
Abstract
Early life adversity (ELA) tends to accelerate neurobiological ageing, which, in turn, is thought to heighten vulnerability to both major depressive disorder (MDD) and Alzheimer’s disease (AD). The two conditions are putatively related, with MDD representing either a risk factor or early symptom of AD. Given the substantial environmental susceptibility of both disorders, timely identification of their neurocognitive markers could facilitate interventions to prevent clinical onset. To this end, we analysed multimodal data from the Adolescent Brain and Cognitive Development study (ages 9–10 years). To disentangle genetic from correlated genetic-environmental influences, while also probing gene-adversity interactions, we compared adoptees, a group generally exposed to substantial ELA, with children raised by their biological families via genetic risk scores (GRS) from genome-wide association studies. AD and MDD GRSs predicted overlapping and widespread neurodevelopmental alterations associated with superior fluid cognition. Specifically, among adoptees only, greater AD GRS were related to accelerated structural maturation (i.e., cortical thinning) and higher MDD GRS were linked to delayed functional neurodevelopment, as reflected in compensatory brain activation on an inhibitory control task. Our study identifies compensatory mechanisms linked to MDD risk and highlights the potential cognitive benefits of accelerated maturation linked to AD vulnerability in late childhood.
Similar content being viewed by others

Introduction
Early life adversity (ELA; e.g., poverty, maltreatment or neglect by a caregiver) is likely to require significant adaptation and can derail neurodevelopmental trajectories because the molecular brakes designed to facilitate normative maturational changes preserve instead the nocive consequences of ELA1,2,3. Systemic low-grade inflammation is a key mechanism through which ELA hinders optimal brain function by accelerating cellular senescence (e.g., DNA methylation for stress-relevant [serotonergic, glucocorticoid signalling] genes), and thus increases longer-term psychiatric and neurodegenerative risk4,5,6,7.
Among the neuropathologies typified by systemic inflammation and accelerated cellular ageing, Major Depressive Disorder (MDD) and Alzheimer’s Disease (AD) figure prominently as leading causes of disability worldwide8,9,10,11,12,13. Although only modestly genetically related, the two conditions are robustly linked to prior stress exposure, with disrupted synaptic transmission in the prefrontal cortex (PFC) being the alleged substrate of the cognitive control deficits that typify the clinical stage of both AD and MDD5,14,15,16,17,18,19. Recent literature suggests that MDD may be a risk factor or even an early symptom of AD20, which could help shed some light on the more subtle cellular changes which unfold decades before the clinical onset of AD21. Since many of the genetic factors linked to AD are under substantial environmental modulation22, characterisation of their early life neurocognitive correlates, including those shared with MDD and those susceptible to ELA exposure, could facilitate timely detection and identify avenues for intervention to decrease the risk for progression to dementia in older adulthood.
To our knowledge, most research on the brain correlates of AD and/or MDD risk has examined individuals raised by their birth families. These investigations cannot separate genetic from correlated ongoing non-genetic contributions to the observed phenotypes, as genetically vulnerable parents may create familial contexts that could either exacerbate (e.g., through reduced cognitive stimulation) or attenuate (via compensatory behaviours) their offspring’s risk for MDD/AD. Hence, any neurodevelopmental deviations in children at risk for AD/MDD who are raised by their birth families reflect both their own genetic vulnerability and their adjustment to the environment created by parents who may share their vulnerabilities.
To address the confounding effect of genetic effects and rearing environment, we characterised the neurocognitive correlates of genetic vulnerability to MDD/AD in late childhood (9–10 years) by comparing the profiles of adoptees and non-adoptees (i.e., children raised by their biological parents) who participated in the Adolescent Brain and Cognitive Development (ABCD) study. Inclusion of the adoptee group allowed us to (1) separate genetic from correlated gene-environment contributions to brain development, and (2) characterise the neurocognitive correlates of AD/MDD risk in a group exposed to environmental conditions thought to precipitate the onset of both disorders. Indeed, there is considerable evidence that adoptees tend to experience substantial ELA, as attested by both European and North American research23,24,25,26,27,28. For instance, according to current US adoption statistics29, of non step-parent adoptions (such as the majority of those herein investigated), approximately 60% involve children who have spent time in foster care, an experience usually preceded by substantial ELA exposure24,26,30. ELA has been linked to accelerated neurobiological maturation in childhood and adolescence31. While theorised to be an adaptative response to adverse rearing circumstances, which may optimise coping in the short-term32, accelerated maturation is likely to prevent fine-tuning of the slower developing brain circuits relevant to cognitive control, thereby increasing longer-term psychological vulnerability33,34, including risk for AD and MDD, respectively19,20,35.
Genetic risk scores (GRS) derived from genome-wide association studies (GWAS) quantified genetic liability to MDD and sporadic AD, respectively36,37,38. Two AD GRSs, one including only the Apolipoprotein E (APOE) region (i.e., APOE AD GRS) and a second excluding the APOE region (no-APOE AD GRS), were computed considering evidence that the two forecast distinguishable trajectories of neurocognitive impairments and differential susceptibility to environmental factors22,39,40. Specifically, APOE-based risk is associated with deviations in normative brain maturation from infancy onwards and predicts primarily memory-related deficits, stemming from progressive (medial) temporal and posterior parietal atrophy22,41. Complementarily, no-APOE-based risk for AD foreshadows a developmental trajectory of relatively greater deficits in cognitive control, language, and visuospatial processing, arising from a much larger progressive pattern of neurodegeneration which encompasses temporal, frontal, and parietal lobe structures22.
Cognitive control abilities, quantified with a so-called fluid cognition battery, constituted our core mental marker of MDD/AD risk due to their direct relevance to both pathologies, as well as their reported impact on lifespan neurogenetic and cardiovascular trajectories19,42. Because AD and MDD are typified by accelerated brain ageing10, developmental timing, estimated relative to other participants of the same chronological age, was examined as a brain marker for both. Neurodevelopmental timing was quantified with both structural and functional indices considering evidence that ELA impacts them differently43.
Cortical thickness was our index of structural neurodevelopmental timing due to its well-defined maturational trajectory, its liability to genetic control, as well as its susceptibility to ELA44,45,46,47. Functional neurodevelopmental timing was inferred from mean levels (BOLDM) and variability (BOLDSV) in blood oxygenation level dependent (BOLD) functional magnetic resonance imaging (fMRI) responses on a task probing inhibitory (attentional) control. Inhibitory control nears maturation in late childhood and is the cornerstone of optimal mental performance48. In contrast, difficulties with inhibitory control, which tend to be observed among adoptees49, constitute a transdiagnostic contributor to psychopathology50. Our interest in BOLDM stemmed from its relevance to functional maturation, since similar behavioural performance on identical tasks is linked to greater BOLDM in children relative to adults51. Complementarily, our focus on BOLDSV was prompted by its susceptibility to developmental changes and its transdiagnostic involvement in psychopathology and remission following treatment52,53,54,55. To account for mental state-specific effects and their differential relevance to MDD/AD risk56, BOLDSV was assessed during the externally oriented inhibitory control task and during wakeful rest. The latter tends to trigger an internally oriented attentional focus, which is key to MDD, while also evoking mind wandering episodes, which reportedly capture AD-linked deficits in spontaneous cognition57,58,59. Greater BOLDSV during rest, particularly for brain regions implicated in externally oriented processing56, and reduced BOLDSV, operationalised as reduced variability in task-evoked activation during inhibitory control performance, were regarded as indices of greater functional maturation.
