoon in Fig. 3b). The SSO of NAA (Hb, Hb') was prepared in the three-spin system, and the signals of Hb and Hb' in the CH2 group were selected afterward. Given the maximum achievable B1 field strength and limitations on specific absorption rate (RF energy deposition), the decoupling pulse partially reserved the 1H signals of CH3 (20% routine 1H spectrum). The signals of endogenous NAA molecule (Hb and Hb’) in NAA targeted 1H MRS spectrum have been observed distinctly in vivo while the signals of biomolecules such as lipids, Glutamate (Glu), Glutamine (Gln), γ-Aminobutyric acid (GABA), total Choline (choline-containing compounds, tCho) and total Creation (sum of creatine and phosphocreatine, tCr) are significantly suppressed. Analyzing the numerical integration shows that the signal intensity of NAA-CH2 in the NAA-targeted spectrum is ⁓ 30% of the signal intensities in the routine MRS spectrum, and the signal intensities of tCr and tCho are less than 5% of the original signal. The pulse sequence has been applied on three healthy subjects and the yielded spectra are quite similar, demonstrating the feasibility of SFOC-MRS-3T (see Fig. S13). Note that the J-coupling between the two 1H spin pairs, i.e., (Ha, Hb), and (Ha, Hb’), can be easily measured via the signal splitting in the spectrum. The accurate measurement of the NAA signals (i.e., the chemical shifts) provides a way to probe the pH values of the local environment of NAA, which will be discussed in detail later.

(a) Routine MRI images showing the voxel position in the human brain. The following 1H MRS spectra were acquired in this voxel with a size of 20 × 20 × 20 mm3; (b) the routine 1H MRS spectrum (black) and the NAA targeted 1H MRS spectrum (red) acquired by using SFOC-MRS-3T (see Fig. S11). The amplitude and phase of the OC pulses of NAA in the experiments are shown in the Fig. S12. The scan time of 64 scan accumulation in NAA-targeted experiments is about 3.2 min. The nutation frequency of the decoupling pulse in SFOC-MRS-3T sequence is 200 Hz (about 200 W for quadrature body RF coil) and the duration is 5 ms. More experimental details can be found in Supporting Information. The experiments were carried out on the Siemens 3T Prisma MRI scanner.
Selectively probing endogenous biomolecules with MRI/MRS can have many promising applications in molecular biology and medicine. For example, it may be an easy way to measure pH. In 1H NMR, the chemical shift of a molecule often depends on the pH of the local environment44. Probing the signal of a pH sensitive molecule in vivo can thus measure the local pH values in vivo. Figure 4a shows the 1H MRS spectra of the NAA samples with different pH values. To facilitate this comparison, the in vivo 1H MRS spectrum of the human brain is also shown in Fig. 4a (grey line). Clearly, the chemical shift difference (δ) between the rightmost peak of the Hb’ signals and the CH3 peak changes upon the pH value variation. The 1H MRS spectrum of the human brain nicely matches that from the NAA aqueous solution with a pH value of 7.4. Therefore, the NAA-targeted 1H MRS provides an easy way to probe the pH in human brains in vivo.

(a) The 1H MRS spectra of NAA aqueous solution containing buffers at the various pH values (black, purple, blue, and green lines) and the NAA-targeted 1H MRS spectrum of a human brain (the grey line); (b) the plot of the frequency difference between the CH3 signal and the first peak of Hb’ signals of NAA (see a), δ, as a function of the pH values. The full width at half maximum (FWHM) of NAA signals in NAA-targeted spectrum is approximately 3 Hz. The error margin of the pH measurement using SFOC-MRS-3T sequence is ~ 0.1 when the frequency uncertainty of the NAA signals is 0.5 Hz (up limit).
We also measured the frequency difference, δ, between the rightmost peak of the Hb’ signals and the CH3 peak of NAA (see Fig. 4a) in the 1H MRS spectra of the NAA aqueous solution samples with different pH values. Figure 4b plots the frequency difference, δ, and the pH values. The 1H MRS spectra of the NAA solution samples having different pH values can be found in Figure S14 in Supporting Information. This plot is a reference curve to measure the local pH values in vivo with NAA-targeted 1H MRS. Note that in this plot, the frequency difference, δ, is not very sensitive to the pH values ranging from 6.8 to 7.5. However, in principle the sensitivity can be easily improved when the external field, B0, increases. Furthermore, this sensitivity can also be improved via alternative pH-sensitive biomolecules. This might lead to a new avenue for the in vivo pH measurement.