Parenting can either accentuate or dampen the impact of ELA and/or genetic vulnerability to psychopathology23,25,60,61,62. Indeed, affective enrichment in childhood, including responsive parenting, can lessen the sequelae of earlier ELA exposure, and adoptive parental warmth reportedly fosters superior cognitive functioning, including inhibitory control63,64,65,66,67,68,69. Consequently, we investigated whether children’s perceptions of parental warmth would moderate the impact of AD and/or MDD genetic risk on neurodevelopmental timing and fluid cognitive abilities.
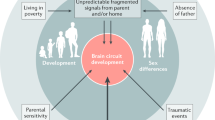
In sum, because accelerated brain ageing typifies both AD and MDD10, we tested whether genetic loading for either disorder is linked to earlier structural and/or functional neurodevelopment in late childhood. The hypothesised accelerated maturation associated with AD/MDD risk was expected to hinder the fine-tuning of the slower developing neurocognitive circuits relevant to inhibitory control33,34, and, thus, in turn, predict poorer fluid cognition (see Fig. 1 for a representation of our model). The inclusion of adoptees allowed us to disentangle gene-environment correlations and characterise “purer” neurocognitive correlates of AD/MDD genetic risk in a group in which their likely substantial ELA exposure may have amplified the impact of genes on vulnerability to accelerated neurobiological ageing31 and stress-linked pathologies, such as AD and MDD19,20,35. Secondly, we probed whether perceptions of high parental warmth would attenuate the adverse effect of MDD/AD risk on neurodevelopment and fluid cognition skills.

Outline of our conceptual model. Genetic risk for AD and/or MDD was predicted to accelerate structural and functional neurodevelopment, as reflected in patterns of cortical thickness and BOLD fMRI signal values during rest and performance of an inhibitory control task, respectively (see “Method” for details). Accelerated structural and/or functional neurodevelopment was expected to be linked to poorer fluid cognition scores. Adoption status and parental warmth were tested as potential moderators of the genetic risk-neurodevelopment-fluid cognition inter-relationships. AD Alzheimer’s Disease, MDD Major Depressive Disorder.
Results
Partial least squares (PLS) results
PLS 1: neurodevelopmental patterns differentiate between genetic risk for AD vs. MDD
The first PLS analysis revealed one significant latent variable (LV) (p = 0.0004), accounting for 53.93% of the GRS-brain data covariance. This LV distinguished AD from MDD GRS-linked brain markers (Fig. 2b) and was most strongly expressed in frontal, insular, parieto-occipital, and temporal areas (see Fig. 2a). Higher AD GRS was associated with greater cortical thinning among adoptees but increasing run 1 to run 2 BOLDSV on the inhibitory control task (i.e., the stop-signal task [SST]) among non-adoptees. Complementarily, in both groups, higher MDD GRS was linked to increasing run 1 to run 2 BOLDSV on the SST, as well as increasing cross-run BOLDM and greater overall BOLDM among adoptees only.

Results of the behavioural-PLS analyses. Panels (b) and (d) show the correlations between the LV brain scores and the GRSs. Error bars are the 95% CIs from the bootstrap procedure. CIs that do not include zero reflect robust correlations between the respective GRS and the brain score in a given condition (i.e., data type) across all participants. Panels (a) and (c) depict the Destrieux ROIs with robust loadings on the LVs in panels (b) and (d), respectively, and visualized with the Freesurfer Surface (https://chrisadamsonmcri.github.io/freesurfer_statsurf_display). In the brain figures in panels (a) and (c), absolute BSR values lower than 3 have been set to zero. Rest BOLDSV = amplitude index of resting state low frequency fluctuations in BOLD signal. SST BOLDM Run 1/2 Up = difference between the GLM-derived run 2 and run 1 betas, based on the Correct Stop > Correct Go contrast. SST BOLDSV Run 1/2 Up = difference between the GLM-derived standard errors associated with the run 2 and run 1 beta, respectively, based on the Correct Stop > Correct Go contrast. SST Run 1/2 BOLDM = average of the GLM-derived run 2 and run 1 betas, based on the Correct Stop > Correct Go contrast. SST Run 1/2 BOLDSV = average of the GLM-derived standard errors associated with the run 2 and run 1 beta, respectively, based on the Correct Stop > Correct Go contrast. LV latent variable, CI confidence interval, LH left hemisphere, RH right hemisphere, AD Alzheimer’s Disease, APOE Apolipoprotein E, MDD Major Depressive Disorder, GRS genetic risk score, SST Stop-Signal Task, GLM general linear model, BOLD blood oxygenation level dependent.
PLS 2: APOE- vs. no-APOE-based genetic vulnerability to AD is linked to distinct neurodevelopmental markers
The second PLS analysis identified a sole significant LV (p = 0.0002), which accounted for 47.18% of the covariance in the GRS-brain data and differentiated among brain markers of genetic risk for MDD, as well as APOE- vs no-APOE-linked vulnerability to AD (Fig. 2d). The associated brain LV was most strongly expressed in frontal, parietal, superior temporal, mid-posterior cingulate and parahippocampal gyri, as well as in occipito-temporal areas (Fig. 2c). The neural markers of MDD GRS comprised the same functional data types as those observed in the first PLS analysis. However, distinguishable brain correlates were observed for the two AD GRSs. Specifically, higher APOE GRS predicted increasing run 1 to run 2 BOLDSV on the SST among non-adoptees, whereas higher no-APOE GRS predicted greater cortical thinning among adoptees.
Supplemental tests
PLS analyses including only White children confirmed that racial differences in genetic architecture and risk loci70,71,72 did not impact our reported results (see Supplemental Materials).
Moderated mediation analyses: genetic risk effects on brain and cognition among adoptees vs. non-adoptees
To investigate whether the PLS-identified neural correlates of AD and MDD risk mediate the genetic vulnerability-fluid cognition links, and whether any observed associations differ by adoption status, we conducted a series of moderated mediation analyses in which adoption status and parental warmth were entered as moderators of the GRS-brain, brain-fluid cognition and GRS-fluid cognition links, whereas crystallised cognition was introduced as a covariate. The predictors, mediators, moderator (parental warmth only) and outcomes of these analyses were residualised by the variables detailed in section “Residualisation for confounding variables” below and standardised separately within the adoptee and non-adoptee group, respectively. Our moderated mediational analyses focused on the GRS-brain associations identified by PLS to be significant in the adoptee group. However, in supplemental analyses, we verified that increasing run 1 to run 2 BOLDSV on the SST did not mediate the link between the full or APOE-based AD GRS and fluid cognition (see Fig. 2b,d).
Composite AD GRS: cortical thickness as a mediator
Our analysis revealed that adoption status (but not parental warmth) partially moderated the indirect effect of AD GRS on fluid cognition via cortical thickness (see Fig. 3). Specifically, among adoptees (but not non-adoptees), higher AD GRS was related to greater cortical thinning (than expected by chronological age), suggestive of accelerated structural neurodevelopment, which, in turn, predicted superior fluid cognition scores among both adoptees and non-adoptees.

Results of the moderated mediation analysis comparing the effect of the composite AD GRS on fluid cognition via cortical thickness among adoptees and non-adoptees. AD Alzheimer’s Disease, GRS genetic risk score. Coefficients in red font describe the adoptees, those in blue font, the biological offspring, whereas those in black font apply to the full sample.
Explained variance in fluid cognition scores
Follow-up correlation-based analyses revealed that the composite AD GRS explained 0.4% (adoptees) and 0.01% (non-adoptees), respectively, whereas cortical thickness explained 0.2% (no statistically significant group difference as per the moderated mediation results above) of the variance in fluid cognition scores.
No-APOE AD GRS: cortical thickness as a mediator
Adoption status, but not parental warmth, partially moderated the link between no-APOE AD risk and fluid cognition via cortical thickness (see Fig. 4). Thus, replicating the results obtained with the composite AD GRS, we found that among adoptees (but not non-adoptees), higher no-APOE-based AD GRS was linked to greater cortical thinning, which, in turn, predicted higher fluid cognition scores among both adoptees and non-adoptees.

Results of the moderated mediation analysis comparing the effect of the no-APOE AD GRS on fluid cognition via cortical thickness among adoptees and non-adoptees. AD Alzheimer’s Disease, APOE Apolipoprotein E, GRS genetic risk score. Coefficients in red font describe the adoptees, those in blue font, the biological offspring, whereas those in black font apply to the full sample.
Explained variance in fluid cognition scores
Follow-up correlation-based analyses revealed that the no-APOE AD GRS explained 0.2% (adoptees) and 0.02% (non-adoptees), respectively, whereas cortical thickness explained 0.2% (no statistically significant group difference as per the moderated mediation results above) of the variance in fluid cognition scores.
MDD GRS: SST BOLDM (average and cross-run increase) and BOLDSV (cross-run increase) as parallel mediators
We observed a moderated mediation effect by adoption status (but not parental warmth) indicating that the indirect effect of MDD GRS on fluid cognition via SST BOLDM was significant among adoptees but not among non-adoptees (see Fig. 5). Specifically, among adoptees only, higher MDD GRS values predicted higher SST BOLDM, which, in turn, was associated with superior fluid cognition scores.

Results of the moderated mediation analysis comparing the effect of the MDD GRS on fluid cognition via average SST BOLD activation among adoptees and non-adoptees. MDD Major Depressive Disorder, GRS genetic risk score, SST Stop-Signal Task, BOLD blood oxygenation level dependent. Coefficients in red font describe the adoptees, those in blue font, the biological offspring, whereas those in black font apply to the full sample.
Explained variance in fluid cognition scores
Follow-up correlation-based analyses revealed that the MDD GRS explained 1% (adoptees) and 0.01% (non-adoptees), respectively, whereas SST BOLDM explained 8.4% (adoptees) and 0.0004% (non-adoptees), respectively, of the variance in fluid cognition scores.
Discussion
The present study provides novel evidence of late childhood neurodevelopmental alterations which are related to genetic vulnerability for AD and MDD, respectively. Contrary to our hypotheses, the observed maturational deviations were linked to superior, rather than poorer, fluid cognitive performance and emerged only among adoptees, a group likely to have experienced substantial ELA26,30. As such, our findings reaffirm the role of stress exposure in both MDD and AD19,20,35, as well as the importance of disentangling correlated gene-environment influences to better characterise the intergenerational transmission of adaptive and pathological functional profiles.
In line with the proposed role of synaptic dysregulation in the PFC as a key contributor to both AD- and MDD-related pathologies19, we report a link between genetic risk for either disorder and developmental alterations in this region. Reinforcing the key role of cognitive control in both pathologies19, we show that the broader maturational brain profiles linked to AD and MDD vulnerability overlap with regions robustly implicated in intentional decision-making, working memory performance and lifespan fluctuations in cognitive flexibility73,74. Furthermore, complementing prior investigations on the relevance of APOE-based AD risk to brain development from infancy onwards41, we demonstrate that no-APOE-based genetic risk accounts for the observed overlap in the neurodevelopmental alterations related to MDD and AD GRSs in late childhood, at least among children not raised by their biological families. As the strongest effects emerged in frontal and parietal regions and were related to performance on a fluid cognition battery, our findings corroborate the documented neurodegenerative and mental (i.e., memory- vs cognitive control-related) profile that distinguishes no-APOE from APOE-based risk22.
It is important to note that the neural patterns linked to AD risk reflected structural developmental deviations, whereas those associated with MDD risk indicated functional alterations. The stronger heritability of structural (relative to functional) brain indices75 raises the possibility that our AD-related findings may reflect genetic influences to a greater extent than our MDD-linked results. For AD, this interpretation is bolstered by the fact that the GRS-accelerated structural neurodevelopment link replicates the genetically influenced brain print of this pathology10, implying that accelerated brain maturation linked to AD vulnerability may yield some cognitive benefits, albeit modest, in late childhood.
As for MDD, the GRS-brain associations were in the opposite direction to those generally documented for this disorder in adulthood8,9,10,12. One possibility is that the brain print of MDD varies by age, with accelerated/decelerated development/ageing both likely to typify this disorder, but at different life stages and yielding distinct functional outcomes. An alternative interpretation, based on the specificity of the GRS-brain-cognition relationships to the adoptees, is that the MDD-related findings reflect compensatory mechanisms arising from an interaction between genetic vulnerability and prior ELA exposure, which tends to be higher in this group26,30. Although adverse life experiences were predicted to accentuate vulnerability to AD and MDD by accelerating brain ageing and interfering with the fine tuning of brain circuits involved in cognitive control19,20,33,34, this may not always be the case. Indeed, there is suggestive evidence that exposure to harsh and unpredictable circumstances, as it plausibly applies to adoptees26,30, can also foster the development of certain cognitive control components76, some of which were captured by the ABCD fluid cognition battery. In this context, the MDD-associated GRS-brain-cognition relationships may be accommodated within the framework of differential susceptibility77, as genetic risk for MDD among adoptees may accentuate responsiveness not only to the adverse, but also to the beneficial (e.g., mental flexibility-promoting76) aspects of their early life environments.
Although our main analyses showed AD GRS-linked structural and MDD GRS-related functional developmental alterations, our supplemental tests using more lenient significance thresholds for the GRS-contributing single nucleotide polymorphisms (SNPs) identified accelerated cortical thinning as a common feature of both AD and MDD genetic vulnerability, thereby replicating the brain print of both disorders10. Taken together, our results highlight the interplay between genetic and environmental (i.e., adversity-related) contributions to MDD-related developmental deviations in function versus structure, some of which appear to play a compensatory role.
Among children raised by their birth parents, the only significant GRS-brain association suggested that greater vulnerability to AD and/or MDD is linked to decreased robustness in neural activity on the inhibitory control task. Although the underpinnings of this effect need to be probed in more depth, it seems plausible that decreased attentional focus may, at least, partially explain our finding, in line with the broadly posited association of poorer cognitive control with both AD and MDD19. It is worth noting that, consonant with prior rodent findings78, among non-adoptees, an overlap in neurodevelopmental alterations was detected for MDD and APOE-, rather than no-APOE-, based AD risk. These results suggest that overlapping maturational deviations linked to MDD and no-APOE-based AD vulnerability may indicate primarily direct genetic effects, while shared neural alterations associated with MDD and APOE-based AD liability may stem from correlated gene-environment influences, reflecting both the direct and indirect impact of genes.