It is worthy of noting that B0 and B1 inhomogeneity may have influences on the performance of the OC pulse sequences. We have considered such influences during the design of the OC pulse sequence. Some simulations were performed to study the robustness of the OC pulse sequence upon the B0 and B1 inhomogeneity. Figure S15a shows the frequency offset dependence of the OC pulse. The carrier frequency was centered at the Hb’ signals of NAA and varied between − 7.5 Hz and 7.5 Hz, which is in the range of the typical frequency variation induced by the B0 inhomogeneity in a 3T scanner. The simulation results show that this causes at most 5% loss of the transfer efficiency of SSO in our pulse sequence. Figure S15b shows the B1 amplitudes dependence of the OC pulse. The B1 amplitudes of OC varies from − 10% Amax to 10% Amax. The simulation indicates that this causes at most 10% loss of the transfer efficiency of SSO in our pulse sequence. Based on these results, we thus conclude that B0 and B1 inhomogeneity will not cause significant influences on the performance of the OC pulse sequences in this work.
Conclusion
In this work, based on OC of the singlet order, we developed a series of novel MR methods to implement molecularly targeted MRI and MRS. OC was used to precisely control the evolution of multiple-spin systems, and to prepare the SSOs with high preparation efficiency. Several molecules, including NAA, DA, and AGG, have been used as the targeted probes in the MRI and MRS experiments. Taking advantage of the high efficient preparation of SSOs by using OC pulses, we reported the first in vivo NAA- targeted 1H MRS spectra of a human brain, demonstrating the feasibility of in vivo probing of varied metabolites in humans. It is worthy of noting that the high-resolution molecularly targeted 1H MRS spectrum may provide many important information of the local environment of the targeted molecule. The in vivo probing of the local pH value in Fig. 4 is considered as a primary example. Compared with the other MR approaches for in vivo pH measurement such as hyperpolarized CO213, 31P MRS45 and chemical exchange saturation transfer (CEST)46, the method in this work relies on the signals of the endogenous molecule (i.e., the signals of CH2 of NAA) and may provide a non-invasive and quick way to in vivo measure the local pH value of a living tissue. Moreover, it is expected that the high-resolution targeted 1H MRS spectrum also can provide the information of molecular organization in a specific local environment. For example, we found that the NAA-targeted 1H MRS spectrum of the healthy human brain nicely matches that from the high concentration NAA aqueous solution with a pH value of 7.4. It indicates that, although the total amount of NAA in a human brain might be low, the local concentration of NAA in a tissue most likely is very high.
We thus believe that the schemes developed in this work will have a profound influence on cell/molecular biology, brain science, medicine, pharmacology, medical physics, chemistry, physics, and MRI/NMR methodology; this might inspire many innovative ideas in the study of the chemical and physical processes in the metabolism. For example, magnetic resonance molecular imaging of biomarkers often involves using contrast agent with high specificity. Peptides, antibodies, or small ligands can be used to achieve targeting, but they must be linked to MRI contrast containing Gd or micron-sized particles of iron oxide (MPIO). This complicates the synthesis and can lead to safety concerns. With the selective imaging/probing approach shown in this work, it is possible to image specific endogenous biomarkers, thereby directly eliminating the need for exogenous contrast agents. Even when direct selective imaging of the biomarker is not possible, we can still use external targeting agents without binding them to MRI contrast agents. This can lead to new possibilities for magnetic resonance molecular imaging with implications for metabolism, early diagnosis, and drug development.
Data availability
The datasets used and/or analysed during the current study available from the corresponding author on reasonable request.
References
Gallagher, F. A. An introduction to functional and molecular imaging with MRI. Clin. Radiol. 65, 557–566 (2010).
van der Graaf, M. In vivo magnetic resonance spectroscopy: Basic methodology and clinical applications. Eur. Biophys. J. 39, 527–540 (2010).
Blümich, B. In Essential NMR (eds Gerlind B. & Kawarpal S.) Chapter 4 73–109 (Springer International Publishing, 2019).
Ma, J. Dixon techniques for water and fat imaging. J. Magn. Reson. Imaging 28, 543–558 (2008).
Mullins, P. G. et al. Current practice in the use of MEGA-PRESS spectroscopy for the detection of GABA. Neuroimage 86, 43–52 (2014).