Contrary to our predictions, parental warmth did not moderate the association between genetic risk and neurocognitive development among either adoptees or non-adoptees. However, in our sample, children’s ratings of parental warmth were very high and showed relatively little variability. Consequently, more in-depth longitudinal investigations with measures spanning cellular to functional systems levels are warranted to elucidate the buffering role of parent–child relationship quality, including parental warmth and responsiveness79. Mounting evidence testifies to the profound impact of parenting on child development. For instance, child maltreatment is linked to epigenetic changes in oxytocin function, which, in turn, trigger structural and functional brain alterations relevant to reward and external attention processes80. Conversely, both rodent and human (adoption) studies indicate that child and adolescent exposure to enriched and emotionally responsive environments can reverse the sequelae of prior ELAs (e.g., restored adult hippocampal neurogenesis, reductions in stress reactivity and biological ageing64,65,67,81). Consequently, the neurobiological mechanisms through which enriched social environments may compensate for the damage inflicted by earlier exposure to harsher milieus, as well as the potential age-specificity of any detected pathways would be certainly worth exploring in the future.
Our research paves the way for several lines of inquiry. First, use of recently discovered genetic risk loci and more liberal significance thresholds for the GRS-contributing SNPs82,83,84 could help elucidate the clinical symptoms and underlying neural circuitry linked to accelerated brain ageing as a function of MDD and/or APOE vs no-APOE AD risk (e.g., anhedonia85,86; sleep disturbances87). Such investigations could also characterise the molecular pathways through which APOE variants (APOE 2 vs APOE 4) may either protect against or increase vulnerability to ageing-related cognitive decline88, thereby optimising screening and intervention for at-risk individuals.
Second, the cellular substrates of gene-perinatal environment interactions in neurodegeneration and psychopathology, particularly those relevant to fluid cognition, warrant further investigation (e.g., E/I imbalance, neurotransmitter-specific alterations3,89). Such inquiries are well-justified by evidence that pre-/perinatal stressors (e.g., malnutrition, maternal trauma, inflammation, psychopathology, substance use) interact with the offspring’s genetic profile to shape their lifespan development, including biological ageing rate90,91,92.
Third, given the domain-specificity of inhibitory control93, the differential neurocognitive alterations on general vs. emotional context-specific tasks, linked to AD vs. MDD risk, are worth probing. Fourth, the relatively small sample of adoptees did not provide us with sufficient statistical power to test for sex differences in any of the documented gene-brain-cognition relationships. Such investigations are worth pursuing because there are sex differences in the prevalence of both AD and MDD94,95, metabolic brain senescence, age-, APOE-4 status-related AD risk, as well as in the neuro(epi)genetic profiles and intergenerational transmission patterns of psychopathology88,96,97,98.
Fifth, the well-documented neurogenetic and adverse life outcome overlap in psychiatric and degenerative brain disorders highlight the importance of disentangling the alterations in neurocognitive development specific to AD and MDD, respectively, from those associated with global vulnerability to psychopathology10,99,100,101. For instance, in our study, the brain areas linked to AD/MDD risk overlap with those of a recently characterised “vulnerability network” implicated in global psychiatric risk, substance use and educational attainment102. Thus, although some regionally constrained effects may exist, disorder-specific pathological profiles (including MDD/AD) may reflect not variability in the topography of the affected brain network, but rather fluctuations in the precise combination of cross-modality (structure/function) deficits.
Sixth, there is a need for systematic investigations of a wider range of ELAs (e.g., poor sleep quality, air pollution, urbanicity) and of likely buffers, be they dispositional/person-related (e.g., purpose in life, self-control, emotion regulation skills, educational attainment) or environmental/lifestyle-related (e.g., green space exposure, nutritional supplement use, aerobic engagement)103,104,105,106,107. Seventh, although we controlled for many demographic variables, lingering differences between adoptees and non-adoptees could have still impacted our reported findings. Our present results based on a relatively small adoptee sample need to be replicated and extended in studies using a combined twin/GRS approach with biological and adoptive families, including an in-depth demographic assessment, well-documented adoption route (e.g., foster care/international/step-family) and pre-adoptive history of adversity. Such investigations could elucidate direct vs. indirect genetic, as well as bidirectional parent–offspring effects on the AD- and MDD-relevant phenotypes108,109.
In sum, we identified overlapping neurodevelopmental mechanisms linked to superior fluid cognition among adoptees at genetic risk for AD and MDD, respectively. The AD-related structural profile replicated the accelerated brain ageing print of this disorder, while highlighting its unexpected, albeit modest, cognitive benefits in early life. The MDD-linked functional profile was the reverse of its neuropathological print and reflected compensatory mechanisms likely related to prior adversity exposure.
Methods
Participants
We used data pre-processed by the ABCD study team (ABCD data release 3.0) from 117 adoptees and 4382 non-adoptees, aged 9–10 years, who were biologically unrelated and provided high-quality data on all scrutinised measures (see Table 1 for detailed demographic information). The majority (72%) were confirmed non step-parent adoptions. In the remaining 28% of the cases, the mother was confirmed to be not biologically related to the child (i.e., adoptive), whereas information on the biological relatedness of the father to the child was missing.
Out-of-scanner measures
Scores on all the measures were released by the ABCD team and are described in detail in110 (see Supplementary Materials 1.2–1.4). Fluid and crystallised cognition were assessed with the National Institutes of Health (NIH) Toolbox. Unadjusted (rather than age-/sex-adjusted) scores were used for both to avoid interference with our numerous confounding variables, which included age and sex (see section on confound residualisation below). Fluid cognition scores reflected average performance on inhibitory control, cognitive flexibility, working memory, processing speed and episodic memory tests. Crystallised cognition scores indicated average performance on receptive vocabulary and oral reading tasks.
Children’s perceptions of parental warmth were gauged with the Acceptance subscale from the Child Report of Behavior Inventory. Concurrent adversity was quantified through parental responses on a measure of unmet material needs, as well as parent and child responses on two measures assessing family conflict and neighbourhood crime.
Neuroimaging data
We used tabulated structural (i.e., cortical thickness) and functional magnetic resonance imaging (fMRI: resting state and task-related) data pre-processed by the ABCD team and mapped onto the 148 regions-of-interest (ROIs) in the Destrieux anatomical atlas (see Supplementary Materials 1.5–1.7). The task fMRI data had been collected during performance of a stop-signal task (SST) which measures the ability to inhibit an ongoing speeded motor response to a “Go” signal110. Our SST analyses focused on the beta and associated standard error (SEM) values derived from the Correct Stop > Correct Go contrast, as estimated with a general linear model (GLM) in the Analysis of Functional NeuroImages (AFNI111). There was no reason to control for behavioural performance because: (1) we only analysed correct trials, and (2) task difficulty was dynamically adjusted to maintain a set number of correct responses across participants110.
Two BOLDM -related estimates were computed for each of the 148 Destrieux ROIs based on the Correct Stop > Correct Go contrast. The first was the difference in standardised GLM beta values between the second and the first run of the SST task. Lower values on this measure typified individuals who became more “brain-efficient” with practice (i.e., correct performance was linked to less neural activity on run 2 relative to run 1, cf.112,113,114). The second BOLDM -related estimate was the average GLM beta value across the two runs of the SST task. Lower values on this measure characterised participants with overall greater neural efficiency, likely indicative of greater functional maturation (cf.51).
Based on the Correct Stop > Correct Go contrast, two BOLDSV-related indices were also estimated for each of the 148 Destrieux ROIs. Both were based on the SEM associated with the GLM beta coefficient for the Correct Stop > Correct Go contrast. The first BOLDSV-related index was computed as the difference between the run 2 and run 1 standardised SEM values. Lower values on this measure identified participants who showed greater stabilisation of the task-related response from run 1 to run 2 (relative to the sample mean). The second index was the average run 1 and run 2 SEM, with lower values typical of participants with a more consistent response to the task-relevant information. Resting state BOLDSV was estimated as an amplitude index of low frequency fluctuations.
Genetic risk scores (GRS)
MDD and AD (full, APOE [chromosome 19:44.4–46.5 Mb] and no-APOE) GRSs were each computed as the weighted sum of risk alleles, significant at GWAS level p ≤ 5 × 10–8. These were derived from the summary statistics of two large GWASs focused on each disorder36,37 (see Supplementary Materials 1.8). The absence of the relevant SNPs (rs7412 and rs429358) from the quality controlled ABCD genetic data prevented us from computing the APOE AD GRS as the sum of e4/e2 alleles40. Hence, as stated above, the APOE AD GRS was estimated as the weighted sum of risk alleles in the APOE region, thereby using the same procedure as for the other GRSs.
Residualisation for confounding variables
To minimise bias in our multivariate brain-behaviour analyses115, only the non-imaging variables were residualised for the following confounders: sex, race (separate dummy-coded variables for “Black”, “Asian”, ‘Mixed Race” regressed simultaneously from the non-imaging variables to account for potential differences between these racial groups and White participants), handedness, serious medical problems, scanner site, material deprivation, family conflict, neighbourhood crime, age at adoption, average modality-specific motion per participant, and chronological age (in order to estimate accelerated/decelerated neurodevelopment relative to the other participants) (see Supplementary Materials 1.9). Due to data (un)availability, only the non-adoptee data were residualised for perinatal adversity, as indexed by a summary score released by the ABCD team and reflecting maternal prenatal care, maternal substance use during pregnancy, prenatal maternal health conditions, prematurity, birth complications and developmental milestones116. The adoptee and non-adoptee data were residualised separately.
MRI and GRS data analysis
To characterise the relationship between MDD/AD risk and neurodevelopmental timing, we used partial least squares correlation (PLS117), a multivariate technique that can identify in a data-driven manner neural patterns (i.e., latent variables or LVs) related to different conditions (task PLS) and/or individual differences variables (behavioural PLS) (see Supplementary Materials 1.10.1). We conducted two behavioural PLS analyses featuring the MDD GRS (both analyses) and either the composite AD GRS (analysis 1) or the APOE- vs no-APOE AD GRSs (analysis 2) in the “behavioural” set. The brain matrix contained the coefficients corresponding to each brain data type, which was modelled as a separate condition (i.e., “cortical thickness”, “Rest BOLDSV” [= amplitude of resting state low frequency fluctuations in BOLD signal, as released by the ABCD team], “SST BOLDM Run 1/2 Up” [= difference between the GLM-derived run 2 and run 1 betas, based on the Correct Stop > Correct Go contrast], “SST BOLDSV Run 1/2 Up” [= difference between the GLM-derived SEMs associated with the run 2 and run 1 beta, respectively, based on the Correct Stop > Correct Go contrast], “SST Run 1/2 BOLDM” [= average of the GLM-derived run 2 and run 1 betas, based on the Correct Stop > Correct Go contrast, as released by the ABCD team], “SST Run 1/2 BOLDSV” [= average of the GLM-derived SEMs associated with the run 2 and run 1 beta, respectively, based on the Correct Stop > Correct Go contrast, as released by the ABCD team]). The adoptees and non-adoptees were modelled as separate groups. LV significance was determined with 5000 permutations, whereas the reliability of each ROI’s contribution to a particular LV was quantified based on the standard error estimates (SEs) from 1000 bootstraps117,118. A bootstrap ratio (BSR) (weight/SE) of 3 in absolute value (conceptually similar to an associated p-value < 0.003) was used as a robustness threshold for all ROIs (cf.117,118).
Three moderated mediation analyses using Hayes’ PROCESS 3.5 macro for the Statistical Package for the Social Sciences (SPSS119) probed whether MDD, composite AD and APOE vs no-APOE AD GRSs predicted distinct neurocognitive developmental patterns among adoptees versus non-adoptees (see Supplemental Materials 1.10.2). Mediation models were tested employing 95% CI (percentile bootstrap, 50,000 bootstraps) with a heteroscedasticity consistent standard error and covariance matrix estimator.
Data availability
The raw data are available at https://nda.nih.gov/abcd upon completion of the relevant data use agreements. The ABCD data repository grows and changes over time. The ABCD data used in this report came from Adolescent Brain Cognitive Development Study (ABCD)—Annual Release 3.0 #901. DOIs can be found at https://doi.org/10.15154/1519007.
Code availability
We used existing code, as specified in the main text with links for free download.
References
Duffy, K. A., McLaughlin, K. A. & Green, P. A. Early life adversity and health-risk behaviors: Proposed psychological and neural mechanisms. Ann. N. Y. Acad. Sci. 1428, 151–169 (2018).
Nelson, C. A. 3rd. & Gabard-Durnam, L. J. Early adversity and critical periods: Neurodevelopmental consequences of violating the expectable environment. Trends Neurosci. 43, 133–143 (2020).
Adjimann, T. S., Argañaraz, C. V. & Soiza-Reilly, M. Serotonin-related rodent models of early-life exposure relevant for neurodevelopmental vulnerability to psychiatric disorders. Transl. Psychiatry 11, 280 (2021).
Beurel, E., Toups, M. & Nemeroff, C. B. The Bidirectional Relationship of Depression and Inflammation: Double Trouble. Neuron 107, 234–256 (2020).
Ho, T. C. & King, L. S. Mechanisms of neuroplasticity linking early adversity to depression: Developmental considerations. Transl. Psychiatry 11, 517 (2021).
Parade, S. H. et al. A systematic review of childhood maltreatment and DNA methylation: candidate gene and epigenome-wide approaches. Transl. Psychiatry 11, 134 (2021).
Yuan, N., Chen, Y., Xia, Y., Dai, J. & Liu, C. Inflammation-related biomarkers in major psychiatric disorders: A cross-disorder assessment of reproducibility and specificity in 43 meta-analyses. Transl. Psychiatry 9, 233 (2019).
Han, L. et al. Brain aging in major depressive disorder: Results from the ENIGMA major depressive disorder working group. Mol. Psychiatry 26, 5124–5139 (2021).
Han, L. et al. Contributing factors to advanced brain aging in depression and anxiety disorders. Transl. Psychiatry 11, 402 (2021).
Kaufmann, T. et al. Common brain disorders are associated with heritable patterns of apparent aging of the brain. Nat. Neurosci. 22, 1617–1623 (2019).
Lyra E Silva, N. M. et al. Pro-inflammatory interleukin-6 signaling links cognitive impairments and peripheral metabolic alterations in Alzheimer’s disease. Transl. Psychiatry 11, 251 (2021).
Cole, J. J. et al. No evidence for differential gene expression in major depressive disorder PBMCs, but robust evidence of elevated biological ageing. Transl. Psychiatry 11, 404 (2021).