Weissleder, R. & Mahmood, U. Molecular imaging. Radiology 219, 316–333 (2001).
Gambhir, S. S. Molecular imaging of cancer with positron emission tomography. Nat. Rev. Cancer 2, 683–693 (2002).
Massoud, T. F. & Gambhir, S. S. Molecular imaging in living subjects: Seeing fundamental biological processes in a new light. Genes Dev. 17, 545–580 (2003).
Reinfeld, B. I. et al. Cell-programmed nutrient partitioning in the tumour microenvironment. Nature 593, 282–288 (2021).
Ding, F., Zhan, Y., Lu, X. & Sun, Y. Recent advances in near-infrared II fluorophores for multifunctional biomedical imaging. Chem. Sci. 9, 4370–4380 (2018).
Tempany, C. M. & McNeil, B. J. Advances in biomedical imaging. JAMA 285, 562–567 (2001).
Puts, N. A. & Edden, R. A. In vivo magnetic resonance spectroscopy of GABA: A methodological review. Prog. Nucl. Magn. Reson. Spectrosc. 60, 29–41 (2012).
Gallagher, F. A. et al. Magnetic resonance imaging of pH in vivo using hyperpolarized 13C-labelled bicarbonate. Nature 453, 940–943 (2008).
Golman, K., Zandt, R. I., Lerche, M., Pehrson, R. & Ardenkjaer-Larsen, J. H. Metabolic imaging by hyperpolarized 13C magnetic resonance imaging for in vivo tumor diagnosis. Cancer Res. 66, 10855–10860 (2006).
Jasanoff, A. MRI contrast agents for functional molecular imaging of brain activity. Curr. Opin. Neurobiol. 17, 593–600 (2007).
Lee, T., Cai, L. X., Lelyveld, V. S., Hai, A. & Jasanoff, A. Molecular-level functional magnetic resonance imaging of dopaminergic signaling. Science 344, 533–535 (2014).
Viale, A. & Aime, S. Current concepts on hyperpolarized molecules in MRI. Curr. Opin. Neurobiol. 14, 90–96 (2010).
Lee, J. H. et al. Artificially engineered magnetic nanoparticles for ultra-sensitive molecular imaging. Nat. Med. 13, 95–99 (2007).
Weissleder, R. Molecular imaging in cancer. Science 312, 1168–1171 (2006).
Woods, M., Woessner, D. E. & Sherry, A. D. Paramagnetic lanthanide complexes as PARACEST agents for medical imaging. Chem. Soc. Rev. 35, 500–511 (2006).
Kleimaier, D. et al. The cellular heat shock response monitored by chemical exchange saturation transfer MRI. Sci. Rep. 10, 11118 (2020).
Ward, K. M., Aletras, A. H. & Balaban, R. S. A new class of contrast agents for MRI based on proton chemical exchange dependent saturation transfer (CEST). J. Magn. Reson. 143, 79–87 (2000).
Mescher, M., Merkle, H., Kirsch, J., And, M. G. & Gruetter, R. Simultaneous in vivo spectral editing and water suppression. NMR Biomed. 11, 266–272 (1998).
Choi, I. Y., Lee, S. P., Merkle, H. & Shen, J. Single-shot two-echo technique for simultaneous measurement of GABA and creatine in the human brain in vivo. Magn. Reson. Med. 51, 1115–1121 (2004).
McLean, M. A. et al. In vivo GABA+ measurement at 1.5T using a PRESS-localized double quantum filter. Magn. Reson. Med. 48, 233–241 (2002).
Shen, J., Shungu, D. C. & Rothman, D. L. In vivo chemical shift imaging of gamma-aminobutyric acid in the human brain. Magn. Reson. Med. 41, 35–42 (1999).
Brown, T. R., Kincaid, B. M. & Ugurbil, K. NMR chemical shift imaging in three dimensions. Proc. Natl. Acad. Sci. USA 79, 3523–3526 (1982).
Carravetta, M. & Levitt, M. H. Long-lived nuclear spin states in high-field solution NMR. J. Am. Chem. Soc. 126, 6228–6229 (2004).
DeVience, S. J., Walsworth, R. L. & Rosen, M. S. Preparation of nuclear spin singlet states using spin-lock induced crossing. Phys. Rev. Lett. 111, 173002 (2013).
Levitt, M. H. Singlet nuclear magnetic resonance. Annu. Rev. Phys. Chem. 63, 89–105 (2012).