Guerrero, A., De Strooper, B. & Arancibia-Cárcamo, I. L. Cellular senescence at the crossroads of inflammation and Alzheimer’s disease. Trends Neurosci. 44, 714–727 (2021).
Brainstorm Consortium et al. Analysis of shared heritability in common disorders of the brain. Science 360, 8757 (2018).
Lutz, M. W., Luo, S., Williamson, D. E. & Chiba-Falek, O. Shared genetic etiology underlying late-onset Alzheimer’s disease and posttraumatic stress syndrome. Alzheimer’s Dementia 16, 1280–1292 (2020).
Norton, M. C. et al. Early parental death and remarriage of widowed parents as risk factors for Alzheimer disease: The Cache County study. Am. J. Geriatr. Psychiatry 19, 814–824 (2011).
Radford, K. et al. Childhood stress and adversity is associated with late-life dementia in aboriginal Australians. Am. J. Geriatr. Psychiatry 25, 1097–1106 (2017).
Tani, Y., Fujiwara, T. & Kondo, K. Association between adverse childhood experiences and dementia in older Japanese adults. JAMA Netw. Open 3, e1920740 (2020).
Yan, Z. & Rein, B. Mechanisms of synaptic transmission dysregulation in the prefrontal cortex: Pathophysiological implications. Mol. Psychiatry 27, 445–465 (2022).
Dafsari, F. S. & Jessen, F. Depression: An underrecognized target for prevention of dementia in Alzheimer’s disease. Transl. Psychiatry 10, 160 (2020).
De Strooper, B. & Karran, E. The cellular phase of Alzheimer’s disease. Cell 164, 603–615 (2016).
Frisoni, G. B. et al. The probabilistic model of Alzheimer disease: The amyloid hypothesis revised. Nat. Rev. Neurosci. 23, 53–66 (2022).
Anthony, R. E., Paine, A. L. & Shelton, K. H. Adverse childhood experiences of children adopted from care: The importance of adoptive parental warmth for future child adjustment. Int. J. Environ. Res. Public Health 16(12), 2212. https://doi.org/10.3390/ijerph16122212 (2019).
Nadeem, E. et al. Long-term effects of pre-placement risk factors on children’s psychological symptoms and parenting stress among families adopting children from foster care. J. Emot. Behav. Disord. 25, 67–81 (2017).
Paine, A. L., Perra, O., Anthony, R. & Shelton, K. H. Charting the trajectories of adopted children’s emotional and behavioral problems: The impact of early adversity and post-adoptive parental warmth. Dev. Psychopathol. 33, 922–936 (2021).
Turney, K. & Wildeman, C. Adverse childhood experiences among children placed in and adopted from foster care: Evidence from a nationally representative survey. Child Abuse Negl. 64, 117–129 (2017).
Simmel, C., Barth, R. P. & Brooks, D. Adopted foster youths’ psychosocial functioning: A longitudinal perspective. Child Fam. Soc. Work 12, 336–348 (2007).
Vandivere, S. & McKlindon, A. The well-being of U.S. children adopted from foster care, privately from the United States and internationally. Adopt. Q. 13, 157–184 (2010).
Adoption Network. US Adoption Statistics. (2021). https://adoptionnetwork.com/adoption-myths-facts/domestic-us-statistics/.
Lawrence, C. R., Carlson, E. A. & Egeland, B. The impact of foster care on development. Dev. Psychopathol. 18, 57–76 (2006).
Belsky, J. Early-life adversity accelerates child and adolescent development. Curr. Dir. Psychol. Sci. 28, 241–246 (2019).
Miller, J. G. et al. Early life stress, frontoamygdala connectivity, and biological aging in adolescence: A longitudinal investigation. Cereb. Cortex 30, 4269–4280 (2020).
Tooley, U. A., Bassett, D. S. & Mackey, A. P. Environmental influences on the pace of brain development. Nat. Rev. Neurosci. 22, 372–384 (2021).
McLaughlin, K. A., Colich, N. L., Rodman, A. M. & Weissman, D. G. Mechanisms linking childhood trauma exposure and psychopathology: A transdiagnostic model of risk and resilience. BMC Med. 18, 96 (2020).
Shen, X. et al. A phenome-wide association and Mendelian randomisation study of polygenic risk for depression in UK Biobank. Nat. Commun. 11, 2301 (2020).
Howard, D. M. et al. Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat. Neurosci. 22, 343–352 (2019).
Kunkle, B. W. et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 51, 414–430 (2019).
Young, A. I., Benonisdottir, S., Przeworski, M. & Kong, A. Deconstructing the sources of genotype-phenotype associations in humans. Science 365, 1396–1400 (2019).
Sierksma, A., Escott-Price, V. & De Strooper, B. Translating genetic risk of Alzheimer’s disease into mechanistic insight and drug targets. Science 370, 61–66 (2020).
Leonenko, G. et al. Genetic risk for Alzheimer Disease is distinct from genetic risk for amyloid deposition. Ann. Neurol. 86, 427–435 (2019).
Dean, D. C. 3rd. et al. Brain differences in infants at differential genetic risk for late-onset Alzheimer disease: A cross-sectional imaging study. JAMA Neurol. 71, 11–22 (2014).
Stevenson, A. J. et al. Childhood intelligence attenuates the association between biological ageing and health outcomes in later life. Transl. Psychiatry 9, 323 (2019).
Colich, N. L., Rosen, M. L., Williams, E. S. & McLaughlin, K. A. Biological aging in childhood and adolescence following experiences of threat and deprivation: A systematic review and meta-analysis. Psychol. Bull. 146, 721–764 (2020).
Mills, K. L. et al. Inter-individual variability in structural brain development from late childhood to young adulthood. Neuroimage 242, 118450 (2021).
Monninger, M. et al. The long-term impact of early life stress on orbitofrontal cortical thickness. Cereb. Cortex 30, 1307–1317 (2020).
Parker, N. et al. Corticosteroids and regional variations in thickness of the human cerebral cortex across the lifespan. Cereb. Cortex 30, 575–586 (2020).
Whitaker, K. J. et al. Adolescence is associated with genomically patterned consolidation of the hubs of the human brain connectome. Proc. Natl. Acad. Sci. USA. 113, 9105–9110 (2016).
Constantinidis, C. & Luna, B. Neural substrates of inhibitory control maturation in adolescence. Trends Neurosci. 42, 604–616 (2019).
Paine, A. L., Burley, D., Anthony, R., Van Goozen, S. & Shelton, K. H. The neurocognitive profiles of children adopted from care and their emotional and behavioral problems at home and school. Child Neuropsychol. 27, 17–36 (2021).
Romer, A. L. & Pizzagalli, D. A. Is executive dysfunction a risk marker or consequence of psychopathology? A test of executive function as a prospective predictor and outcome of general psychopathology in the adolescent brain cognitive development study®. Dev. Cogn. Neurosci. 51, 100994 (2021).
Pas, P., Hulshoff Pol, H. E., Raemaekers, M. & Vink, M. Self-regulation in the pre-adolescent brain. Dev. Cogn. Neurosci. 51, 101012 (2021).
Kebets, V. et al. Fronto-limbic neural variability as a transdiagnostic correlate of emotion dysregulation. Transl. Psychiatry 11, 545 (2021).
Nomi, J. S., Bolt, T. S., Ezie, C., Uddin, L. Q. & Heller, A. S. Moment-to-moment BOLD signal variability reflects regional changes in neural flexibility across the lifespan. J. Neurosci. 37, 5539–5548 (2017).
Månsson, K. N. T. et al. Moment-to-moment brain signal variability reliably predicts psychiatric treatment outcome. Biol. Psychiat. https://doi.org/10.1016/j.biopsych.2021.09.026 (2021).
Wang, H. et al. The longitudinal relationship between BOLD signal variability changes and white matter maturation during early childhood. Neuroimage 242, 118448 (2021).
Waschke, L., Kloosterman, N., Obleser, J. & Garrett, D. D. Behaviour needs neural variability. Neuron 109, 1–16 (2021).
Kaiser, R. H., Andrews-Hanna, J. R., Wager, T. D. & Pizzagalli, D. Large-scale network dysfunction in major depressive disorder: Meta-analysis of resting-state functional connectivity. JAMA Psychiat. 72, 603–611 (2015).
Kvavilashvili, L., Niedźwieńska, A., Gilbert, S. J. & Markostamou, I. Deficits in spontaneous cognition as an early marker of alzheimer’s disease. Trends Cogn. Sci. 24, 285–301 (2020).
Zabelina, D. L. & Andrews-Hanna, J. Dynamic network interactions supporting internally-oriented cognition. Curr. Opin. Neurobiol. 40, 86–93 (2016).
Hyde, L. W. et al. An ecological approach to understanding the developing brain: Examples linking poverty, parenting, neighborhoods, and the brain. Am. Psychol. 75, 1245–1259 (2020).
Marceau, K. et al. Parenting and prenatal risk as moderators of genetic influences on conduct problems during middle childhood. Dev. Psychol. 55, 1164–1181 (2019).
Pace, C. S., Di Folco, S., Guerriero, V. & Muzi, S. Late-adopted children grown up: A long-term longitudinal study on attachment patterns of adolescent adoptees and their adoptive mothers. Attach. Hum. Dev. 21, 372–388 (2019).
Cioffi, C. C. et al. Does maternal warmth moderate longitudinal associations between infant attention control and children’s inhibitory control?. Infant Child Dev. 29, e2147 (2020).
Gapp, K. et al. Potential of environmental enrichment to prevent transgenerational effects of paternal trauma. Neuropsychopharmacology 41, 2749–2758 (2016).
Gunnar, M. R., DePasquale, C. E., Reid, B. M., Donzella, B. & Miller, B. S. Pubertal stress recalibration reverses the effects of early life stress in postinstitutionalized children. Proc. Natl. Acad. Sci. USA. 116, 23984–23988 (2019).
Humphreys, K. L. et al. High-quality foster care mitigates callous-unemotional traits following early deprivation in boys: A randomized controlled trial. J. Am. Acad. Child Adolesc. Psychiatry 54, 977–983 (2015).
Luby, J. L., Baram, T. Z., Rogers, C. E. & Barch, D. M. neurodevelopmental optimization after early-life adversity: Cross-species studies to elucidate sensitive periods and brain mechanisms to inform early intervention. Trends Neurosci. 43, 744–751 (2020).
Reuben, J. D. et al. Warm parenting and effortful control in toddlerhood: Independent and interactive predictors of school-age externalizing behavior. J. Abnorm. Child Psychol. 44, 1083–1096 (2016).
Schloesser, R. J., Lehmann, M., Martinowich, K., Manji, H. K. & Herkenham, M. Environmental enrichment requires adult neurogenesis to facilitate the recovery from psychosocial stress. Mol. Psychiatry 15, 1152–1163 (2010).
Abdellaoui, A. & Verweij, K. Dissecting polygenic signals from genome-wide association studies on human behaviour. Nat. Hum. Behav. 5, 686–694 (2021).
Nievergelt, C. M. et al. International meta-analysis of PTSD genome-wide association studies identifies sex- and ancestry-specific genetic risk loci. Nat. Commun. 10, 4558 (2019).
Wojcik, G. L. et al. Genetic analyses of diverse populations improves discovery for complex traits. Nature 570, 514–518 (2019).
Si, R., Rowe, J. B. & Zhang, J. Functional localization and categorization of intentional decisions in humans: A meta-analysis of brain imaging studies. Neuroimage 242, 118468 (2021).
Kupis, L. et al. Brain dynamics underlying cognitive flexibility across the lifespan. Cereb. Cortex 31, 5263–5274 (2021).
Arnatkeviciute, A., Fulcher, B. D., Bellgrove, M. A. & Fornito, A. Where the genome meets the connectome: Understanding how genes shape human brain connectivity. Neuroimage 244, 118570 (2021).
Mittal, C., Griskevicius, V., Simpson, J. A., Sung, S. & Young, E. S. Cognitive adaptations to stressful environments: When childhood adversity enhances adult executive function. J. Pers. Soc. Psychol. 109, 604–621 (2015).
Homberg, J. R. & Jagiellowicz, J. A neural model of vulnerability and resilience to stress-related disorders linked to differential susceptibility. Mol. Psychiatry 27, 514–524 (2022).
Fang, W. et al. APOE4 genotype exacerbates the depression-like behavior of mice during aging through ATP decline. Transl. Psychiatry 11, 507 (2021).
Toumbelekis, M., Liddell, B. J. & Bryant, R. A. Secure attachment priming protects against relapse of fear in Young adults. Transl. Psychiatry 11, 584 (2021).
Nishitani, S. et al. A multi-modal MRI analysis of brain structure and function in relation to OXT methylation in maltreated children and adolescents. Transl. Psychiatry 11, 589 (2021).
Rule, L., Yang, J., Watkin, H., Hall, J. & Brydges, N. M. Environmental enrichment rescues survival and function of adult-born neurons following early life stress. Mol. Psychiatry 26, 1898–1908 (2021).
Bellenguez, C., Grenier-Boley, B. & Lambert, J. C. Genetics of Alzheimer’s disease: Where we are, and where we are going. Curr. Opin. Neurobiol. 61, 40–48 (2020).
Wightman, D. P. et al. A genome-wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer’s disease. Nat. Genet. 53, 1276–1282 (2021).
Fulton-Howard, B. et al. Greater effect of polygenic risk score for Alzheimer’s disease among younger cases who are apolipoprotein E-ε4 carriers. Neurobiol. Aging 99, 101.e1-101.e9 (2021).
Chen, X. et al. Kynurenines increase MRS metabolites in basal ganglia and decrease resting-state connectivity in frontostriatal reward circuitry in depression. Transl. Psychiatry 11, 456 (2021).
Guffanti, G. et al. Depression genetic risk score is associated with anhedonia-related markers across units of analysis. Transl. Psychiatry 9, 236 (2019).
Winer, J. R. et al. Tau and β-amyloid burden predict actigraphy-measured and self-reported impairment and misperception of human sleep. J. Neurosci. 41, 7687–7696 (2021).
Zhao, N. et al. Alzheimer’s risk factors age, APOE genotype, and sex drive distinct molecular pathways. Neuron 106, 727-742.e6 (2020).
Tochitani, S. et al. GABAA receptors and maternally derived taurine regulate the temporal specification of progenitors of excitatory glutamatergic neurons in the mouse developing cortex. Cereb. Cortex 31, 4554–4575 (2021).