Levitt, M. H. Long live the singlet state!. J. Magn. Reson. 306, 69–74 (2019).
Mamone, S., Rezaei-Ghaleh, N., Opazo, F., Griesinger, C. & Gloggler, S. Singlet-filtered NMR spectroscopy. Sci. Adv. 6, eaaz1955 (2020).
Pravdivtsev, A. N., Kiryutin, A. S., Yurkovskaya, A. V., Vieth, H.-M. & Ivanov, K. L. Robust conversion of singlet spin order in coupled spin-1/2 pairs by adiabatically ramped RF-fields. J. Magn. Reson. 273, 56–64 (2016).
Mamone, S. et al. Localized singlet-filtered MRS in vivo. NMR Biomed. 34, e4400 (2021).
Pravdivtsev, A. N., Sonnichsen, F. D. & Hovener, J. B. In vitro singlet state and zero-quantum encoded magnetic resonance spectroscopy: Illustration with N-acetyl-aspartate. PLoS ONE 15, e0239982 (2020).
Devience, S. J., Walsworth, R. L. & Rosen, M. S. Nuclear spin singlet states as a contrast mechanism for NMR spectroscopy. NMR Biomed. 26, 1204–1212 (2013).
Huang, C. et al. Adaptable singlet-filtered nuclear magnetic resonance spectroscopy for chemical and biological applications. Anal. Chem. 94, 4201–4208 (2022).
Jenne, A. et al. DREAMTIME NMR spectroscopy: Targeted multi-compound selection with improved detection limits. Angew. Chem. Int. Ed. 134, e202110044 (2022).
Khaneja, N., Reiss, T., Kehlet, C., Schulte-Herbruggen, T. & Glaser, S. J. Optimal control of coupled spin dynamics: Design of NMR pulse sequences by gradient ascent algorithms. J. Magn. Reson. 172, 296–305 (2005).
Silberman, A. et al. Acid-induced downregulation of ASS1 contributes to the maintenance of intracellular pH in cancer. Cancer Res. 79, 518–533 (2019).
Galenkamp, K. M. O. et al. Golgi acidification by NHE7 regulates cytosolic pH homeostasis in pancreatic cancer cells. Cancer Discov. 10, 822–835 (2020).
Carravetta, M. & Levitt, M. H. Theory of long-lived nuclear spin states in solution nuclear magnetic resonance. I. Singlet states in low magnetic field. J. Chem. Phys. 122, 214505 (2005).
Bengs, C., Sabba, M., Jerschow, A. & Levitt, M. H. Generalised magnetisation-to-singlet-order transfer in nuclear magnetic resonance. Phys. Chem. Chem. Phys. 22, 9703–9712 (2020).
Vamvakaki, M., Palioura, D., Spyros, A., Armes, S. P. & Anastasiadis, S. H. Dynamic light scattering vs 1H NMR investigation of pH-responsive diblock copolymers in water. Macromolecules 39, 5106–5112 (2006).
Lutz, N. W., Le Fur, Y., Chiche, J., Pouyssegur, J. & Cozzone, P. J. Quantitative in vivo characterization of intracellular and extracellular pH profiles in heterogeneous tumors: A novel method enabling multiparametric pH analysis. Cancer Res. 73, 4616–4628 (2013).
Wu, Y., Soesbe, T. C., Kiefer, G. E., Zhao, P. & Sherry, A. D. A responsive europium(III) chelate that provides a direct readout of pH by MRI. J. Am. Chem. Soc. 132, 14002–14003 (2010).
Acknowledgements
This work was supported by Xing-Fu-Zhi-Hua Foundation of ECNU and Microscale Magnetic Resonance Platform of ECNU.
Author information
Authors and Affiliations
Contributions
Y.-F.Y. conceived the project and designed the experiments. J.-X.X., D.-X.W. Y.-F.Y. developed the pulse sequences. J.-X.X., J.L., R.T. and C.F. translate the pulse sequences for the MRI scanner. J.-X.X., Y.R. and J.W. did the experiments. G. Y., H.Z., J.L., Y.R. and Y.-F.Y. analyzed the MRI and MRS data. J.-X.X., D.-X.W., H.Z. and Y.-F.Y. wrote the manuscript. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xin, JX., Yang, G., Zhang, H. et al. Using optimal controlled singlet spin order to accurately target molecular signal in MRI and MRS. Sci Rep 13, 2212 (2023). https://doi.org/10.1038/s41598-023-28425-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-28425-2
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