Krontira, A. C., Cruceanu, C. & Binder, E. B. Glucocorticoids as mediators of adverse outcomes of prenatal stress. Trends Neurosci. 43, 394–405 (2020).
Sawyer, K. M., Zunszain, P. A., Dazzan, P. & Pariante, C. M. Intergenerational transmission of depression: Clinical observations and molecular mechanisms. Mol. Psychiatry 24, 1157–1177 (2019).
Coley, E. & Hsiao, E. Y. Malnutrition and the microbiome as modifiers of early neurodevelopment. Trends Neurosci. 44, 753–764 (2021).
Zhuang, Q. et al. Segregating domain-general from emotional context-specific inhibitory control systems: Ventral striatum and orbitofrontal cortex serve as emotion-cognition integration hubs. Neuroimage 238, 118269 (2021).
Bangasser, D. A. & Cuarenta, A. Sex differences in anxiety and depression: circuits and mechanisms. Nat. Rev. Neurosci. 22, 674–684 (2021).
Ferretti, M. T. et al. Sex differences in Alzheimer disease: The gateway to precision medicine. Nat. Rev. Neurol. 14, 457–469 (2018).
Goyal, M. S. et al. Persistent metabolic youth in the aging female brain. Proc. Natl. Acad. Sci. USA. 116, 3251–3255 (2019).
Issler, O. et al. Sex-specific role for the long non-coding RNA LINC00473 in depression. Neuron 106, 912–926 (2020).
Paden, W. et al. Sex differences in adult mood and in stress-induced transcriptional coherence across mesocorticolimbic circuitry. Transl. Psychiatry 10, 59 (2020).
Blanco, C. et al. Psychiatric disorders and risk for multiple adverse outcomes: A national prospective study. Mol. Psychiatry 26, 907–916 (2021).
Cross-Disorder Group of the Psychiatric Genomics Consortium. Genomic relationships, novel loci, and pleiotropic mechanisms across eight psychiatric disorders. Cell 179, 1469–1482 (2019).
Radonjić, N. V. et al. Structural brain imaging studies offer clues about the effects of the shared genetic etiology among neuropsychiatric disorders. Mol. Psychiatry 26, 2101–2110 (2021).
Taquet, M. et al. A structural brain network of genetic vulnerability to psychiatric illness. Mol. Psychiatry 26, 2089–2100 (2021).
Andersen, S. H., Richmond-Rakerd, L. S., Moffitt, T. E. & Caspi, A. Nationwide evidence that education disrupts the intergenerational transmission of disadvantage. Proc. Natl. Acad. Sci. USA 118, e2103896118 (2021).
Cheng, W. et al. Sleep duration, brain structure, and psychiatric and cognitive problems in children. Mol. Psychiatry 26, 3992–4003 (2021).
Madison, A. A. et al. Omega-3 supplementation and stress reactivity of cellular aging biomarkers: An ancillary substudy of a randomized, controlled trial in midlife adults. Mol. Psychiatry 26, 3034–3042 (2021).
Miller, J. G., Dennis, E. L., Heft-Neal, S., Jo, B. & Gotlib, I. H. Fine particulate air pollution, early life stress, and their interactive effects on adolescent structural brain development: A longitudinal tensor-based morphometry study. Cerebr. Cortex 1, 346 (2021).
Xiao, K. et al. Beneficial effects of running exercise on hippocampal microglia and neuroinflammation in chronic unpredictable stress-induced depression model rats. Transl. Psychiatry 11, 461 (2021).
Friedman, N. P., Banich, M. T. & Keller, M. C. Twin studies to GWAS: There and back again. Trends Cogn. Sci. 25, 855–869 (2021).
McAdams, T. A. et al. The relationship between parental depressive symptoms and offspring psychopathology: evidence from a children-of-twins study and an adoption study. Psychol. Med. 45, 2583–2594 (2015).
Casey, B. J. et al. The adolescent brain cognitive development (ABCD) study: Imaging acquisition across 21 sites. Dev. Cogn. Neurosci. 32, 43–54 (2018).
Cox, R. W. AFNI: Software for analysis and visualization of functional magnetic 377 resonance neuroimages. Comput. Biomed. Res. 29, 162–173 (1996).
Gold, B. T., Kim, C., Johnson, N. F., Kryscio, R. J. & Smith, C. D. Lifelong bilingualism maintains neural efficiency for cognitive control in aging. J. Neurosci. 33, 387–396 (2013).
Heinzel, S. et al. Working memory load-dependent brain response predicts behavioral training gains in older adults. J. Neurosci. 34, 1224–1233 (2014).
Neubauer, A. C. & Fink, A. Intelligence and neural efficiency. Neurosci. Biobehav. Rev. 33, 1004–1023 (2009).
Winkler, A. M., Renaud, O., Smith, S. M. & Nichols, T. E. Permutation inference for canonical correlation analysis. Neuroimage 220, 117065 (2020).
Barch, D. M. et al. Demographic, physical and mental health assessments in the adolescent brain and cognitive development study: Rationale and description. Dev. Cogn. Neurosci. 32, 55–66 (2018).
Krishnan, A., Williams, L. J., McIntosh, A. R. & Abdi, H. Partial least squares (PLS) methods for neuroimaging: A tutorial and review. Neuroimage 56, 455–475 (2011).
McIntosh, A. R. & Lobaugh, N. J. Partial least squares analysis of 399 neuroimaging data: applications and advances. Neuroimage 23(Suppl 1), S250–S263 (2004).
Hayes, A. F. Introduction to Mediation, Moderation, and Conditional Process Analysis: A Regression-Based Approach 2nd edn. (New York, 2018).
Acknowledgements
Data used in the preparation of this article were obtained from the Adolescent Brain Cognitive Development (ABCD) Study (https://abcdstudy.org), held in the NIMH Data Archive (NDA). This is a multisite, longitudinal study designed to recruit more than 10,000 children age 9–10 and follow them over 10 years into early adulthood. The ABCD Study is supported by the National Institutes of Health and additional federal partners under Award Numbers U01DA041048, U01DA050989, U01DA051016, U01DA041022, U01DA051018, U01DA051037, U01DA050987, U01DA041174, U01DA041106, U01DA041117, U01DA041028, U01DA041134, U01DA050988, U01DA051039, U01DA041156, U01DA041025, U01DA041120, U01DA051038, U01DA041148, U01DA041093, U01DA041089, U24DA041123, U24DA041147. A full list of supporters is available at https://abcdstudy.org/federal-partners.html. A listing of participating sites and a complete listing of the study investigators can be found at https://abcdstudy.org/consortium_members/. ABCD consortium investigators designed and implemented the study and/or provided data but did not necessarily participate in analysis or writing of this report. This manuscript reflects the views of the authors and may not reflect the opinions or views of the NIH or ABCD consortium investigators. Amy L. Paine was funded by the Economic and Social Research Council (Grant Reference: ES/T00049X/1).
Author information
Authors and Affiliations
Contributions
R.P., A.P., V.E.P. and K.S. designed the research and interpreted the data; R.P. analysed the data and wrote the first draft; A.P., V.E.P. and K.S. edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Petrican, R., Paine, A.L., Escott-Price, V. et al. Overlapping brain correlates of superior cognition among children at genetic risk for Alzheimer’s disease and/or major depressive disorder. Sci Rep 13, 984 (2023). https://doi.org/10.1038/s41598-023-28057-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-28057-6
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
